
Method Article
Activated Cross-linked Agarose for the Rapid Development of Affinity Chromatography Resins - Antibody Capture as a Case Study
In This Article
Summary
In this procedure, a DsRed-based epitope ligand is immobilized to produce a highly selective affinity resin for the capture of monoclonal antibodies from crude plant extracts or cell culture supernatants, as an alternative to Protein A.
Abstract
The purification of monoclonal antibodies (mAbs) is commonly achieved by Protein A affinity chromatography, which can account for up to 25% of the overall process costs. Alternative, cost-effective capture steps are therefore valuable for industrial-scale manufacturing, where large quantities of a single mAb are produced. Here we present a method for the immobilization of a DsRed-based epitope ligand to a cross-linked agarose resin allowing the selective capture of the HIV-neutralizing antibody 2F5 from crude plant extracts without using Protein A. The linear epitope ELDKWA was first genetically fused to the fluorescent protein DsRed and the fusion protein was expressed in transgenic tobacco (Nicotiana tabacum) plants before purification by immobilized metal-ion affinity chromatography. Furthermore, a method based on activated cross-linked agarose was optimized for high ligand density, efficient coupling and low costs. The pH and buffer composition and the soluble ligand concentration were the most important parameters during the coupling procedure, which was improved using a design-of-experiments approach. The resulting affinity resin was tested for its ability to selectively bind the target mAb in a crude plant extract and the elution buffer was optimized for high mAb recovery, product activity and affinity resin stability. The method can easily be adapted to other antibodies with linear epitopes. The new resins allow gentler elution conditions than Protein A and could also reduce the costs of an initial capture step for mAb production.
Introduction
Biopharmaceutical products are important for the treatment of a broad spectrum of diseases in nearly every branch of medicine1. Monoclonal antibodies (mAbs) dominate the biopharmaceutical market, with worldwide sales expected to reach almost €110 billion in 20202. The favored expression platform for mAbs are Chinese hamster ovary cell lines, which typically produce high mAb titers of up to 10 g∙L-1 in the culture supernatant3,4. However, the production of mAbs in mammalian cell cultures is expensive due to the high cost of the medium and the need for sterile fermentation5. Alternative expression platforms such as plants potentially offer a faster, simpler, less expensive and more scalable approach for industrial manufacturing6,7.
In addition to the costs associated with mammalian cell cultures, the widespread use of Protein A affinity chromatography for product capture is a major cost driver for the industrial production of mAbs. Protein A is naturally found on the surface of Staphylococcus aureus cells and it binds to the fragment crystallizable (Fc) region of certain murine and human antibodies, thereby acting as a defense mechanism to evade the humoral immune system8. Protein A has become the industry gold standard for the capture of mAbs from cell culture supernatants and is also widely used by the research community because it is highly selective, typically achieving mAb purities of ~95% in a single step8. Unsurprisingly, sales of Protein A over the last two decades have closely mirrored the sales of mAbs8. Depending on the production scale, the costs of Protein A can correspond to more than 25% of the total process costs and thereby affect the market price of therapeutic mAbs, which can be up to €2,000 g-1 5,9. Therefore, alternative chromatography resins with a similar purification performance have the potential to substantially reduce production costs, making antibody-based therapies accessible for a larger number of patients10,11,12. Such alternatives may also circumvent the disadvantages of Protein A chromatography, including the harsh elution conditions at low pH (typically <3.5) that can potentially cause mAbs to undergo conformational changes that promote aggregation13. Importantly, Protein A is selective only for the Fc region of certain IgG subclasses, so non-functional molecules with truncated binding domains may co-purify with the intact product5, whereas mAb derivaties such as single-chain variable fragments do not bind to Protein A at all.
Here, we describe an alternative affinity chromatography resin for the capture of the HIV-neutralizing mAb 2F5 using its linear epitope ELDKWA (one letter amino acid code)5,14. We genetically fused the 2F5 epitope to the C-terminus of the fluorescent protein DsRed, which functioned as a carrier and reporter molecule, and produced the resulting protein DsRed-2F5-Epitope (DFE) in transgenic tobacco (Nicotiana tabacum) plants. DFE was purified by single-step immobilized metal-ion affinity chromatography (IMAC). The immobilization of the purified DFE affinity ligand onto a cross-linked agarose resin was achieved by chemical coupling using N-hydroxysuccinimide (NHS)-activated cross-linked agarose columns. Statistical experimental designs were then used to optimize the immobilization procedure and coupling efficiency15. The purification strategy for mAb 2F5 was evaluated in terms of antibody purity, yield and ligand stability. In contrast to Protein A, which binds the Fc region, DFE bound to the complementarity-determining region of 2F5, ensuring the purification of molecules with an intact paratope. Our concept can easily be adapted to any mAb with a linear epitope or to other peptide-based affinity ligands which can be easily identified by microarray studies16.

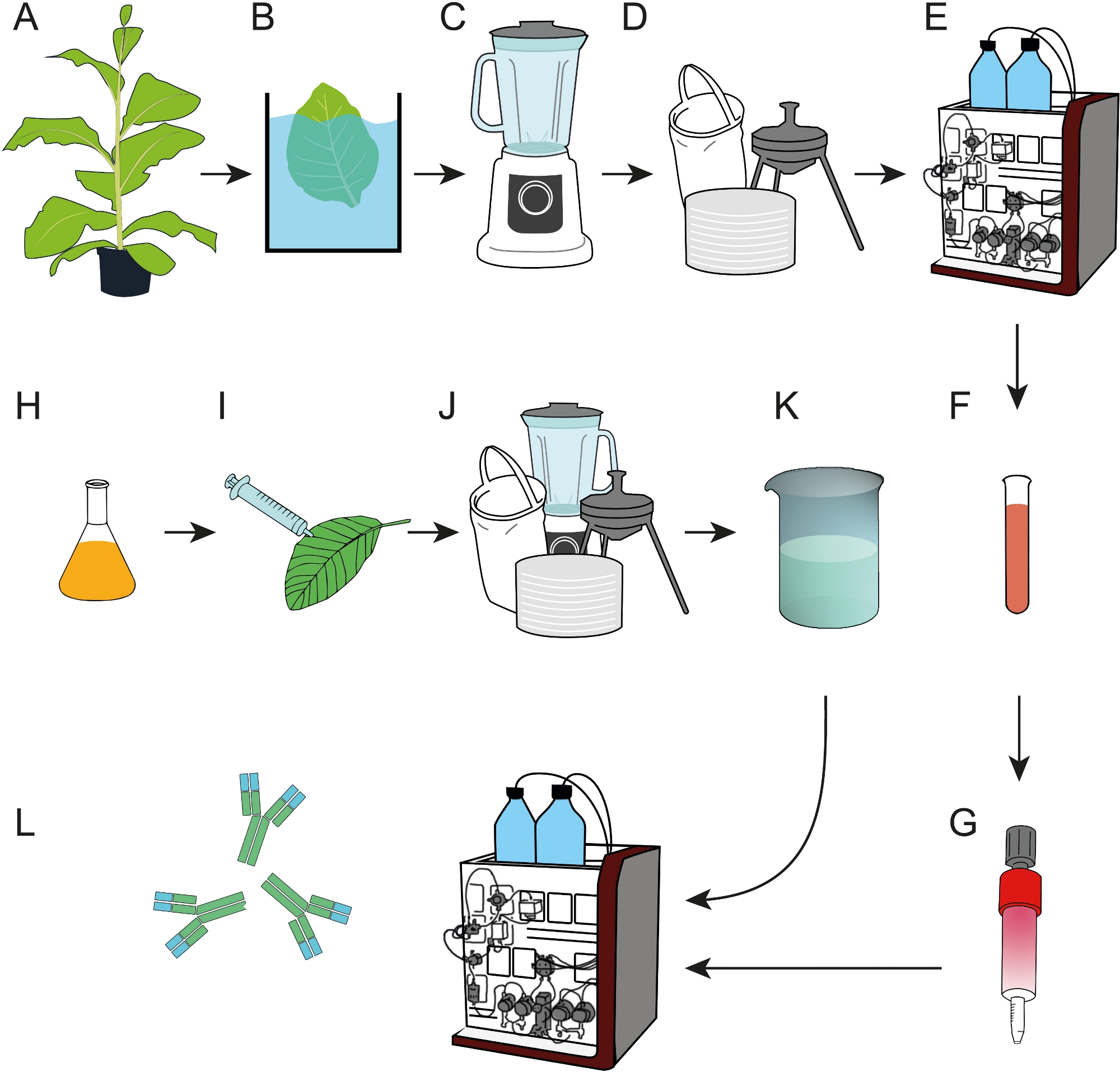
Figure 1: Process flow scheme for the preparation of epitope affinity resins that can be used for the capture of mAbs from crude plant extracts or cell culture supernatants. (A) The affinity ligand DFE was expressed in transgenic tobacco plants. A heat precipitation step (B) was included before harvested leaves were homogenized (C). (D) The crude plant extract was clarified by bag filtration, depth filtration and 0.2 µm sterile filtration. (E) DFE was then purified by IMAC. (F, G) The purified DFE affinity ligand was immobilized on EDC/NHS-activated cross-linked agarose columns. (H) Bacterial cultures carrying T-DNA encoding antibody 2F5 were used for transient expression in N. benthamiana plants (I) grown in a phytotron. (J) N. benthamiana leaves were harvested and processed as described in D. (K) mAb 2F5 was purified from the clarified extract using the DFE affinity columns (L). Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. Cultivate the Transgenic Tobacco Plants
NOTE: The design of the DFE fusion protein and the generation of transgenic plants are described elsewhere5,17.
- Germinate the transgenic tobacco seedlings in soil. Irrigate the plants with a 1.0 g·L-1 fertilizer solution. Transfer the plants to single, 100 mm x 100 mm x 60 mm (length, width, height) pots when they grow to a diameter of ~0.04 m.
- Cultivate the transgenic tobacco plants in a greenhouse with a 16-h photoperiod (25/22 °C light/dark temperature regime), with 70% relative humidity and automated fertilization with a 1.0 g·L-1 fertilizer solution for 15 min every hour.
- After 7 weeks, harvest all leaves except the four cotyledon leaves at the base of the plant stem. Immediately process the harvested leaves as described below.
2. Heat Precipitation of Host Cell Proteins
- Set up a water bath with a 20 L working volume in a stainless-steel heated vessel (0.3 m x 0.3 m x 0.3 m). At the start, place a magnetic pump in the water bath to agitate the liquid permanently with a flow of 5 L min-1. Mount an adjustable thermostat on the water bath for temperature control (steps 2.4 and 2.8).
CAUTION: All the following steps up to 2.10 involve the handling of hot liquid. Wear appropriate personal protective equipment including thermally insulated gloves and goggles. - Add 15 L of deionized water to the water bath and heat the liquid to 70 °C.
- Place a 10 L bucket filled with 5 L of deionized water on a magnetic stirrer. Add sodium phosphate to a final concentration of 120 mM. Adjust the pH to 8.14 using 10 M hydrochloric acid. When all the components have dissolved, add the resulting concentrated blanching buffer (5 L) to the water bath from step 2.2.
- Agitate the water bath until the temperature reaches 70 °C. Use 10 M hydrochloric acid to adjust the pH to 8.14 if necessary. Continue agitating for at least 15 min after the required temperature and pH are reached to ensure that the entire assembly is in thermal equilibrium.
- Prepare a 20 L bucket (0.3 m x 0.3 m x 0.3 m) filled with deionized water at a temperature of ~17 °C (required for step 2.9).
- Prepare 400 g aliquots of the harvested tobacco leaves from step 1.3. Place one aliquot in a perforated basket (0.2 m x 0.2 m x 0.2 m; pore width at least 0.02 m x 0.02 m). Avoid overfilling the basket with plant material or packing the leaves too tightly to avoid damaging the tissue.
- Fully submerge the basket in the hot blanching buffer from step 2.4 and make sure all leaves remain under the liquid surface. Use a thermostable silicone spoon to hold the leaves under the surface if necessary.
- Incubate the tobacco leaves for 1.5 min in the blanching fluid while the pump is still agitating the liquid. Monitor the liquid temperature during the entire incubation period. Avoid blocking the pump inlet with tobacco leaves.
- Remove the basket of tobacco leaves from the blanching buffer and drain residual buffer from the leaves for 30 s. Transfer the basket to the bucket filled with cold water (from step 2.5) and immerse the leaves for 30 s. Remove the basket and drain residual water from the leaves for 30 s before homogenization (step 3).
- Repeat steps 2.6 to 2.9 with fresh aliquots from 2.6 until the entire harvested biomass is processed. Constantly monitor and adjust the pH and the temperature of the blanching buffer in the water bath.
NOTE: Blanched plant material can be stored on ice for up to 30 min when several blanching cycles are required to process the entire biomass. Blanched leaves can also be stored in vacuum-sealed bags at –80 °C for at least 3 weeks. However, immediate processing is recommended because prolonged storage can reduce the DFE yield.
3. Protein Extraction and Clarification
CAUTION: The following steps involve a blender with rotating blades. Do not work in the blender vessel when powered or mounted on the motor unit.
- Place 150 g (wet mass) of blanched tobacco leaves (step 2.9) in the blender vessel and add 450 mL of extraction buffer (50 mM sodium phosphate, 500 mM sodium chloride, 10 mM sodium disulfite, pH 7.5). Close the blender cap tightly to prevent the spilling of plant material or buffer.
- Homogenize the leaves for 3x for 30 s, with 30 s breaks between each pulse. Ensure that all leaves are homogenized and that none stick to the top of the blender vessel. If necessary, open the blender during the breaks and push down leaves that stick at the upper part of the vessel, using a clean silicone spoon.
- After homogenization, take a 1.0 mL sample of the homogenate for subsequent analysis (step 7).
- Collect the homogenate in a vessel of adequate size (e.g. if working with a total biomass of 1.0 kg, use a vessel with a capacity of at least a 5 L). Repeat steps 3.1 to 3.3 until all of the blanched biomass is homogenized.
- Mount a bag filter into a filter mount and place another adequately sized vessel (see 3.4) beneath the assembly. Apply the homogenate to the bag filter at a rate of ~0.15 L min-1. Take samples of the bag filtrate after each liter for subsequent analysis (step 7) and measure the turbidity of the bag filtrate pool as a 1:10 dilution in extraction buffer using a turbidimeter or similar device.
- Further clarify the bag filtrate pool using 0.02 m2 of a K700 (top) and KS50 (bottom) depth filter layer combination per liter of bag filtrate pool. Apply a flow rate of 3.0 L min-1·m-2 up to a maximum pressure of 0.2 MPa. Then remove residual particles by passing the clarified filtrate through a 0.2 µm sterile filter as previously described5.
4. Purification of the DFE Affinity Ligand
- Prepare a fast protein liquid chromatography system by flushing with elution buffer (10 mM sodium phosphate, 300 mM imidazole, pH 7.4), wash buffer (10 mM sodium phosphate, pH 7.4) and equilibration buffer (20 mM sodium phosphate, 500 mM sodium chloride, pH 7.4). Mount a column containing ~50 mL of chelating cross-linked agarose resin per kilogram of leaf biomass (~2.5 L depth filtrate).
- Charge the column with nickel ions by applying 5 column volumes (CV) of 200 mM nickel sulfate solution and wash it with 5 CV of elution buffer. Use a flow rate of 50 cm h-1 and monitor the UV signals at 260, 280 and 558 nm for all subsequent chromatography steps.
- Equilibrate the column with 10 CV of equilibration buffer. Then load 50 CV of the clarified plant extract (from step 3.6) onto the conditioned column.
- Wash the column with 10 CV of wash buffer. Elute the DFE fusion protein with 5 CV elution buffer and collect the product-containing fraction once the UV signals at 280 nm and 558 nm have increased to more than 5 mAU above the baseline.
- Take a 0.2 mL sample from the elution fraction in order to measure the total soluble protein (TSP) concentration (step 7.1), DFE concentration (steps 7.2 and 7.3), and DFE purity (step 7.4).
- Mount a column containing ~50 mL of cross-linked dextran resin on a chromatography system. Equilibrate the column with 5 CV of coupling buffer (200 mM HEPES, 500 mM sodium chloride, pH 8.5). Inject 10 mL of the DFE elution fraction (step 4.4) for buffer exchange and monitor the UV absorbance at 280 and 558 nm.
- Collect the DFE-containing fraction once the UV signals at 280 nm and 558 nm have increased to more than 5 mAU above the baseline. Take a 0.2 mL sample and determine the TSP concentration, DFE concentration, and DFE purity (step 7).
- Concentrate the purified DFE sample (from step 4.7) to 15 g L-1 using a centrifugal concentrator tube at 3,000 x g at 4 °C for 30 min in a centrifuge. Continue with the coupling reaction (step 5).
NOTE: Store the DFE solution at 4 °C if the concentration or coupling steps cannot be carried out immediately.
5. Coupling DFE to the Activated Cross-linked Agarose Resin
NOTE: Do not replace the isopropanol used for the storage of NHS-activated columns until all equipment and solutions for coupling are ready. Never let the columns run dry.
- Setup a design of experiments (DoE) model to optimize the coupling of DFE to the activated resin, with pH, buffer composition and DFE concentration as factors. The details of the DoE method are described elsewhere15.
- Prepare the affinity ligand solution (from step 4.8) in a concentration range from 0.5–15 g·L-1 or as defined in the DoE and store it on ice until the coupling reaction is ready (step 5.5). Fill at least ten 2 mL syringes with the DFE solution and prepare an adapter to mount the syringes on NHS-activated cross-linked agarose columns with a bed volume of 1.0 mL.
- For every 10 columns used for coupling, prepare 30 mL of deactivation solution (0.5 M ethanolamine, 0.5 M sodium chloride, pH 8.3), 30 mL of low-pH solution (0.1 M sodium acetate, 0.5 M sodium chloride, pH 4.0) and 10 mL of storage solution (0.05 M disodium phosphate, 0.1% (m/v) sodium azide, pH 7.0).
- Prepare 20 mL of 1 mM hydrochloric acid in a tube and incubate it on ice for at least 20 min. Prepare a precision scale to monitor the flow-through fractions for all steps during the coupling reaction (step 5.7).
- Open a sealed NHS-activated cross-linked agarose column and mount the syringe adapter at the column inlet. Prevent any air entering the column by applying a drop of buffer onto the adapter inlet before connecting it to the syringe.
- Wash the column with 6 mL of ice-cold 1 mM hydrochloric acid (from step 5.4) at a flow rate of <1 mL min-1 and immediately proceed to step 5.7.
- Inject 1.5 mL of DFE solution (step 5.2) using a 2 mL syringe at a flow rate of <1 mL min-1 and collect the flow-through fraction on a precision scale (step 5.4) for subsequent analysis (step 7). Seal the column at both ends and incubate for 15–45 min at 22 °C, depending on the DoE setup.
- Inject 6 mL of deactivation solution followed by 6 mL of low-pH solution at a flow rate of <1 mL min-1 to remove non-covalently bound ligands from the resin. Then inject 6 mL of deactivation solution and incubate the column for 15 min.
- Inject 6 mL of low-pH solution into the column, followed by 6 mL of deactivation solution. Then inject another 6 mL of low-pH solution into the column.
- Inject 2 mL of storage solution into the column and store at 4 °C.
6. Testing the Purification of mAbs from Clarified Plants Extracts
- Prepare 100 mL of clarified plant extract containing 2F55 or the supernatant from the preferred cell-based expression system, also containing 2F5.
- Prepare equilibration buffer (20 mM sodium phosphate, 500 mM sodium chloride, pH 7.4), low-pH elution buffer (0.05 M citrate, 0.05 M sodium chloride, pH 4.0–3.25), and high-ionic-strength elution buffer (1.0–4.0 M magnesium chloride, 0.1 M HEPES, pH 8.0)
- Flush the chromatography system with the buffers. Mount a DFE affinity column (from step 5.10) on the chromatography system and equilibrate with 5 CV of equilibration buffer at a flow rate of 1.0 mL min-1. Monitor the UV absorbance at 280 nm.
NOTE: Loading plant extract or cell culture supernatant onto the column can cause an increase in backpressure. Set a high-pressure alert at 0.2 MPa to avoid damage to the chromatography system or DFE column. - Load 80 mL of the clarified plant extract or supernatant (step 6.1) onto the column at a flow rate of 0.5 mL min-1 to guarantee a contact time of 2 min. Collect the flow-through samples in 2 mL fractions for breakthrough curve reconstruction (step 7.3). Store the flow-through samples at 4 °C if immediate sample analysis is not possible.
- Wash the column with 6 CV of equilibration buffer. Collect a sample of the wash at the beginning, middle and end of this step.
- Elute mAb 2F5 with 5 CV of low-pH elution buffer or high-ionic-strength elution buffer (0.1 M HEPES, 1.25 M magnesium chloride, pH 8.0). Collect the DFE fraction when the UV 280 nm signal has increased to 5 mAU above the baseline.
- Optimize the elution buffer for each epitope–antibody pair. For 2F5, 1.25 M magnesium chloride achieved an optimal balance between product recovery and ligand stability.
NOTE: The magnesium chloride solution is prone to precipitation. Therefore, dissolve the magnesium chloride in ~700 mL of water. Separately dissolve the HEPES in 100 mL of water and adjust the pH to 8.0. Add the dissolved magnesium chloride solution to the HEPES solution and add water to a final volume of 1.0 L. Do not adjust the pH after dissolving the magnesium chloride because this will cause precipitation.
- Optimize the elution buffer for each epitope–antibody pair. For 2F5, 1.25 M magnesium chloride achieved an optimal balance between product recovery and ligand stability.
- Analyze all samples taken during steps 6.4–6.6 using the Bradford method, lithium dodecylsulfate polyacrylamide gel electrophoresis (LDS-PAGE) and surface plasmon resonance (SPR) spectroscopy (step 7).
7. Sample Analysis
- Measure the TSP concentration using the Bradford method18,19.
- In triplicate, pipette 2.5 µL of each sample into a single well of a 96-well plate. Use eight bovine serum albumin (BSA) standards in triplicate covering the range 0–2,000 mg·L-1.
- Add 200 µL of Bradford reagent to each well and mix by gently pipetting up and down. Keep the pipette level to avoid the formation of bubbles that distort the subsequent readout.
- Incubate the plate for 10 min at 22 °C and measure the absorbance at 595 nm in a spectrophotometer. Calculate the TSP concentration in the samples based on a standard curve through the BSA reference points.
- Quantify DFE by fluorimetry
- In triplicate, pipette 50 µL of each sample into single wells of a black 96-well half-area plate. Include six DsRed standards covering the range 0–225 mg·L-1.
- Measure the fluorescence twice using a 530 ± 30 nm excitation filter and a 590 ± 35 nm emission filter in a spectrophotometer. Calculate the DFE concentration in the samples based on a standard curve through the DsRed reference points.
- Measure the 2F5 concentration by SPR spectroscopy20.
- Prepare the HBS-EP running buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 3 mM ethylenediaminetetraacetic acid (EDTA), 150 mM sodium chloride, 0.005% v/v Tween-20, pH 7.4) and filter sterilize it by passing it through a 0.2 µm vacuum bottle-top filter. Degas the buffer for 15 min.
- Connect the HBS-EP running buffer to the SPR instrument before docking a carboxymethylated dextran surface chip and equilibrate the system using the prime function. Start a manual run with a flow rate of 30 μL min-1 and condition the chip surface by alternately injecting 30 mM hydrochloric acid and 25 mM sodium hydroxide (two injections each) over flow cells 1 and 2 at a flow rate of 30 μL min-1 for 1 min.
- Prepare 300 µL of a 500 mg·L-1 Protein A solution in 10 mM sodium acetate (pH 4.0). Thaw vials containing 0.4 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 0.1 M NHS and centrifuge at 16,000 x g for 1 min before mixing 70 μL of EDC and 70 μL of NHS.
- Transfer the EDC/NHS mixture and the Protein A solution to 7 mm plastic sample vials and place in the sample rack. Activate the carboxymethylated dextran chip surface by injecting the EDC/NHS mixture over flow cells 1 and 2 at a flow rate of 10 μL min-1 for 10 min.
- Couple Protein A to the activated carboxymethylated dextran surface by injecting the Protein A solution over flow cell 2 at a flow rate of 15 μL min-1 for 15 min.
- While coupling the Protein A, thaw a vial of 1.0 M ethanolamine hydrochloride (pH 8.5) and centrifuge at 16,000 x g for 1 min. Transfer 150 µL to a 7-mm plastic vial and place into the instrument. When step 7.3.5 is complete, deactivate the chip surface by injecting the ethanolamine solution over flow cells 1 and 2 at a flow rate of 10 μL min-1 for 7 min.
- Condition the chip surface by alternately injecting 30 mM hydrochloric acid and 25 mM sodium hydroxide (two injections each) over flow cells 1 and 2 at a flow rate of 30 μL min-1 for 0.5 min.
- Prepare standards of antibody 2F5 in HBS-EP running buffer at a concentration of 500 µg·L-1 and dilute samples containing 2F5 in HBS-EP to a final concentration in the range 20–1,000 µg·L-1. Inject samples and standards over flow cells 1 and 2 at a flow rate of 30 μL min-1 for 3 min. Inject a 2F5 standard after every 15 samples.
- Subtract the relative units (RU) signal of flow cell 1 (reference cell) from the signal of flow cell 2 (measurement cell) for each sample and calculate the antibody concentration based on the RU signal of 2F5 standard injections.
- After every sample or standard injection, regenerate the chip surface by injecting 30 mM hydrochloric acid at a flow rate of 30 μL min-1 for 0.5 min over flow cells 1 and 2.
- Plot the 2F5 concentration for each flow-through sample from step 6.4 against the flow through volume to obtain the 2F5 breakthrough curve. Calculate the amount of injected 2F5 when 10% of the loading 2F5 concentration was reached to obtain the 10% dynamic binding capacity (DBC).
- Analyze protein samples by LDS-PAGE.
- Open a ready-to-use 4–12% BisTris LDS polyacrylamide gel and place it in an electrophoresis module. Transfer 800 mL of running buffer (50 mM MES, 50 mM Tris base, 0.1% (m/v) SDS, 1 mM EDTA, pH 7.3) into the module and add 0.5 mL of antioxidant solution.
- In a 1.5 mL reaction tube, mix 10 µL of loading buffer with 4 µL of reducing agent and 26 µL of the protein sample. Incubate the tube in a heat block for 10 min at 80 °C. Centrifuge the tube at 500 x g for 30 s.
- Load the LDS polyacrylamide gel (10 µL of sample per lane) and load 5 µL of pre-stained protein standard (10–180 kDa) in a separate lane.
- Close the electrophoresis chamber and connect the power supply. The electrophoresis should run for 40 min and at constant 200 V.
- Remove the gel from the electrophoresis chamber. Open the gel sheathing and wash the gel for 15 min in water on a shaker at 19 rpm. Stain the gel for 1 h in staining solution on a shaker at 19 rpm.
- Destain the gel for 1 h in water on a shaker at 19 rpm. Scan the gel using a film scanner.
Results
Expression and purification of the affinity ligand
The fusion protein DFE was expressed in transgenic tobacco plants grown in a greenhouse. The yield was ~120 mg·kg-1 leaf mass with an average biomass of ~130 g per plant. The DFE purity was <5% of TSP in crude plant extracts before blanching but increased to ~40% after heat treatment at 70 °C for 1.5 min, which precipitated >97% of the host cell proteins. The blanching step was easily integrated into the harvesting and extraction routine (Figure 1) and took less than 2 h of extra time, including setting up the water bath. The overall recovery of DFE was 23.5 mg kg1 with a purity of >90%. The steps responsible for product loss were blanching, depth filtration and IMAC, with specific losses of 40%, 27% and 45%, respectively. The depth filter capacity was on average 135 ± 36 L m-2 (±SD, n=3) and thus in the upper range of values reported in the literature21. The DFE yield increased with plant age (Figure 2).
Immobilization of the affinity ligand on NHS-activated chromatography columns
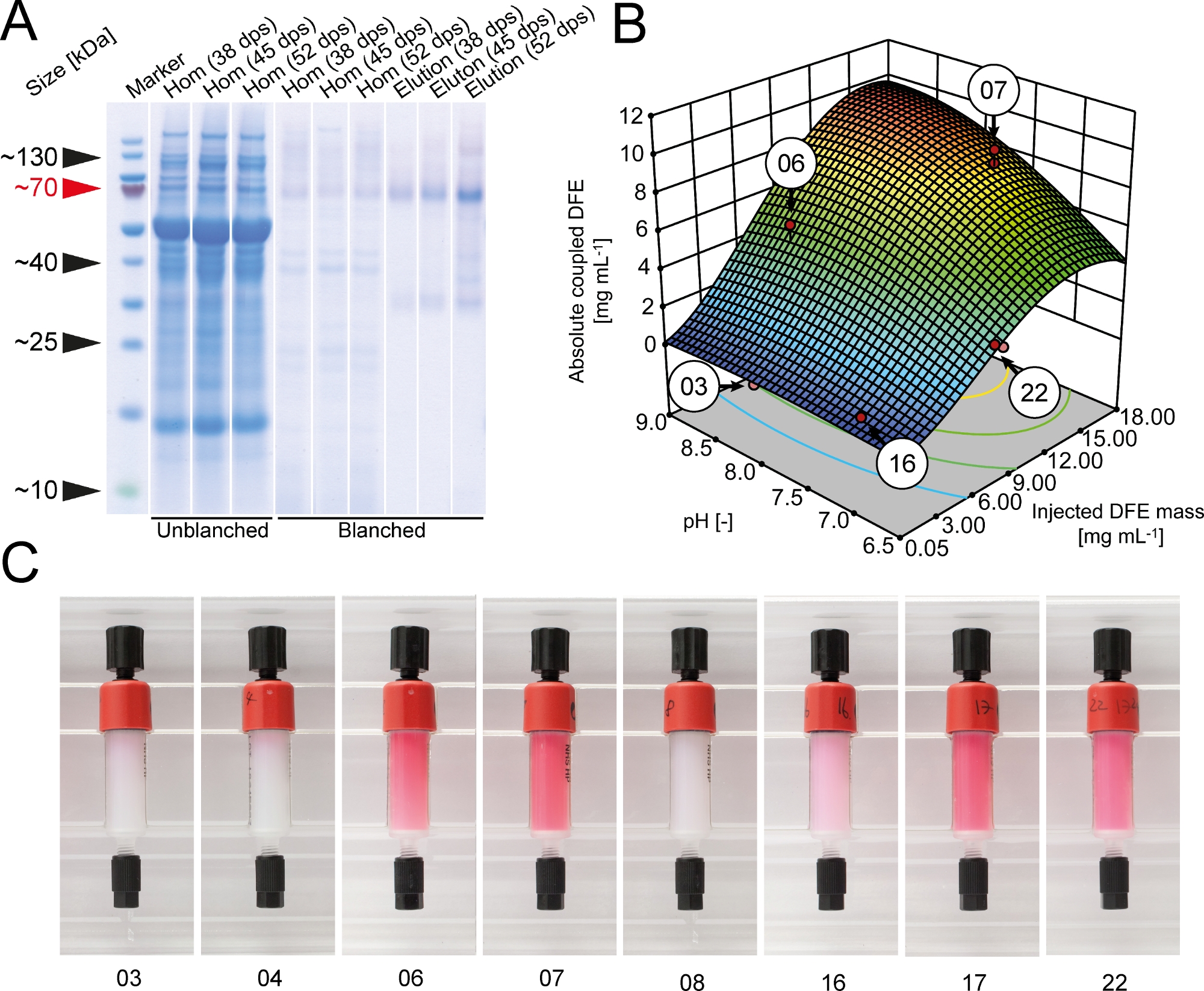
During initial coupling tests, we found that HEPES buffer (pH 8.3) increased the coupling efficiency to 89 ± 6% (±SD, n=3) compared to 78 ± 9% (±SD, n=3) for the bicarbonate buffer recommended by the manufacturer. Therefore, HEPES was used for all subsequent coupling experiments. A DoE approach was selected to optimize the coupling efficiency of DFE to NHS-activated cross-linked agarose resin. The absolute amount of DFE immobilized on the resin increased with the mass of DFE injected into the column and plateaued at ~15 g·L-1 whereas the coupling yield declined continuously as more DFE was injected (Figure 2). The coupling yield was also >50% lower in an acidic buffer, indicating the need to screen for suitable coupling conditions for each ligand on a case-by-case basis. Ideal conditions in terms of coupling yield, absolute quantity of immobilized DFE and column costs were identified using the numerical optimization tool of the DoE software. The most desirable conditions (pH 9.0 and 7.0 mg of DFE per 1 mL of cross-linked agarose resin) were located on a large plateau and were therefore robust. The DFE molecules retained their red fluorescence even after coupling, and the color intensity corresponded to the total amount of immobilized DFE (Figure 2). Therefore, column color can be used as a simple quality control parameter to estimate the coupling efficiency and column quality. The fluorescence also confirmed that DFE fusion protein assembled in the tetrameric state of native DsRed.

Figure 2: Optimization of DFE immobilization to NHS-activated cross-linked agarose resin. (A) LDS-PAA gel with western blot overlay of homogenate and elution samples from unblanched and blanched transgenic DFE plant extracts. Harvest of plants was performed 38, 45 or 52 days after seeding. Western blots were performed using an anti-His6-antibodies5. (B) Total amount of coupled DFE affinity ligand in dependence if coupling pH and overall mass of purified DFE injected onto NHS-activated cross-linked agarose columns. Red dots indicate the actual experiments performed to build the response surface model. (C) DFE affinity columns after the coupling procedure. The numbers correspond to the coupling conditions highlighted in panel B. dps = days post seeding. Please click here to view a larger version of this figure.
{kind=link}
Testing 2F5 isolation using the DFE affinity resin
The recombinant 2F5 antibody was transiently produced in Nicotiana benthamiana plants grown in a phytotron5. The capture of 2F5 from the crude plant extract was tested using affinity columns coupled with ~7.0 mg purified DFE (step 6). Elution from Protein A resins usually involves an acidic buffer (pH ~3.3)13. Therefore, we initially evaluated different low-pH elution buffers (pH 6.0–3.25) for the DFE columns. The elution of 2F5 was successful at pH values below 4.5 with the highest recovery of ~35% at pH 3.25. However, low-pH elution inactivated both the antibody (as confirmed by SPR spectrometry) and the DFE ligand (as indicated by the loss of color and the lower DBC, Figure 3). The latter was anticipated given that native DsRed denatures at pH <4.022,23. To avoid product and ligand denaturation, we tested magnesium chloride as an alternative elution agent because it has previously been used to elute mAbs from other affinity resins24. A magnesium chloride concentration of 1.25 M was sufficient to elute 2F5 from the DFE affinity resin with a recovery of 105 ± 11% (±SD, n=3) and a purity of 97 ± 3% (±SD, n=3). This performance was comparable to Protein A resins25,26. The equilibrium dissociation constant (KD) of DFE-purified 2F5 antibody and the synthetic ligand Fuzeon was 791 pM whereas that of a Protein A-purified counterpart was 763 pM5. Furthermore, no substantial color loss was observed in the resin over a total of 25 bind-and-elute cycles. The DBC of the DFE affinity resin at 10% 2F5 breakthrough declined linearly over the course of 25 cycles to ~15% of the initial value (Figure 3).

Figure 3: Testing the isolation of 2F5 from clarified plant extracts using the DFE affinity resins. (A) DFE affinity resins after one elution cycle using buffers with different pH values in the range 4.0–3.0 and the same resins after a neutralization step at pH 6.0. (B) DFE affinity resins after one and six cycles of 2F5 purification using 1.25 M or 4.00 M magnesium chloride as eluent. (C) Chromatograms of frontal loading experiments (break-through curves) to determine the cycle-dependent dynamic binding capacity of DFE resin using magnesium chloride as eluent. The break-through curves were measured for 4.0 M and 1.25 M magnesium chloride elution buffers and various cycle numbers. (D) Cycle-dependent dynamic binding capacity of DFE using 1.25 M magnesium chloride as eluent. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Applications of the novel affinity resin
We have shown that custom affinity chromatography resins for the capture of mAbs can be manufactured by immobilizing a ligand containing a mAb-specific epitope to NHS-activated cross-linked agarose. To design such a resin, it was necessary to know the epitope sequence and to use a linear epitope. The resulting resins are advantageous for the capture of mAbs because they could potentially replace expensive Protein A affinity chromatography steps. The interaction between 2F5 and DFE in our case study was mediated by epitope–paratope binding, so our ligand should be more selective than Protein A, which binds to the Fc region of most murine and human IgGs. Because individual ligands are needed for each mAb, our method may initially seem suitable mainly for antibodies that are produced on a very large scale. However, by combining our approach with rapid plant-based transient protein expression, new affinity ligands can be prepared in less than 2 weeks27 with minimal effort28. Hence, the method is also suitable for small-scale mAb purification.
Production and potential improvements of the affinity ligand
Plants offer a fast and safe production platform for affinity ligands5,29,30, such as the DFE fusion protein featured in our case study. Blanching the plant material greatly reduced the quantity of host cell proteins in a single step and was easily integrated into a standard clarification routine. However, the recovery of the ligand was low in the current setup, probably due to its moderate thermal stability and some non-specific binding to the filter layers, as reported for other products31,32,33. Engineering the carrier to increase its thermal stability may therefore help to improve the ligand yield in the future, as described for the malaria vaccine candidate CCT, the antitumor enzyme PpADI or a mesophilic β-glucosidase34,35,36. The same holds true for the depth filtration step, where protein engineering may help to reduce non-specific binding to the filter material37. The production costs for DFE and similar ligands could also be reduced by improving the overall efficiency of clarification using flocculants or filter additives38,39.
When DsRed is used as a carrier, it forms a tetrameric complex. This is advantageous because it increases the number of epitopes per ligand, but it may also render the ligand more susceptible to disassembly or denaturation during affinity chromatography. A monomeric carrier protein such as mCherry may therefore be preferable, because it is stable at low pH40, and the inclusion of tandem repeats of the epitope would increase the avidity of the ligand and thus increase resin capacity5,26,41. For simple carrier-epitope proteins (i.e., those with no disulfide bonds or post-translational modifications) microbial production systems may reduce the manufacturing costs and make the ligands more competitive with Protein A. For example, green fluorescent protein has been expressed in bacterial cells with a yield of ~1 g·kg-1 biomass, which would significantly reduce ligand production costs42.
Regardless of the expression host, a purified affinity ligand was required during coupling to minimize the immobilization of host cell proteins or media components that can otherwise reduce resin selectivity and capacity. The inclusion of a poly-histidine tag for IMAC purification increased the purity to ~90% in a single step, facilitating rapid and inexpensive ligand production5,43,44. However, the position of the fusion tag is important because it has the potential to sterically hinder epitope binding or to induce the cleavage of either the tag or the epitope from the carrier45,46.
Immobilization of the affinity ligand on NHS-activated chromatography columns
Immobilization was carried out manually or using a chromatography system. The small buffer volumes per column seemed to favor manual handling (e.g., due to the minimal waste volumes). However, if multiple/larger columns are needed, the chromatography system makes the coupling conditions easier to control (e.g., regulated flow rates) and is therefore more likely to achieve reproducible results in terms of DBC. Our data suggest that the coupling buffer and pH have an important effect on the coupling efficiency and overall column costs. Screening factors that influence the coupling reaction and adjusting them for each carrier protein (or even for each carrier–ligand fusion) could therefore improve coupling efficiency and resin performance, and we recommend this approach.
Testing 2F5 isolation using the DFE affinity resin
Product yield and purity are important aspects of resin performance, and in the case of DFE we achieved a yield of 105 ± 11% (±SD, n=3) and a purity of 97 ± 3% (±SD, n=3), which is comparable with the performance of benchmark Protein A resins25,26. Another key performance indicator for resins in general (and particularly for those based on affinity ligands) is the DBC at 10% product breakthrough, because this parameter affects the amount of resin required for a specific process and thus the costs. For the DFE ligand, the initial DBC was ~4 g·L-1 resin, which is ~13% of the corresponding value for Protein A under similar conditions (only 2 min contact time)25,47 but about 15-fold higher compared to other custom affinity resins such as the anti-FSH-immunoaffinity ligand using the same residence time of 2 min48. The DBC of DFE declined to 15% of the starting value after 25 bind-and-elute cycles, whereas more than 50 cycles are required for the same loss of DBC in commercial Protein A resins49. However, it is important to note that our carrier has not yet been optimized to the same extent as Protein A, which has been comprehensively investigated and improved over the last four decades8.
Thus far we have improved the resin stability and product recovery by switching from a low-pH elution buffer to a high concentration of magnesium chloride (Figure 3), as recommended in earlier studies13. The characteristic red color of the affinity ligand did not fade substantially during the 25 bind-and-elute cycles, so we speculate that endogenous plant proteases in the clarified plant extracts31 may have truncated and thus inactivated the epitope of the ligand. Therefore, designing protease-resistant linkers to connect the carrier and epitope may help to maintain the initial DBC over an extended number of cycles. Given the rapid and simple expression and purification of the DFE ligand, its straightforward coupling to commercial chromatography resins, and its excellent product yield and purity, we believe that our method offers a suitable alternative to Protein A for the purification of mAbs and antibody derivatives which do not bind to Protein A, especially if improvements to the carrier and linker can improve the DBC and ligand stability. This assumption was supported by the small difference in the dissociation constant of DFE-purified and Protein A-purified 2F5 antibody5, indicating that our new affinity ligand allows the recovery of high-quality mAbs.
Benefits and current limitations of the method
Producing the affinity ligand as a genetic fusion with a carrier protein increases solubility in aqueous buffers and thus compatibility with typical ligand coupling conditions. In contrast, blank peptides derived from solid phase peptide synthesis may have limited solubility under these conditions due to their sequence50, which cannot be changed because it is dictated by the amino acid sequence of epitope recognized by the mAb to be purified. Others have therefore used an on-resin synthesis of peptide ligands51. The static binding capacity of the resulting resin was high (~80 g·L-1), but the process of resin preparation is lengthy, a dynamic binding capacity was not reported and the obtained purity and recovery were lower than in our approach. An additional advantage of a fusion protein ligand in laboratory scale is that the ligand and variants thereof can be rapidly produced, purified and tested with minimal effort in an easy-to-use high-through expression system52.
The two current limitations of the method presented here are the low dynamic binding capacity of 3 g·L-1 and its 90% reduction over the course of 25 bind-and-elute cycles5. These limitations can be addressed in the future by applying less stringent loading conditions and replacing the current DsRed carrier with an engineered, more stable variant respectively. For example, doubling the current contact time from 2 to 4 minutes has the potential to double the dynamic binding capacity as was shown for some Protein A resins26.
Troubleshooting
The following table highlights potential problems that can be encountered during this protocol and provides hints on how to solve them (Table 1).
| Table 1: Potential problems that can be encountered and possible fixes. | |||
| Protocol step | Problem | Couse | Fix |
| 1 | Plants do not grow | Compromised growth conditions | Check the pH and conductivity of the fertilizer |
| Check the temperature and light conditions | |||
| 2 and 3 | Large quantities of host cell proteins are present after extraction | Incomplete precipitation | Check the temperature during blanching |
| Check the agitation in the blanching bath | |||
| 2 and 3 | No product found in the plant extract | Blanching temperature too high | Check temperature and pH during blanching |
| pH in blanching buffer too low | |||
| 3 | Large stem or leaf parts remain after extraction | Incomplete mixing in blender | Make sure the plant material does not form a plug in the blender |
| 3 | Rapid pressure increase during depth filtration | Incorrect filter selection and/or orientation | Check the filter type and orientation |
| 4 | Little fusion protein during elution / a lot of fusion protein in flow-through | IMAC resin was not charged with metal ions | Check if the IMAC resin was correctly charged with ions |
| Fusion protein lost the affinity tag | Avoid intense sunlight and high temperatures during plant cultivation | ||
| 4 | Fusion protein lost during concentration | Fusion protein bound to the membrane | Check the membrane type |
| Make sure the concentration factor was not too high | |||
| 5 | Low coupling yield | Incorrect sequence of coupling reagent addition | Check the reagents labels and sequence of addition |
| Incorrect preparation of the columns before coupling | Check the conditions of column preparaiton | ||
| 5 and 6 | Low mAb yield | Low mAb expression in the plant biomass | Test mAb expression in biomass |
| Low ligand density | Check the purity of the fusion protein preparation | ||
| 7 | Very low/high protein concentrations in Bradford assay | Bubble formation during pipetting | Check for bubbles in the 96-well palte |
| 7 | Low mAb concentration during SPR measurement | Compromised Protein A chip | Compare with results of standard mAb with known concentration |
| Incorrect sample dilution | Check the dilution rate and buffer | ||
Table 1: Trouble-shooting.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
We would like to acknowledge Ibrahim Al Amedi for cultivating the transgenic tobacco plants and Dr. Thomas Rademacher for providing the tobacco expression vector. The authors wish to thank Dr. Richard M. Twyman for editorial assistance and Markus Sack for fruitful discussions about the DFE affinity ligand structure. This work was funded in part by the Fraunhofer-Gesellschaft Internal Programs under Grant No. Attract 125-600164 and the state of North-Rhine-Westphalia under the Leistungszentrum grant no. 423 “Networked, adaptive production”. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Research Training Group “Tumor-targeted Drug Delivery” grant 331065168. GE healthcare supported the open-access publication of this article.
Materials
| Name | Company | Catalog Number | Comments |
| 10 L/20 L Bucket | n/a | n/a | Blanching equipment |
| 2100P Portable Turbidimeter | Hach | 4650000 | Turbidimeter |
| ÄKTApure | GE Helthcare | 29018226 | Chromatography system |
| Allegra 25R | Beckman Coulter | 369434 | Centrifuge |
| Amine Coupling Kit | GE Healthcare | BR100050 | SPR chip coupling kit |
| Amine Coupling Kit | GE Healthcare | BR100050 | SPR chip coupling kit |
| Antibody 2G12 | Fraunhofer IME | n/a | Standard for SPR quantification |
| Blender | Waring | 800EG | Blender |
| BP-410 | Fuhr | 2632410001 | Bag filter |
| CanoScan 5600F | Canon | 2925B009 | Scanner |
| Centrifuge tube 50 mL self-standing | Labomedic | 1110504 | Reaction tube |
| Chelating Sepharose FF | GE Helthcare | 17-0575-01 | Chromatography resin |
| Cond 3320 | WTW | EKA 3338 | Conductometer |
| Design-Expert(R) 8 | Stat-Ease, Inc. | n/a | DoE software |
| Discovery Compfort | Gilson | F81029 | Multichannel pipette |
| Disodium phosphate | Carl Roth GmbH | 4984.3 | Media component |
| Diverse bottles | Schott Duran | n/a | Glas bottles |
| Dri Block DB8 | Techne | Z381373 | Heat block |
| DsRed | Fraunhofe IME | n/a | Standart |
| EDTA | Carl Roth GmbH | 8043.2 | Buffer component |
| EnSpire | Perkin Elmer | 2390-0000 | Plate reader |
| ETHG-912 | Oregon Scientific | 086L001499-230 | Thermometer |
| F9-C | GE Helthcare | 29027743 | Fraction collector |
| Ferty 2 Mega | Kammlott | 5.220072 | Fertilizer |
| Forma -86C ULT freezer | ThermoFisher | 88400 | Freezer |
| HEPES | Carl Roth GmbH | 9105.3 | Buffer component |
| Hettich Centrifuge Mikro 200 | Hettich | 2400 | Centrifuge |
| HiPrep 26/10 | GE Helthcare | GE17-5087-01 | Chromtography column |
| HiTrap NHS-activated Sepharose HP, 1 mL | GE Helthcare | 17-0716-01 | Chromatography columns |
| Hydrochloric acid | Carl Roth GmbH | 4625.1 | Buffer component |
| Imidazole | Carl Roth GmbH | 3899.2 | Buffer component |
| K700 | Pall | 5302305 | Depth filter layer |
| KM02 basic | IKA | n/a | Magnetic stirrer |
| KS50P 60D | Pall | B12486 | Depth filter layer |
| L/S 24 | Masterflex | SN-06508-24 | Tubing |
| Lauda E300 | Lauda Dr Wobser GmbH | Z90010 | Immersion circulator |
| Magnesium chloride | Carl Roth GmbH | KK36.2 | Buffer component |
| Masterflex L/S | Masterflex | HV-77921-75 | Peristaltic pump |
| Minisart 0.2 µm | Sartorius | 16534K | Filter unit |
| Nalgene Rapid-Flow PES bottle top filter | Thermo Fischer Scientific | 595-4520 | Vacuum filtration of SPR buffers |
| Nickel sulphate | Carl Roth GmbH | T111.1 | Buffer component |
| Novex NuPAGE 4-12% BisTris LDS gels | Invitrogen | NP0336BOX | LDS-PAA gels |
| Novex X-cell Mini Cell | Invitrogen | EI0001 | PAGE chamber |
| NuPAGE 20x running buffer | Invitrogen | NP0002 | Buffer concentrate |
| NuPAGE antioxidant | Invitrogen | NP0005 | Antioxidant |
| PageRuler protein ladder (10-180 kDa) | Invitrogen | 26616 | Protein standart |
| Perforated bucked | n/a | n/a | Blanching |
| PH 3110 | WTW | 2AA110 | PH meter |
| PowerPac HC | Biorad | 1645052 | Electrophoresis module |
| Protein A from Staphylococcus aureus | Sigma-Aldrich | P7837-5MG | Coating of SPR chips |
| Sephadex G-25 fine, cross linked dextran | GE Helthcare | 17003301 | Chromatography resin |
| Silicone spoon | n/a | n/a | Spoon |
| Simply Blue SafeStain | Invitrogen | LC6060 | Gel staining solution |
| Sodium acetate | Carl Roth GmbH | 6773.1 | Buffer component |
| Sodium acetate | Carl Roth GmbH | X891.1 | Media component |
| Sodium azide | Sigma Aldrich | S2002-100G | Media component |
| Sodium chloride | Carl Roth GmbH | P029.2 | Buffer component |
| Sodium citrate | Carl Roth GmbH | HN13.2 | Buffer component |
| Sodium bisulfite | Carl Roth GmbH | 243973-100G | Media component |
| Sodium phosophate | Carl Roth GmbH | T877.2 | Media component |
| SPR Affinity Sensor - High Capacity Amine | Sierra Sensors GmbH/Bruker Daltonics | SPR-AS-HCA | SPR chip |
| SPR-2/4 Surface Plasmon Resonance Analyzer | Sierra Sensors GmbH/Bruker Daltonics | n/a | SPR device |
| SSM3 | Stuart | 10034264 | Mini Gyro-rocker |
| Heated vessel, 20 L | Clatronic | n/a | Blanching chamber |
| Sterile syringes, 2 mL | B. Braun | 4606027V | Syringes |
| Syringe adpter (Union Luer F) | GE Helthcare | 181112-51 | Syringe adapter |
| TE6101 | Sartorius | TE6101 | Precision scale |
| Tween-20 (Polysorbate) | Merck | 8170721000 | Buffer component |
| Unicorn 6.4 | GE Helthcare | 29056102 | Chromatography software |
| Vacuum bags | Ikea | 203.392.84 | Plant storge |
| VelaPad 60 | Pall | VP60G03KNH4 | Filter housing |
| Vortex-Genie 2 | Scientific industries | SI-0236 | Vortex |
| XK-26/20 column housing | GE Helthcare | 28-9889-48 | Chromtography column |
References
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnology and Applied Biochemistry. 65 (3), 306-322 (2018).
- Ecker, D. M., Jones, S. D., Levine, H. L. The therapeutic monoclonal antibody market. MAbs. 7 (1), 9-14 (2015).
- Jayapal, K., Wlaschin, K. F., Hu, W. S., Yap, M. G. S. Recombinant Protein Therapeutics from CHO Cells - 20 Years and Counting. Chemical Engineering Progress. 103, 40-47 (2007).
- Kunert, R., Reinhart, D. Advances in recombinant antibody manufacturing. Applied Microbiology and Biotechnology. 100 (8), 3451-3461 (2016).
- Rühl, C., Knödler, M., Opdensteinen, P., Buyel, J. F. A linear epitope coupled to DsRed provides an affinity ligand for the capture of monoclonal antibodies. Journal of Chromatography A. 1571, 55-64 (2018).
- Edgue, G., Twyman, R. M., Beiss, V., Fischer, R., Sack, M. Antibodies from plants for bionanomaterials. Nanomedicine and Nanobiotechnology. 9 (6), e1462 (2017).
- Buyel, J. F., Fischer, R. Very-large-scale production of antibodies in plants: The biologization of manufacturing. Biotechnology Advances. 35 (4), 458-465 (2017).
- Bolton, G. R., Mehta, K. K. The role of more than 40 years of improvement in protein A chromatography in the growth of the therapeutic antibody industry. Biotechnology Progress. 32 (5), 1193-1202 (2016).
- Kelley, B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs. 1 (5), 443-452 (2009).
- Brochier, V., Ravault, V. High throughput development of a non protein A monoclonal antibody purification process using mini-columns and bio-layer interferometry. Engineering in Life Sciences. 16 (2), 152-159 (2016).
- Arakawa, T., Futatsumori-Sugai, M., Tsumoto, K., Kita, Y., Sato, H., Ejima, D. MEP HyperCel chromatography II: binding, washing and elution. Protein Expression and Purification. 71 (2), 168-173 (2010).
- Barroso, T. B., Aguiar-Ricardo, R. J., Roque, A. C. Structural evaluation of an alternative Protein A biomimetic ligand for antibody purification. Journal of Computer-aided Molecular Design. 28 (1), 25-34 (2014).
- Mazzer, A. R., Perraud, X., Halley, J., O'Hara, J., Bracewell, D. G. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. Journal of Chromatography A. 1415, 83-90 (2015).
- Sack, M., et al. Functional analysis of the broadly neutralizing human anti-HIV-1 antibody 2F5 produced in transgenic BY-2 suspension cultures. FASEB Journal. 21 (8), 1655-1664 (2007).
- Buyel, J. F., Fischer, R. Characterization of Complex Systems Using the Design of Experiments Approach: Transient Protein Expression in Tobacco as a Case Study. Journal of Visualized Experiments. (83), 51216 (2014).
- Trasatti, J. P., Woo, J., Ladiwala, A., Cramer, S., Karande, P. Rational design of peptide affinity ligands for the purification of therapeutic enzymes. Biotechnology Progress. 34 (4), 987-998 (2018).
- Buyel, J. F., Gruchow, H. M., Boes, A., Fischer, R. Rational design of a host cell protein heat precipitation step simplifies the subsequent purification of recombinant proteins from tobacco. Biochemical Engineering Journal. 88, 162-170 (2014).
- Simonian, M. H., Smith, J. A. Spectrophotometric and colorimetric determination of protein concentration. Current Protocols in Molecular Biology. 76 (Chapter 10), (2006).
- Buyel, F. J., Kaever, T., Buyel, J., Fischer, R. Predictive models for the accumulation of a fluorescent marker protein in tobacco leaves according to the promoter/5'UTR combination. Biotchnology and Bioengeneering. 110 (2), 471-483 (2013).
- Piliarik, M., Vaisocherova, H., Homola, J. Surface plasmon resonance biosensing. Methods in Molecular Biology. 503, 65-88 (2009).
- Buyel, J. F., Fischer, R. Scale-down models to optimize a filter train for the downstream purification of recombinant pharmaceutical proteins produced in tobacco leaves. Biotechnology Journal. 9 (3), 415-425 (2014).
- Baird, G. S., Zacharias, D. A., Tsien, R. Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proceedings of the National Academy of Sciences, USA. 97 (22), 11984-11989 (2000).
- Vrzheshch, P. V., Akovbian, N. A., Varfolomeyev, S. D., Verkhusha, V. V. Denaturation and partial renaturation of a tightly tetramerized DsRed protein under mildly acidic conditions. FEBS Letters. 487 (2), 203-208 (2000).
- Firer, M. A. Efficient elution of functional proteins in affinity chromatography. Journal of Biochemical and Biophysical Methods. 49 (1-3), 433-442 (2001).
- Pabst, T. M., et al. Engineering of novel Staphylococcal Protein A ligands to enable milder elution pH and high dynamic binding capacity. Journal of Chromatography A. 1362, 180-185 (2014).
- Müller, E., Vajda, J. Routes to improve binding capacities of affinity resins demonstrated for Protein A chromatography. Journal of Chromatography B. 1021, 159-168 (2016).
- Shamloul, M., Trusa, J., Vadim, M., Vidadi, Y. Optimization and Utilization of Agrobacterium-mediated Transient Protein Production in Nicotiana. Journal of Visualized Experiments. (86), 51204 (2014).
- Rademacher, T., et al. Plant cell packs: a scalable platform for recombinant protein production and metabolic engineering. Plant Biotechnology Journal. , (2018).
- Saxena, L., Iyer, B. K., Ananthanarayan, L. Purification of a bifunctional amylase/protease inhibitor from ragi (Eleusine coracana) by chromatography and its use as an affinity ligand. Journal of Chromatography. B. 878 (19), 1549-1554 (2010).
- Kurppa, K., Reuter, L. J., Ritala, A., Linder, M. B., Joensuu, J. J. In-solution antibody harvesting with a plant-produced hydrophobin-Protein A fusion. Plant Biotechnology Journal. 16 (2), 404-414 (2018).
- Menzel, S., et al. Optimized Blanching Reduces the Host Cell Protein Content and Substantially Enhances the Recovery and Stability of Two Plant-Derived Malaria Vaccine Candidates. Frontiers in Plant Science. 7 (159), 1-7 (2016).
- Yigzaw, Y., Piper, R., Tran, M., Shukla, A. A. Exploitation of the adsorptive properties of depth filters for host cell protein removal during monoclonal antibody purification. Biotechnology Progress. 22 (1), 288-296 (2006).
- Menzel, S., et al. Downstream processing of a plant-derived malaria transmission-blocking vaccine candidate. Protein Expression and Purification. 152, 122-130 (2018).
- Vöpel, N., Boes, A., Edgü, G., Beiss, V. Malaria vaccine candidate antigen targeting the pre-erythrocytic stage of Plasmodium falciparum produced at high level in plants. Biotechnology Journal. 9 (11), 1435-1445 (2014).
- Lee, C. W., Wang, H. J., Hwang, J. K., Tseng, C. P. Protein thermal stability enhancement by designing salt bridges: a combined computational and experimental study. PLoS ONE. 9 (11), e112751 (2014).
- Zhu, L., Cheng, F., Piatkowski, V., Schwaneberg, U. Protein engineering of the antitumor enzyme PpADI for improved thermal resistance. Chembiochem. 15 (2), 276-283 (2014).
- Khanal, O., et al. Contributions of depth filter components to protein adsorption in bioprocessing. Biotechnology and Bioengineering. 115 (8), 1938-1948 (2018).
- Buyel, J. F., Fischer, R. Downstream processing of biopharmaceutical proteins produced in plants: the pros and cons of flocculants. Bioengineered. 5 (2), 138-142 (2014).
- Buyel, J. F. Procedure to Evaluate the Efficiency of Flocculants for the Removal of Dispersed Particles from Plant Extracts. Journal of Visualized Experiments. (110), e53940 (2016).
- Fink, D., et al. Ubiquitous expression of the monomeric red fluorescent protein mCherry in transgenic mice. Genesis. 48 (12), 723-729 (2010).
- Gagnon, P. Technology trends in antibody purification. Journal of Chromatography A. 1221, 57-70 (2012).
- Figueira, M., Laramée, L., Murrell, J. C., Groleau, D., Míguez, C. Production of green fluorescent protein by the methylotrophic bacterium Methylobacterium extorquens. FEMS Microbiology Letters. 193 (2), 195-200 (2001).
- Bornhorst, J. A., Falke, J. J. Purification of proteins using polyhistidine affinity tags. Methods in enzymology. 326, 245-254 (2000).
- Sainsbury, F., Jutras, P. V., Vorster, J., Goulet, M., Michaud, D. A. Chimeric Affinity Tag for Efficient Expression and Chromatographic Purification of Heterologous Proteins from Plants. Frontiers in Plant Science. 7, 141-141 (2016).
- Krupka, M., et al. The Position of His-Tag in Recombinant OspC and Application of Various Adjuvants Affects the Intensity and Quality of Specific Antibody Response after Immunization of Experimental Mice. PLoS ONE. 11 (2), e0148497 (2016).
- Goel, A., et al. Relative position of the hexahistidine tag effects binding properties of a tumor-associated single-chain Fv construct. Biochimica et Biophysica Acta. 1523 (1), 13-20 (2000).
- Tustian, A. D., et al. Development of a novel affinity chromatography resin for platform purification of bispecific antibodies with modified protein a binding avidity. Biotechnology Progress. , (2018).
- Zandian, M., Jungbauer, A. An immunoaffinity column with a monoclonal antibody as ligand for human follicle stimulating hormone. Journal of Separation Science. 32 (10), 1585-1591 (2009).
- Kelley, B. Very large scale monoclonal antibody purification: the case for conventional unit operations. Biotechnology Progress. 23 (5), 995-1008 (2007).
- Petrou, C., Sarigiannis, Y., S, K. o. u. t. s. o. p. o. u. l. o. s. Ch. 1. Peptide Applications in Biomedicine, Biotechnology and Bioengineering. , 1-21 (2018).
- Menegatti, S., et al. Design of protease-resistant peptide ligands for the purification of antibodies from human plasma. Journal of Chromatography A. 1445, 93-104 (2016).
- Rademacher, T., et al. Plant cell packs: a scalable platform for recombinant protein production and metabolic engineering. Plant Biotechnology Journal. , 1-7 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved