Method Article
Preparing Lamellae from Vitreous Biological Samples Using a Dual-Beam Scanning Electron Microscope for Cryo-Electron Tomography
In This Article
Summary
Using focused ion beam milling to produce vitreous on-grid lamellae from plunge frozen biological samples for cryo-electron tomography.
Abstract
Presented here is a protocol for preparing cryo-lamellae from plunge-frozen grids of Plasmodium falciparum-infected human erythrocytes, which could easily be adapted for other biological samples. The basic principles for preparing samples, milling, and viewing lamellae are common to all instruments and the protocol can be followed as a general guide to on-grid cryo-lamella preparation for cryo-electron microscopy (cryoEM) and cryo-electron tomography (cryoET). Electron microscopy grids supporting the cells are plunge-frozen into liquid nitrogen-cooled liquid ethane using a manual or automated plunge freezer, then screened on a light microscope equipped with a cryo-stage. Frozen grids are transferred into a cryo-scanning electron microscope equipped with a focused ion beam (cryoFIB-SEM). Grids are routinely sputter coated prior to milling, which aids dispersal of charge build-up during milling. Alternatively, an e-beam rotary coater can be used to apply a layer of carbon-platinum to the grids, the exact thickness of which can be more precisely controlled. Once inside the cryoFIB-SEM an additional coating of an organoplatinum compound is applied to the surface of the grid via a gas injection system (GIS). This layer protects the front edge of the lamella as it is milled, the integrity of which is critical for achieving uniformly thin lamellae. Regions of interest are identified via SEM and milling is carried out in a step-wise fashion, reducing the current of the ion beam as the lamella reaches electron transparency, in order to avoid excessive heat generation. A grid with multiple lamellae is then transferred to a transmission electron microscope (TEM) under cryogenic conditions for tilt-series acquisition. A robust and contamination-free workflow for lamella preparation is an essential step for downstream techniques, including cellular cryoEM, cryoET, and sub-tomogram averaging. Development of these techniques, especially for lift-out and milling of high-pressure frozen samples, is of high-priority in the field.
Introduction
Only the cellular contents of biological samples <500 nm thick can be imaged effectively by transmission electron microscopy (TEM) at cryogenic temperatures, limiting the range of specimens to viruses, prokaryotes, simple single-cellular organisms, and thinner regions of larger eukaryotic cells1. On-grid focused ion beam (FIB)-milling enables thicker plunge-frozen biological samples to be thinned to electron transparent lamellae at cryogenic temperatures (< -150 °C). The resulting lamellae are then transferred to a TEM for visualization and tomographic data collection, enabling high-resolution 3D reconstructions of the cellular and molecular features inside of cells (for reviews see Rigort et al., 20122, Oikonomou et al., 20163, and Wagner et al., 20204).
FIB-milling emerged from the field of materials sciences, where samples are routinely thinned to prepare them for downstream analysis5. It is carried out in a scanning electron microscope (SEM), which has two optical columns: conventional scanning electron microscope optics and a second column containing optics able to generate and finely control a focused ion beam (FIB) - called a FIB-SEM. This allows a specific region of the sample to be ablated by ions generated by a gallium source, removing excess material and leaving behind a lamella6. The milling process is guided by SEM imaging of the topography of the sample, which is used to locate regions of interest and monitor milling progress. For biological applications, the basic setup is largely the same, but milling is carried out at cryogenic temperatures. This has required standard FIB-SEMs to be adapted to have cryo-cooled stages that maintain constant temperatures and low surface contamination rates, as well as airlocks to facilitate sample transfer without devitrification or contamination. Sample shuttles have also been modified to permit a range of different carriers to be mounted inside the cryoFIB-SEM, including TEM grids, planchettes, and capillaries. Several key groups of researchers have been central to the development of these methods and the continuing technological advances in this area7,8,9,10,11,12. Commercial solutions are now more widely available for biological FIB-milling at cryogenic temperature and on-grid milling of lamellae is becoming more routine, given an optimized sample.
A range of room temperature and cryoEM techniques can be used to visualize cellular information across all scales of life, from whole multicellular organisms at modest resolution to understanding the context of complex processes at the cellular level and in even greater detail, to the determination of in situ molecular structures13,14,15,16,17,18,19. Classical room temperature techniques include sectioning fixed and stained, resin imbedded cells and tissues by ultramicrotomy for analysis of cellular morphology by TEM (for review see Studer et al., 200820). Alternative techniques have been developed exploiting secondary electron scattering by SEM to image the surface of blocks of preserved cells, before progressively removing material either with a knife (serial block face imaging) or a focused ion beam21,22,23. This technique has also been successfully implemented at cryogenic temperatures (referred to as cryo-volume imaging) with a cryoFIB-SEM on vitreous, unstained blocks of cells or tissues24. Alternatively, thicker lamellae (~15 µm thick) can be milled and studied by STEM imaging25. Using these techniques, whole blocks containing many cells can be imaged to gather population information or a whole organ/organism can be imaged and reconstructed in 3D. However, to access high-resolution molecular information from cells, samples need to be preserved in a near-native, frozen-hydrated state and, therefore, need to be prepared under cryogenic conditions. Cryo-electron microscopy of vitreous sections (CEMOVIS) is a technique whereby high-pressure frozen blocks of biological material are sectioned under cryogenic conditions with an ultramicrotome. This produces ribbons of cryo-sections (40-100 nm thick)26, which are attached to an EM grid and imaged in the TEM. However, the physical interaction of the knife with the vitreous sample causes crevassing and compression artifacts that can severely distort cellular structure27,28,29,30. Thicker sections are more prone to these artefacts, making it impractical to use sections thicker than ~70 nm26. This limitation greatly restricts the 3D view of the biological structure in the tomogram. FIB-milling at cryogenic temperatures does not experience these problems, but has its own artifacts caused by differential milling rates across parts of the specimen, leading to variable thicknesses within a lamella - called curtaining. This problem is mitigated by the application of an organoplatinum coat applied via a gas injection system (GIS), which protects the front edge of the lamella during milling31. The upper limit of sample thickness for on-grid FIB-milling is defined by the ability to plunge freeze the specimen while keeping it vitreous32, although, with the introduction of cryo lift-out techniques and adapted sample carriers for biological specimens, FIB-milling can also be used to process high-pressure frozen samples31,33,34,35. Additionally, plunge frozen samples cannot be too thin as there must be enough biological material to generate a reasonably sized lamella that will provide enough surface area to collect tilt-series at the required magnification. This problem can be alleviated by milling clumps of smaller cells, such as bacteria or yeast. Final lamella thickness (~100-300 nm) is usually dictated by the sample integrity and milling strategy. Thinner lamellae are better for high-resolution structural work, such as sub-tomogram averaging, but thicker lamellae contain much larger cellular volumes than can be achieved by CEMOVIS, providing more cellular context in a near to natively preserved sample. FIB-milling can also be used to thin plunge-frozen protein crystals for electron diffraction studies36.
FIB-milling of vitreous cells is worth the time and effort to carry out if the scientific question requires high-resolution molecular detail of near-native specimens in situ. With access to more facilities for the routine production of lamellae, the rate limiting step is often the optimization of the sample prior to milling, where time must be taken to ensure the sample is vitreous and an appropriate thickness to produce robust and uniformly thin lamellae. Described here is the sample optimization for FIB-milling plunge-frozen human red blood cells infected with Plasmodium falciparum parasites, the causative agent of malaria, but this approach could be adapted for any given sample.
Protocol
Human blood was obtained from anonymized donors through the UK National Blood and Transplant service and is used within 2 weeks of receipt. No ethical approval is required for its use.
1. Preparation and plunge freezing of Plasmodium falciparum infected red blood cells
- Isolate mature schizonts by centrifugation (1,580 x g) over a 70% (v/v) isotonic density gradient medium (for standard procedures for how to culture asexual blood stages of 3D7 Plasmodium falciparum in human erythrocytes see Blackman M.J., 199537).
- Fix the air-dried thin blood films on a glass slide with 100% methanol to check the morphological homogeneity of the schizonts before staining with 10% Giemsa stain in 6.7 mM phosphate buffer, pH 7.1.
NOTE: To enrich the preparation for schizonts stalled at specific points of egress, schizonts can be further synchronized with compound 2 and E64 inhibitors (see Representative Results and Figure 1 for an explanation of the effect of these inhibitors).
CAUTION: Plasmodium falciparum is a human pathogen and must only be handled in a suitable containment facility following local health and safety guidelines. - Gently centrifuge schizonts (240 x g) to pellet them and resuspend in 2x volume of the cell pellet of RPMI media, resulting in a 50% haematocrit suspension.
- On a manual plunge-freezing rig, apply 2.5 μL of schizonts to the carbon side of a glow-discharged (Use glow-discharge unit settings of 60 s, 30 mA in air, treating the carbon side of the grid only) 200 mesh copper grid with a 2/4 holey carbon film and blot for ~20 s from the back of the grid using grade 1 filter paper with a torn edge. Plunge into liquid nitrogen-cooled liquid ethane and transfer the grids to storage (see Table of Materials for equipment used in this study).
NOTE: The experiment can be paused here, and grids can be stored under liquid nitrogen indefinitely.
CAUTION: Liquid nitrogen is an asphyxiant and causes frostbite; handle with care in a suitable environment with oxygen monitoring.
CAUTION: Liquid ethane causes severe burns and is flammable; use in a fume hood away from sources of ignition.
NOTE: Plunge frozen schizonts are no longer viable. This was determined by incubating human blood with several gold grids of air-thawed plunge-frozen schizonts and observing no parasite growth after several days, in comparison to un-frozen controls. Frozen grids of schizonts are therefore safe to handle outside of containment facilities using normal safety and decontamination procedures (gloves, surface/tool sterilization with >70% ethanol and disposal of grids in >70% ethanol). - Screen grids using a cryo-stage for a light microscope, paying particular attention to the gradient of ice across the grid. In the thinner areas of ice, check individual grid squares for cell coverage.
NOTE: The best squares should be one cell thick at the center of the grid square (Figure 2). This ensures that a lamella can be milled across the center of the square without hitting the thicker ice at the edges against the grid bars. The experiment can be paused here, and grids can be stored under liquid nitrogen indefinitely. If cells carry a fluorescent marker, grids can also be screened by cryoCLEM to locate X/Y positions of interest, which can be correlated with grid locations in the cryoFIB-SEM to direct milling.
2. On-grid FIB-milling of plunge frozen cells
- Mark the front of cryoFIB-specific autogrid rims with a black indelible marker pen to indicate the center of the cutaway section and the opposite side of the rim (Figure 3). Setup the clipping station and clip grids into the marked rings carbon side down.
- Load grids into the cryoFIB-SEM shuttle (typically 2 grids, depending on the shuttle) carbon side up and apply a platinum sputter coat in an argon atmosphere (5 x 10-2 mbar) (5 mA for 60 s - thickness is variable) or a carbon/platinum e-beam rotary coat (~4 nm thickness) to the surface of the cells.
NOTE: Both types of coatings aid charge dispersal during SEM imaging. The benefit of the e-beam rotary coater is that the exact thickness of the coating can be specified. - Load the shuttle into the cryoFIB-SEM and assess the cell distribution on each grid by SEM at 5 kV (13 pA or 25 pA). Take a low-magnification overview (~100x) to look at ice gradients across the grid. Then, take higher-magnification (~5,000x) images to look at individual grid squares and identify the areas of the grid with visible cellular features and low surface contamination to mill.
- Apply a >2 µm organoplatinum coat to the surface of each grid using the gas injection system (GIS). To do this, insert the GIS needle into the chamber above the grid and warm the organoplatinum source to a set temperature for a set time (for example, ~27 °C for 3-10 s) to produce a flow of vapor.
NOTE: The angle of application, temperature, and timing of the organoplatinum coat via the GIS needle should be optimized to maximize an even coating. This will depend on the specific cryoFIB-SEM used (see Representative Results for further explanation). - Tilt the sample so that the plane of the grid is ~10° from the incidence angle of the ion beam and move the center of a suitable grid square to be seen in both the SEM and FIB images.
- Survey the grid using the ion beam at low current (nominally 1.5 pA, 30 kV) and go to high enough magnification (~7,000x) to visualize the cells at the center of a grid square. Bring the sample into focus at the first milling current (300 pA) and correct for astigmatism. Adjust the brightness and the contrast, then mark out two rectangular patterns to mill, one above and one below a 3 µm thick protected region, the center of which is the desired location of the final lamella.
NOTE: The chosen width of the patterns will depend on the topography of the surrounding cell layer and the cell size. 7-20 µm is a suitable width for schizonts, but wider lamellae take longer to mill. The chosen height of the patterns for the first milling step depends on the sample thickness, starting around 6 µm; this may need to be adjusted during milling. Milling is carried out directionally from the outside top and bottom edges of the patterns toward the face of the lamella. This can be done in parallel, where both patterns are milled concurrently or sequentially, first removing material from above the lamella and then from underneath. - Start milling at the first current; monitor the live progress in the ion beam view and intermittently by SEM (5 kV, 13 or 25 pA). Check that the ion beam has broken through the sample above and below the protected region. If not, increase the height of the rectangular patterns to remove more material. Stop when the surface above and below the lamella is completely smooth in the ion beam view.
NOTE: The ion beam has broken through the sample above and below the protected region when the inside of the rectangular patterns contains no features in the focal plane. Some features, such as grid bars, may be visible out of focus in the background. - Change to the next milling current (100 pA); focus and adjust the brightness/contrast. Decrease the space between the two rectangular patterns to 1.5 µm and decrease the pattern height to only cover the un-milled material. Begin milling at the new current until the surface above and below the lamella is completely smooth.
- Repeat this process stepwise until a thickness of 0.3 µm is reached, reducing the ion beam current each time according to the milling scheme in Table 1.
- Mill several lamellae (the amount is dependent on the thickness of the sample and the time available) on one or both grids to a thickness of 0.3 µm, record the X/Y/Z position of each lamella and reserve ~1-2 h at the end of the session to polish the lamellae to their final thickness (60-200 nm).
- Take a low-magnification SEM overview of the entire grid and plan a polishing route starting from lamellae at the front of the grid (closer to the ion beam source) to lamellae at the back of the grid (furthest away from the ion beam source) (Figure 5).
NOTE: This reduces redeposition of ablated material back onto the surface of polished lamellae. - Reduce the space between the two rectangular patterns to 100-200 nm and begin the final polishing step at an ion beam current of 30 pA. Monitor the progress by SEM at 2-3 kV (13 pA, dwell = 300 n, 3072 x 2048, ~2 s for a full frame) and stop polishing when the contrast is lost in the lamella by SEM or when the organoplatinum coat of the lamella itself begins to lose integrity.
- Before removing the grids, acquire a low-magnification SEM image of the entire grid and save images of each lamella. Use them to cross-check the grid later in the TEM. Pause the experiment here and store the grids with lamellae under liquid nitrogen - handle with care.
NOTE: Grids can be lightly sputter coated upon removal from the cryoFIB-SEM, which can help limit drift and charging in the TEM at high-magnification, but this should be done cautiously as too much sputter coating can obscure the biological contents inside the lamellae. It is possible to screen lamellae for fluorescence at this stage; however, obtaining enough signal will depend on the abundance of the labeled protein within the thickness of the lamella. Great care must be taken handling the grids to limit damage to the lamellae and prevent surface contamination.
3. Tilt-series acquisition and general overview of data processing
- Load the grids into the TEM, aligning the milling direction perpendicular to the tilt axis of the stage.
NOTE: Alignment of the grids is done by eye using the marks on the autogrid rim. A ~10° margin of error is acceptable, otherwise the walls of the trench either side of the lamella may obscure the lamella as the grid is tilted. - Acquire a low-magnification map (~150x) of the entire grid and locate the lamellae; then, acquire a medium-magnification map (~1,500x, depending on lamella size) of each lamella and locate the areas of interest.
- Pre-tilt the grid by ±10° to make the plane of the lamella (not the grid) perpendicular to the optical axis
NOTE: The direction of the pre-tilt can be determined by the position of the front edge of the lamellae (looking for the left over organoplatinum coat) in the low and medium magnification maps. For the TEM used here, grids require a +10° pre-tilt if the front edges of the lamellae point up in the maps and require a -10° pre-tilt if they point down. Each grid may be different due to how they were picked up and inserted into the autoloader. - Acquire dose-symmetric tilt-series38 (for example, -54° to +54° with an increment of 3-5°) at a pixel size that permits both the field of view and resolution required for the region of interest. Use a range of defocus values between -2 and -5 µm. Collect movies with 3-10 frames at each increment and for this, adjust these parameters depending on the pixel size to accumulate a total dose of ~150 e-/Å2 (for a 300 kV TEM).
NOTE: Cracks in the lamella should be avoided as these regions may drift. Surface contamination should also be avoided as it may obscure the region of interest or the focus area at high-tilt. - Motion correct the movies using a program such as MotionCor239. Apply a dose weighting filter to the corrected images (accumulated e-/image)40 and estimate the defocus of each image using a program such as CTFFIND441.
- Use fiducial-less alignment (patch tracking) in a program such at etomo (IMOD)42 to calculate the alignment and angular relationship of the images in tilt-series.
- Input the alignment and rotation information, along with the defocus values into a program that can apply three dimensional CTF correction, for example, NovaCTF43. Calculate a corrected tomogram to get output tomograms with binning factors relevant to downstream analysis.
- Analyze the reconstructions and prepare for any downstream processing, for example, filtering, segmentation, or sub-tomogram averaging.
Results
Preparing P. falciparum schizonts for plunge freezing
Compound 2 and E64 inhibitors are used to stall schizonts at different stages of egress, generating an enriched population of schizonts for subsequent study. This is important because without a complementary correlative technique, milling specific sub-cellular targets or cell-types is challenging as the process is essentially blind. Compound 2 is a protein kinase inhibitor that stalls egress prior to vacuole rupture. Schizonts can be synchronized on compound 2 for 4 h, then washed with compound 2-free media to remove the inhibitor, at which point schizonts will mature and egress after approximately 30 min. Alternatively, compound 2 synchronized schizonts can be washed into E64, an irreversible broad-spectrum cysteine protease inhibitor, and incubated for approximately 1 h to stall egress after the point of vacuole rupture, but before host cell rupture. The morphology and homogeneity of treated schizonts should be checked by Giemsa-stained blood smears prior to plunge freezing (Figure 1). Schizonts can be plunge frozen in either of these states using the method described in this publication.

Figure 1: The morphology of compound 2 and E64-stalled P. falciparum schizonts by Giemsa-stained blood smears. (A) In the presence of compound 2 the parasitophorous vacuole (PV) is densely packed with merozoites (purple circles) with a single cluster of hemozoin crystals (dark brown circle). The boundary between the PV and the surrounding hemoglobin in the host red blood cell (hRBC) (grey band) is well defined, as well as the hRBC membrane. (B) In the presence of E64, the PV membrane is ruptured and the merozoites spread out within the hRBC. Each schizont still contains a single cluster of hemozoin crystals. The host cell membrane is leaky and partially collapsed, therefore, no hemoglobin is visible within cell periphery and the boundary of the hRBC membrane is not easily visible. Scale bar, 5 µm. Please click here to view a larger version of this figure.
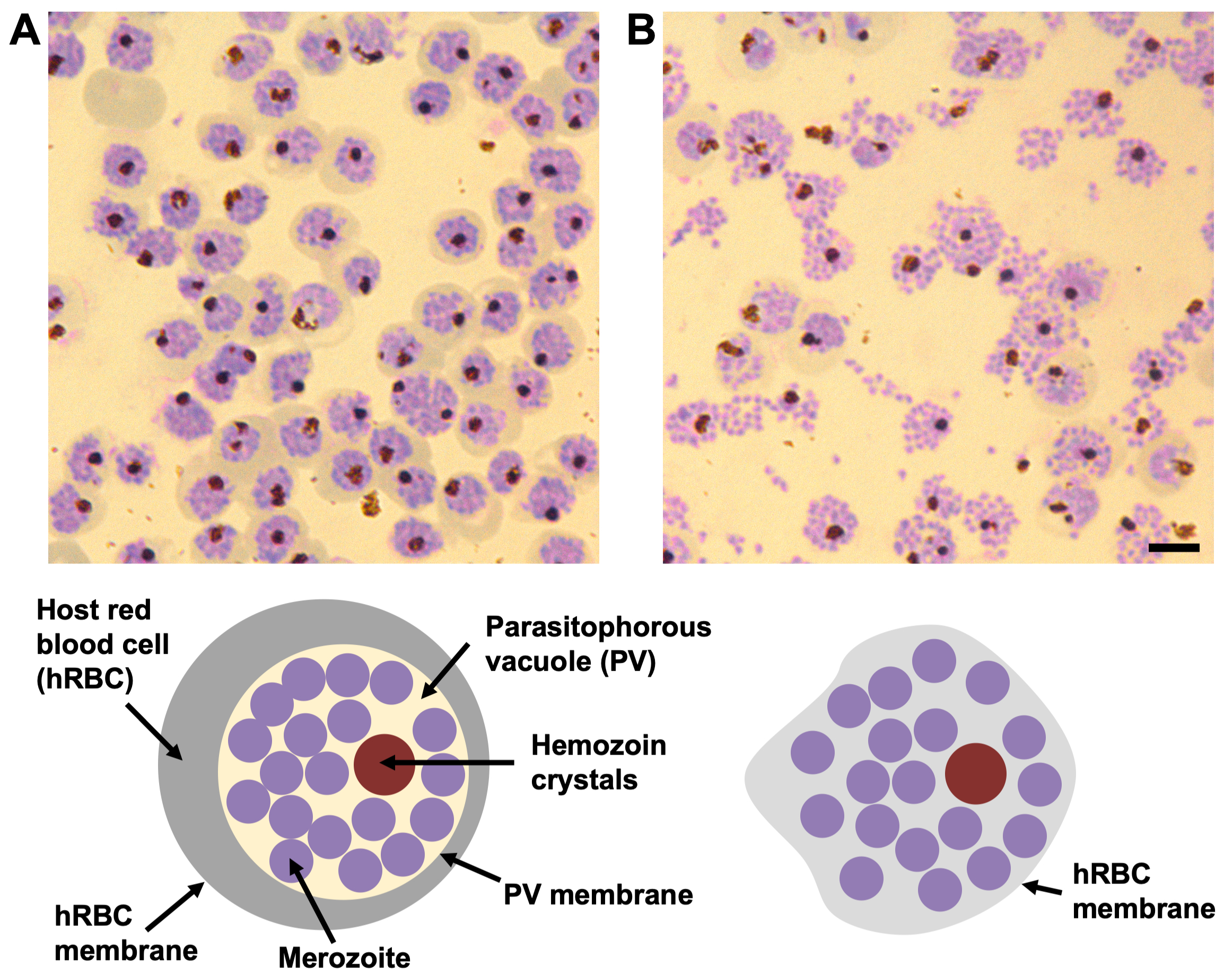{kind=link}
Optimization of plunge freezing
A range of grids with different hole sizes were trialed during the optimization of blotting conditions for P. falciparum infected red blood cells, including 2/2, 3/3, 3.5/1, and 5/2 (square) holey carbon on gold and copper 200 mesh grids. 200 mesh copper finder grids with 2/4 holey carbon film provide a suitably thick layer of cells to mill long vitreous lamellae. Bigger or smaller holes generally resulted in cell layers that were too thin or thick, respectively (Figure 2A-C). With 2/4 holey carbon, schizonts are pulled through the 2 µm holes by blotting the grid from the back (non-carbon side), resulting in cells poking out above and below the carbon film. The 4 µm stretch of carbon between the holes results in a strip of carbon running through the middle of most of the resulting lamellae, adding strength. Finder grids are most suitable for correlative and screening purposes44, but care must be taken to make sure the numbers/letters in the mesh design are not too big as this will block areas for milling.
The blotting time on a manual plunger is approximately 20 s, but the exact point at which to stop blotting was judged by eye when the droplet of liquid being drawn from the grid stopped spreading out on the filter paper. A torn edge was required to break the surface tension of the drop to start the blotting process. An automated plunge freezer was not used in this study, but a reasonable starting point for this sample would be to use the same volume of cells and grid type as used for a manual plunger, making sure to blot the grids from the back in conditions with high humidity (~70%) and ambient temperature (~25 °C). The blotting times and conditions would need to be optimized for the particular automated system used.
Plunge frozen grids of P. falciparum schizonts were screened by a light microscope equipped with a cryo-stage rather than by TEM, because the sample was not transmissible to electrons. For thinner samples, grids can be screened by TEM (whole grid atlas at ~150x magnification) prior to sample transfer into the cryoFIB-SEM, which may be a pre-requisite for accessing a national facility. Special attention should be paid to the gradient of ice thickness across the grid and also within individual grid squares. Good grid squares should be one cell thick or hit the carbon film at their center (Figure 2C). This avoids milling in the thicker ice against the grid bars around the edge of the grid square and will ensure the ion beam breaks through above and below the cell layer, producing a free lamella rather than a wedge. Once reproducible blotting and plunge freezing conditions have been optimized, screening is usually no longer necessary prior to FIB-milling.

Figure 2. Analyzing the cell distribution on grids of plunge frozen P. falciparum schizonts by cryo-light microscopy. (A) An example of ice being too thick across a grid square by cryo-light microscopy, obscuring the cells and the carbon film. Scale bar, 10 µm. (B) An example of the hole size (300 mesh copper grid with 5/2 square holey carbon film) being too big for the cells, resulting in a very thin layer of biological material surrounded by empty regions with no ice. Grids such as this produce very short, unstable lamellae. Scale bar, 10 µm. (C) An example of good cell distribution on a 200-mesh copper grid with 2/4 holey carbon film. These schizonts were treated with E64 inhibitor. The large cells (red box, ~5 µm diameter) with a well-defined periphery are infected red blood cells that still have an intact vacuole membrane. The clusters of small cells (blue box, ~1 µm) are the individual merozoites contained within a partially collapsed host red blood cell. Each schizont has a black spot at its center, indicating the position of the hemozoin crystals (See Figure 1 for additional details). The difference in cell morphology is not as easy to see once inside the cryoFIB-SEM; therefore, pre-screening by cryo-light microscopy is beneficial until reproducible blotting conditions have been reached. The cell coverage around the edges of the grid square next to the grid bars (white-dashed areas) is too thick for milling. The thinner region in the center of the grid square (yellow-dashed area) is an ideal place to mill from, extending the lamella into the surrounding layer of cells. Scale bar, 6 µm. (D) Image of a cryoFIB-specific autogrid rim with two black marks, one within the cutaway section (black bracket) and the other opposite, at 12 o'clock and 6 o'clock. The black arrow represents the milling direction. (E) When the grids are loaded into the autoloader cassette, the marks need to be equidistant either side of the loading tweezers, resulting in the lamellae lying perpendicular to the tilt axis of the stage. Please click here to view a larger version of this figure.
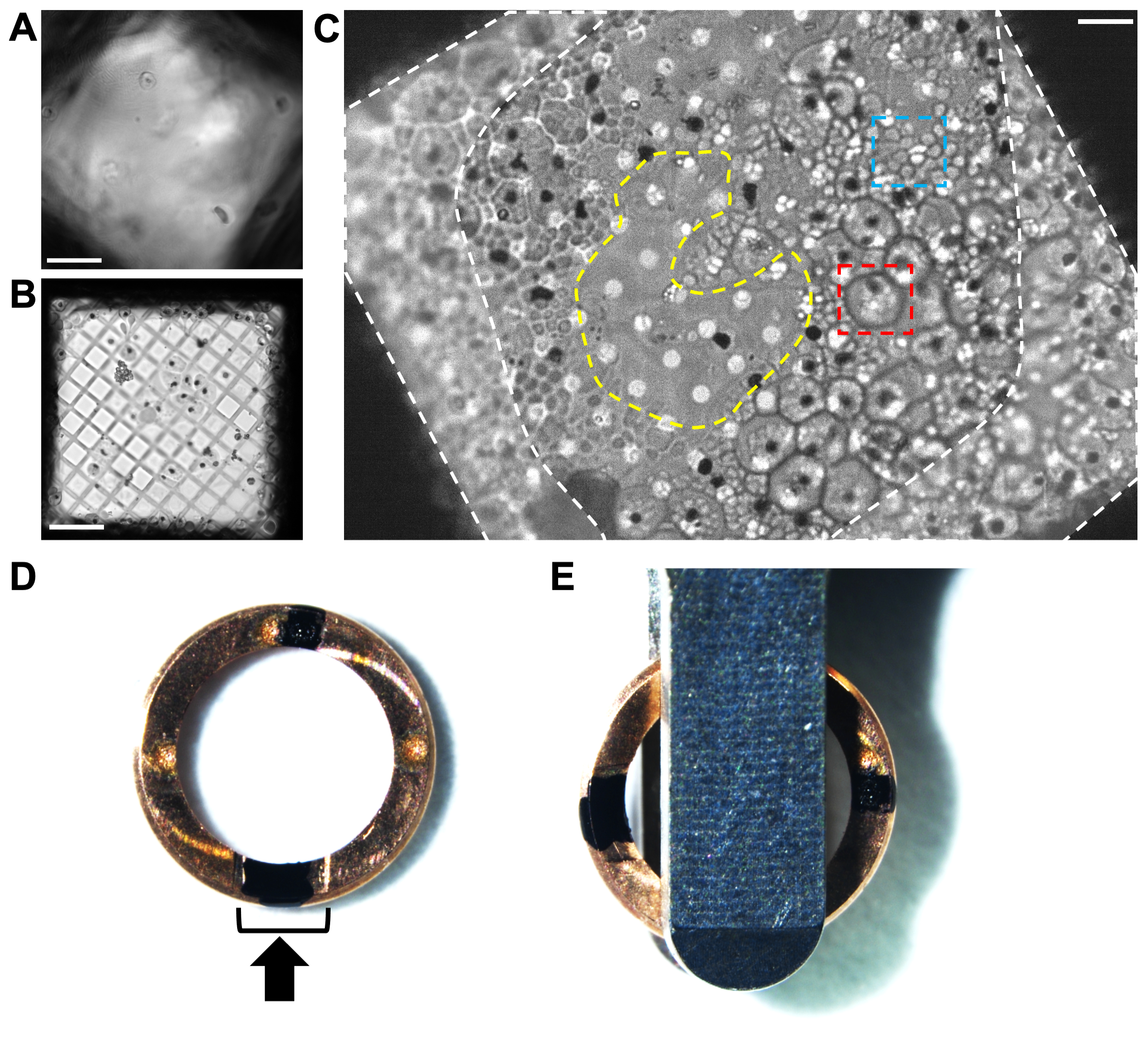{kind=link}
Marking cryoFIB-specific autogrid rims for tomography
Typically, grids are clipped into autogrid rims prior to milling in order to ease handling and provide rigidity, which protects the lamellae from damage during the subsequent transfer steps. CryoFIB-specific autogrid rims have been engineered with a cutaway feature to help access more of the grid face during milling. It is important to orientate the milling direction perpendicular to the tilt-axis of the TEM so that tilt-series acquisition proceeds by rotating the lamella along its length. This ensures that the high walls of the trench surrounding the lamella do not obscure the biological information as the grid is tilted.
Typically, the autogrid rim is marked to help with visual alignment within the cryoFIB-SEM shuttle and later when loading the TEM. For these samples, two marks were applied at 12 o'clock and 6 o'clock with an indelible marker (see Table of Materials), one within the center of the cutaway section of the clip ring and the second directly opposite (Figure 2D). When loading into the TEM (see Table of Materials), both marks should be visible either side of the loading tweezers and aligned 90° to the edge of the tweezers (Figure 2E). It should be noted that the manufacturer recommends aligning grids using the engraved dots on the autogrid rims, as certain inks in proximity to the ion beam can interfere with milling.
Clipped grids can be screened by light microscopy with a modified cassette for a cryo-stage, which can be beneficial to check that the clipping process has not destroyed the carbon film of the grid. Depending on the sample cassette, unclipped grids can also be milled, but great care must be taken during transfer from the cryoFIB-SEM to the TEM to limit any bending of the grid as this will break the lamellae. Unclipped grids can be loaded into the TEM by side-entry cryo-holders, but there is a high chance that the lamellae will get broken if clipping is carried out after milling.
Organoplatinum coating
Organoplatinum coating is carried out on one or both grids once they have been loaded into the cryoFIB-SEM. The needle of the gas injection system (GIS) is inserted into the chamber above the sample to direct a flow of organoplatinum vapor across the surface of the grid from a heated source for a set time. The vapor condenses on cold surfaces and forms a solid layer (~2 µm thick). The integrity of this coat is critical to enable uniformly thin lamellae to be milled. The optimal application conditions for the organoplatinum coat are usually pre-determined by the manufacturer of the instrument, but some optimization may still be required. Most systems either align the GIS needle close to the milling direction or perpendicular to the milling direction, depending on the geometry of ports on the chamber and rotational limits of the stage. Different settings can be trialed by coating the grid, milling a small region, tilting the stage, and measuring the thickness of the coat by SEM.
As well as the setup of the cryoFIB-SEM itself, a number of other factors can affect the application of the organoplatinum coat including 1) the topography of the sample, 2) surface contamination on the grid and 3) the reproducibility of the vapor flow from the GIS needle. As the GIS flow is directional, uneven topography can cause regions in the shadow of cells or surface contamination to be uncoated or have a thinner coat. This can lead to a collapse of the organoplatinum layer during polishing (Figure 3). When selecting an area to mill, it is necessary to be mindful of the surrounding topography, for example, large surface contamination, clumps of cells or broken carbon that project out from the surface of the grid as far as a couple of grid squares away, may block the vapor flow, creating a shadow of thinner organoplatinum that may weaken a lamella. Additionally, very small particles of surface contamination near to the front edge of the lamella should also be avoided as they can pop-off during milling, leaving a weakened patch of bare ice that may result in a hole developing in the lamella during polishing. Finally, if the milling has started and the organoplatinum coat looks unstable, coat again, for longer, or swap onto a back-up grid.

Figure 3: A good quality organoplatinum coating is critical to obtain thin, evenly milled lamellae. (A) A micrograph of the front edge of a lamella where the organoplatinum coating (OP, yellow) has been applied too thinly, leading to a hole in the front edge of the lamella that developed during polishing (green circle) and uneven milling across the whole width of the lamella (streaks). The organoplatinum surface has been sheared off by the ion beam, leading to a spray of material behind the front edge of the coating (yellow-dashed line). (B) The organoplatinum coat (OP) has been applied more thickly, resulting in a more evenly thinned lamella. The integrity of the coat is preserved across the entire width of the lamella and the interface between the organoplatinum coat and the vitrified biological material is well defined (yellow-dashed line). The carbon layer can be seen running through the rear of the lamella (orange). Scale bars, 1 µm. Please click here to view a larger version of this figure.
{kind=link}
Assessing grid quality in the cryoFIB-SEM and the process of milling
Once grids have been transferred to the cryoFIB-SEM the integrity of the carbon film and distribution of the cells on the grids can be screened by SEM (Figure 4A-C). Ice gradients, the positions of grid bars and location of grid numbers on finder grids can be checked by low-magnification SEM at 30 kV (Figure 4B), but the voltage should be reduced back to 5 kV while locating milling positions at high-magnification and monitoring the milling process to increase the contrast from the surface topography.

Figure 4: Assessing grid quality and locating areas to mill in the cryoFIB-SEM. (A) A low-magnification overview of a grid at 5 kV via the SEM. The cutaway section of the autogrid rim is visible at the bottom of the image. Scale bar, 0.5 mm. (B) The same grid at 30 kV by SEM, showing areas of thicker ice (darker grid squares) and thinner ice (lighter grid squares). The inset shows the region in the box, with arrows indicating the numbering on the finder grid, which is visible at 30 kV. Scale bar, 0.5 mm. (C) A medium-magnification overview of two grid squares assessing the distribution of the cells on the carbon film and location of the grid bars. Scale bar, 50 µm. (D) The arrangement of milling patterns for the first cut at high-magnification (ion beam view at 1.5 pA and 30 kV). The red region (3 µm thick) is protected, while the yellow regions will be ablated by the ion beam. The white-dashed line indicates the position of the final lamella. Scale bar, 10 µm. (E) A high-magnification view at 3 kV via the SEM of a polished 200 nm thick lamella (10 µm wide x 15 µm long). The loss of contrast within the lamella at 3 kV indicates that a suitable thickness has been reached. The bright-white front edge is the remaining organoplatinum layer that is applied to the grid via the GIS prior to milling. Scale bar, 5 µm. (F) The same lamella from (E) viewed using the ion beam at 30 kV and 1.5 pA. The thin white line across the black square (white arrow) is the remaining organoplatinum coat on the very front edge of the lamella. Scale bar, 10 µm. Please click here to view a larger version of this figure.
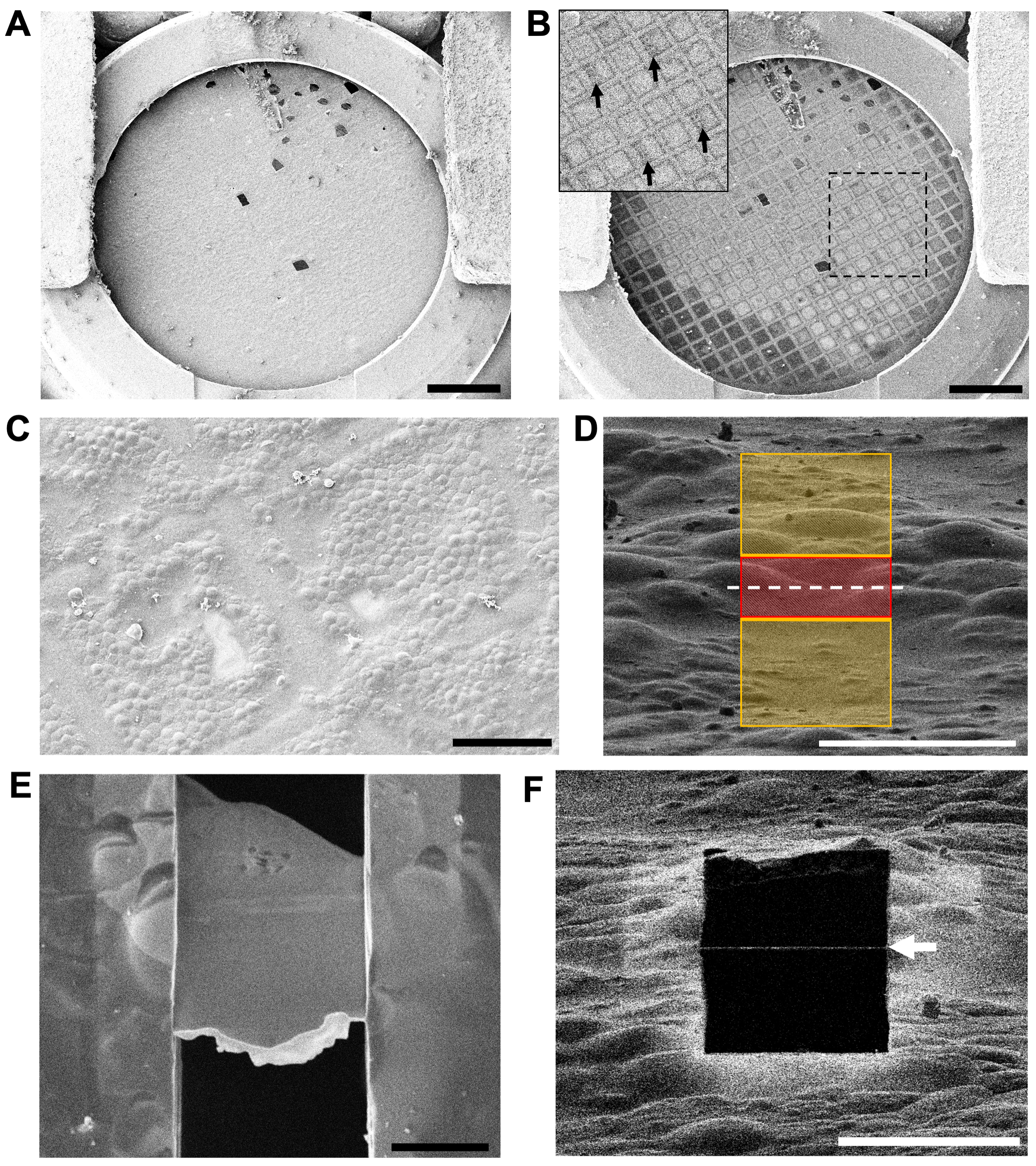{kind=link}
A region to be milled is selected by laying out a pair of rectangular patterns either side of a protected region at high-magnification (~7,000x for schizonts) in the ion beam view (Figure 4D). It is critical that no particles of surface contamination are attached near to the milling region as these may have obscured the application of the protective organoplatinum coat. It is also important that the topography of the region is suitable to support the sides of the lamella once its final thickness has been achieved.
For malarial schizonts (cell size: ~5 µm diameter x 2 µm thick, disc-shape) lamellae between 7-20 µm wide can be milled. If the cell layer is suitably thick, the lamellae will usually end up ~10-15 µm in length, capturing multiple cells above and below the carbon layer (Figure 4E-F). It can be expected to mill 5-10 lamellae in an 8 h session (6-7 h of milling and 1-2 h of polishing). This will vary depending on the thickness of the sample and width of the lamellae, with thicker samples and wider lamellae taking longer to mill. Even damaged grids can be milled as it requires only a handful of good grid squares to generate a set of lamellae (Figure 5A). Additionally, if the sample is thinner than expected, for example, due to over-blotting or variation in the haematocrit of the culture, shorter lamellae can be milled relatively quickly; however, their shorter length will limit the area available for data collection in the TEM (Figure 5B).

Figure 5: Determining when final lamella thickness has been reached during milling. (A) Low-magnification overview of a grid by SEM at 5 kV showing carbon damage during clipping. The undamaged areas contained very thin sample due to over-blotting; however, it was still possible to mill six lamellae on this grid (the region within the white-dashed outline) in a full day session on the cryoFIBSEM. Scale bar, 0.5 mm. (B) A short lamella (~10 µm wide x 3 µm long, not including organoplatinum layer) produced from this grid (SEM at 3 kV), which still provided two regions from which to collect tilt-series. Scale bar, 25 µm. (C) A series of SEM images (3 kV) of a lamella during the final polishing step showing how contrast is lost in the lamella as it is thinned (moving left to right). The dark black line across the middle of the lamella in all images is a strip of carbon film from the grid. Cells in front of this region were vitrified above the carbon film and cells behind this region were vitrified below the carbon film. Milling was stopped when the organoplatinum coat on the left side of the front edge of the lamella was near to losing structural integrity. This stopping point was before the entire lamella was brought to an even thickness, which is why there is still some higher contrast material in the rear of the lamella. (D) An example of a polishing route based on the lamellae milled on the grid shown in (A). A polishing route should start at lamellae nearer the FIB source, moving away from the FIB source to limit redeposition of milled material onto the surface of the lamellae. Please click here to view a larger version of this figure.
{kind=link}
During polishing, the final thickness of a lamella will depend on the structure of the sample in the region being milled, the integrity of the organoplatinum coat, and the time available. Ideally, the sample should be thinned until contrast is lost across the whole surface of the lamella by SEM at 3 kV, suggesting that it is evenly electron transparent and around 150-200 nm thick (Figure 5C). However, it may be necessary to stop milling before this point if the organoplatinum layer develops a hole or the lamella starts to bend. In this case, the lamella may still be thin enough at the front and remain useful for tomography. Conversely, it is possible to thin past the loss of contrast stage if the lamella looks stable, making it even thinner by moving the milling patterns closer together (~100 nm or less). This will depend on what thickness if required for the downstream workflow. A low-magnification SEM image is required to plan a polishing route, starting closer to the ion beam source and working away (Figure 5D). The direction of the polishing route is important to prevent redeposition of ablated material onto lamella that have already been finished.
Collecting and processing tilt-series data
Once loaded into the TEM, a low-magnification full grid montage (~150x) will identify the positions of the lamellae, which can be correlated to the low-magnification SEM image taken at the end of milling. For plunge frozen schizonts, most of the grid is not transparent to electrons, so the lamella positions appear as white notches on a black background (Figure 6A). The angle of the lamellae with respect to the tilt axis of the microscope should be noted, as more than ~10° away from the perpendicular could make tilt-series acquisition difficult. A medium-magnification montage (~1,500x) at the location of each lamella will give an overview of the biological content and check for transfer damage, crystalline ice, or excessive surface contamination (Figure 6B-D). This should also be checked at high-tilt to ensure that no surface contamination obscures the acquisition or focus area. Choosing an acquisition region will depend not only on the biological features, but also the structural integrity of the ice in the surrounding region, for example, avoiding cracks, as these regions will drift, or areas with excessive curtaining, where a lamella will have variable thickness. Before acquiring a tilt-series, a ±10° pre-tilt is applied to the grid to make the plane of the lamellae (not the grid) perpendicular to the optical axis. The direction of the pre-tilt can be determined by the position of the front edge of the lamellae (looking for the left over organoplatinum coat) in the medium-magnification montage. For the TEM used here (300 kV Titan Krios), if the front edges of the lamellae pointed up in the map, this required a +10° pre-tilt and if they pointed down, this required a -10° pre-tilt. Each grid may be different due to the orientation in which they were picked up in the tweezers and inserted into the autoloader (cutaway section facing left or right, produces a 180° rotation). A final consideration is pixel size. Routinely, tilt-series are collected around 2.5-7 Å/pixel, taking into account the size of the feature of interest, the target resolution of the resulting tomographic data and the surface area of the lamella, which may limit the size of acquisition area. It is possible to use a smaller pixel size to obtain high-resolution information and the smallest we have used successfully on these samples is 1.4 Å/pixel (data not shown). Drift will be more apparent at a smaller pixel size and is only really suitable for thin (<100 nm) lamellae where maximizing resolution is of importance in the study.

Figure 6: Choosing accessible regions of interest from which to acquire tilt-series (A) A section of a low-magnification TEM map showing a region containing lamellae, appearing as six white notches on a black background (red arrows). The white squares are broken carbon film. (B) A medium-magnification map of a damaged and devitrified lamella. At the front edge of the lamella (FE) the organoplatinum coat has snapped off (1). There is a clear patch of crystalline ice in the center of the lamella (2). The ion beam has failed to break through at the back edge of the lamella (BE) because the ice is too thick next to the grid bars. Only a small portion of the lamella is free from the surrounding ice at the BE (3), creating a wedge. The thickness has also caused shelves to be cut above the lamella (4), which will most likely block the view of the cells at high-tilt in the TEM. (C) Medium-magnification montage of a whole lamella viewed in the TEM. Typical problems or regions to be avoided when collecting tilt-series from lamellae are areas: covered with surface contamination (SC), covered by the organoplatinum coat (OP, yellow), near cracks (CR and black arrows), with curtaining due to density changes in biological material (CU and region within blue brackets), and areas weakened by breaks in the organoplatinum coat (green circle). The only regions accessible for tilt-series acquisition are the areas within the two black-dashed boxes (labeled 1 and 2). The view must be checked at high-tilt to make sure surface contamination does not obscure the region of interest or the focus area. (D) A medium-magnification montage of a much cleaner lamella, but which still has cracks (CR) caused by thinning of the organoplatinum coat (green circle) during polishing. Here, the region of interest is highlighted by the cell type observed within the lamella, which in this case are the individual merozoites, positioned behind the carbon layer (orange) toward the rear of the lamella (black-dashed box). Scale bars, 3 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Acquiring cryo-ET data from FIB-milled lamella. (A) A micrograph of a region of a lamella containing a red blood cell that has recently been invaded by a P. falciparum merozoite. In (B) the same image is annotated to show the boundary of the red blood cell (red), a number of double-walled intracellular vesicles (purple and blue for the inner and outer membranes, respectively) and the merozoite (green) surrounded by a second membrane derived from the host cell (yellow). A black box shows the area where a tilt-series was acquired. Scale bar, 500 nm. (C) An average of 20 central slices viewed in the XY plane from an 8x binned tomogram (2.4 Å/pixel) acquired at the apical end of the merozoite and in (D) its annotation, showing the two membranes surrounding the cell (green and yellow) and four membranes stacked in the apex of the merozoite (blue) that are associated with an electron dense mushroom-shaped feature (purple). A red arrow indicates the junction of the membrane stacks in the apex and the mushroom-shaped feature (also shown in part F, i). A black arrow indicates a fusion event between the merozoite plasma membrane and one of the multi-layered vesicles within the parasite (also shown in part F, ii). The host red blood cell membrane is shown (red) and a black-dashed line shows the position of a cross-section viewed in the XZ plane, shown in (E). The features in the cross-section (E) are colored and labeled the same as in part (D), with colored arrows pointing to the membranes. A black arrow indicates the position of a sputter coat applied to the lamella after milling and the thickness of the lamella in indicated. For parts (C-E), scale bar, 500 nm. (F) A more detailed view of the features indicated by the red and black arrow heads in part (C), showing the definition in the lipid bi-layers of the membrane stack in the apex of the merozoite (i) and the fusion event between the multi-layered vesicle with the merozoite plasma membrane. Scale bar, 75 nm. Please click here to view a larger version of this figure.
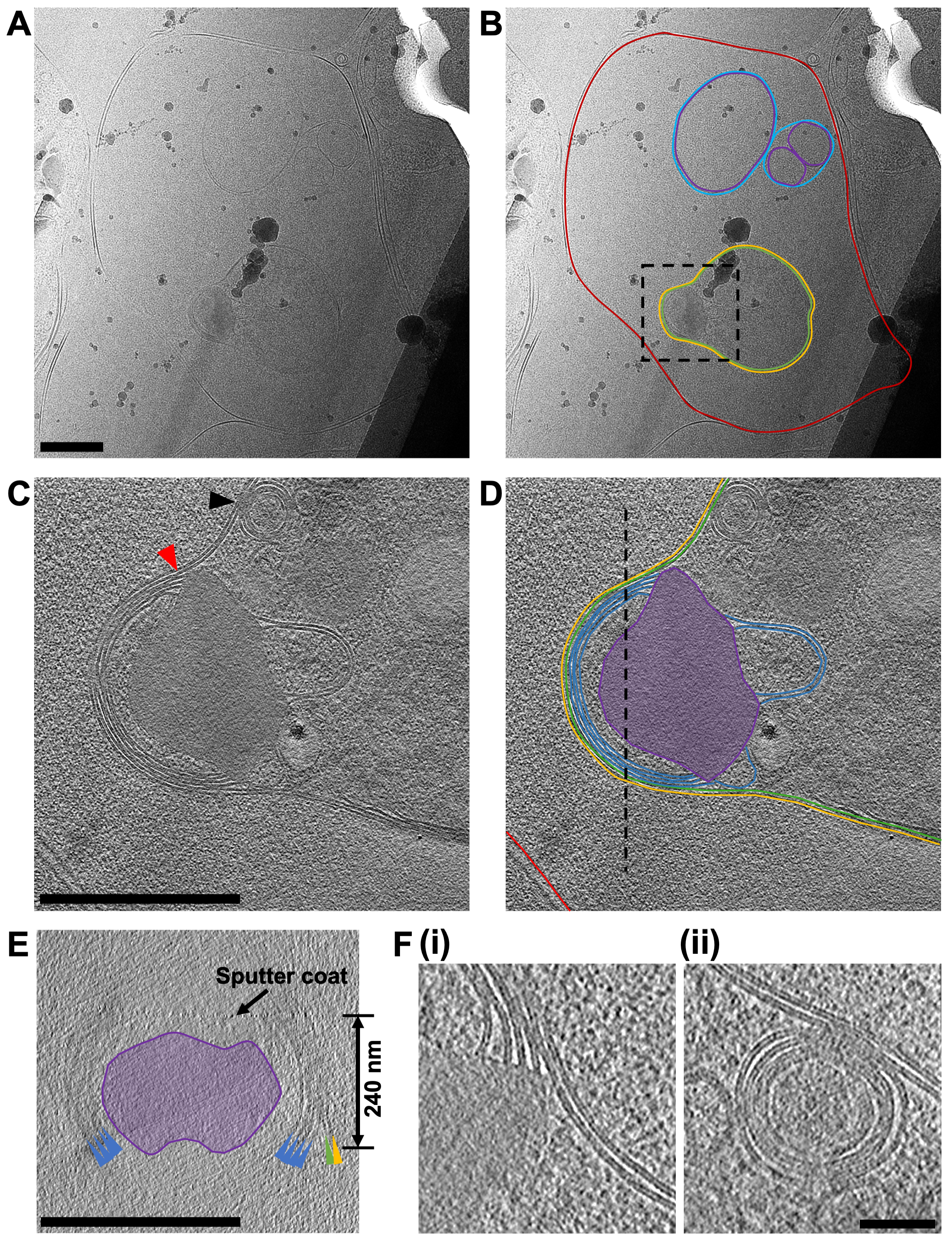{kind=link}
The primary focus of the work is to dissect the pathway of merozoite egress in P. falciparum but given that the population of cells used in the study can never be fully homogenous, often other stages of cell development within the lamellae were observed. The example shown here (Figure 7) contains a red blood cell that has recently been invaded by a P. falciparum parasite (merozoite). The lamella used was 240 nm thick and the grid had been lightly sputter coated with platinum in an argon atmosphere prior to insertion into the TEM, resulting in slightly less contrast than would normally be expected. The red blood cell membrane could be traced in its entirety surrounding the cell. Within the red blood cell were three enclosed membrane-bound structures, two vesicles, each surrounded by a double membrane and a lightbulb-shaped feature (1.2 μm x 0.9 μm at its widest parts), which is consistent with it being a merozoite (Figure 7A-B). The contents of the two vesicles seemed to be similar in contrast to that of the contents of the red blood cell, suggesting that these vesicles may contain hemoglobin. The presence of vesicles within the host red blood cell after invasion has been observed previously45. They are thought to arise from the secretion of lipids and other virulence factors from secretory organelles called rhoptries in the merozoite, which discharge into the host red cell during invasion. The merozoite is surrounded by two closely associated membranes, the inner-most of which is presumably the native plasma membrane of the merozoite, on which there is no visible surface coat, and the outer-most must derive from the host red cell membrane that envelopes the merozoite as it invades. The cytoplasm of the merozoite contains many multi-layered vesicles and an electron dense mushroom-shaped feature adjacent to a stack of membranes at the apex. A tilt-series acquired over this region (2.4 Å/pixel) shows that it contains a stack of four membranes, which appear to be connected to the mushroom-shaped feature (Figure 7C-E). Strikingly, this morphology is quite different to the normal arrangement of organelles and cellular structures in the apical end of a mature merozoite. To assess this, a comparative tilt-series (2.74 Å/pixel) over the apical end on a mature merozoite was obtained from a schizont that had been treated with E64 prior to plunge freezing and milling (Figure 8). This shows that the apical end of a merozoite contains two prominent club-shaped secretory organelles called rhoptries nestled within a set of three polar rings at the apical tip of the cell and surrounded by a number of smaller secretory organelles called micronemes. The inner-most polar ring is attached to a double membrane structure that underlies the merozoite plasma membrane, called the inner membrane complex, which contains motor proteins that drive invasion. It has been shown that during invasion the contents of the secretory organelles are discharged onto the merozoite surface and into the host red blood cell, facilitating attachment of merozoites to host red cells and initiating the motor complex that drives invasion45. The data here shows that after invasion, the merozoite contains no observable structures that resemble rhoptries, micronemes, or polar rings, suggesting that the morphology of the apical end of the merozoite dramatically changes. Fusion of rhoptries prior to invasion has been shown previously by TEM of fixed, room temperature sections46, which is consistent with the production of the mushroom-shaped feature we observe in our post-invasion state merozoite. The stacks of membranes connected to this feature have not been observed previously and since there is no indication of an IMC present in the newly invaded merozoite, we hypothesize that the membrane stacks may be the remnants of the IMC machinery left over after invasion is completed, but this remains to be confirmed.

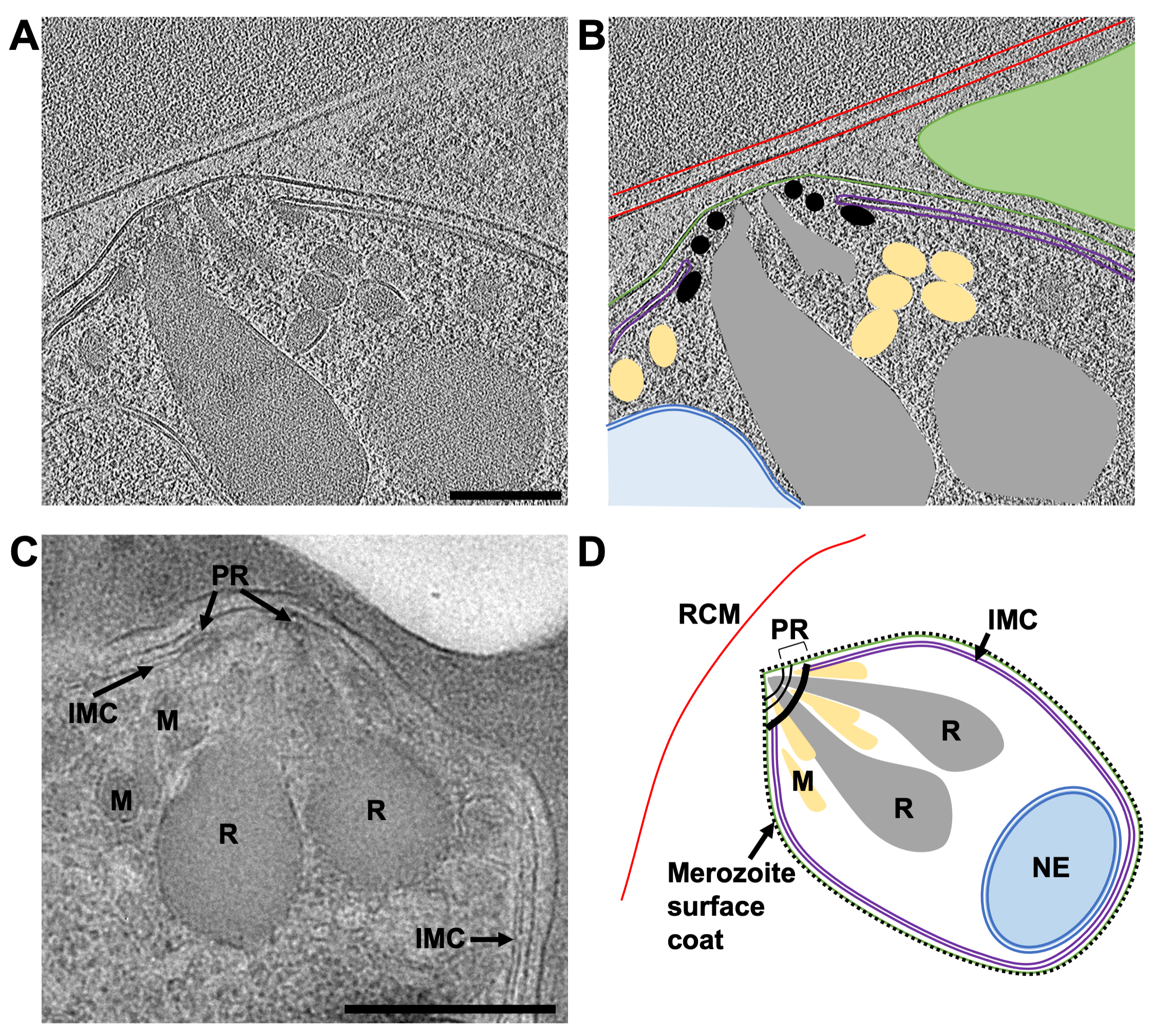
Figure 8: The apical end of mature merozoites by cryo-ET of a FIB-milled lamella and TEM of plastic sections. (A) An average of 20 central slices from an 8x binned tomogram (2.74 Å/pixel) from a 230 nm thick lamella showing the typical morphology of the apical end of a mature merozoite. The schizonts were treated with E64 prior to plunge freezing, thus the PV membrane has ruptured and the merozoites are contained within the host red cell. (B) shows the same as in (A), but with annotations to indicate the host red blood cell membrane (RCM, red), the merozoite plasma membrane (green), the inner membrane complex (IMC, purple), polar rings (PR, black), micronemes (M, yellow), rhoptries (R, grey), and the nuclear envelope (NE, blue). A neighboring merozoite, which is out of plane in this region of tomogram is indicated by a green triangle. Scale bar, 200 nm. (C) The cellular features observed by cryoET are consistent with those from TEM of schizonts preserved in plastic sections. Similar cellular features are indicated in a mature merozoite from a schizont that has been treated with compound 2, stalling egress prior to PV membrane rupture. Scale bar, 500 nm. (D) An annotated schematic of a merozoite showing the cellular features in parts (A-C). Please click here to view a larger version of this figure.
{kind=link}
| Thickness of protected region between the rectangular milling patterns (µm) | Nomical ion beam current (pA) at 30 kV |
| 3 | 300 |
| 1.5 | 100 |
| 0.75 | 50 |
| 0.3 | 30 |
| 0.2 - 0.06 (final polishing) | 30* |
Table 1: Milling strategy to produce thin lamella. Step-wise milling is carried out by reducing the ion beam current corresponding to the thickness of the protected region. This limits sample heating, preventing devitrification. *Polishing should be performed in parallel to evenly apply heat to the lamellae from both sides, reducing the risk of them bending or bowing as they reach their final thickness. Other steps can be done sequentially, milling the pattern above the lamella and then below the lamella, before moving to the next beam current. Surveying the grid to locate milling positions should be carried out at 1.5 pA beam current to minimize sample heating. Please click here to download this Table.
Discussion
While cryoFIB-milling is becoming more routine, preparation of optimal samples for milling has not; therefore, most of the critical steps in this protocol occur before the sample reaches the cryoFIB-SEM. Sample optimization by multiple rounds of cell preparation, plunge freezing, and screening by light and electron microscopy are required to produce the best possible sample before milling is attempted. Having the best sample possible not only increases the chances of success but also optimizes the use of the equipment. For this reason, most national facilities require evidence that sufficient optimization has been carried out before granting time on their machines. Once inside the cryoFIB-SEM, maximizing the effectiveness of the organoplatinum coat is critical in order to produce uniformly thin lamellae. Finally, patience and good sample handling is a prerequisite skill from the user, who will be required to sit for multiple days producing and then transferring grids containing delicate lamellae. This may change with the introduction of automated milling strategies47,48, but as yet, the entire milling process is still largely manual at most facilities.
While the work focused solely on P. falciparum schizonts, the method presented here could be easily modified to optimize grid preparation and milling for other cell types. Important factors to consider are the density of the cells being applied to the grid, the grid type (copper is toxic to some cells), the size and spacing of holes in the carbon film, the blotting time, and the method of blotting (single/double sided, manual, or automated). Additionally, if the cells are being grown on the grids (usually gold grids with a holey carbon film), confluency can be checked by light microscopy before blotting and freezing. The milling strategy depends on how the cells are deposited on the grid. For malaria infected red blood cells, the grid is essentially coated by an unbroken layer of cells, thicker in some areas and thinner in other areas. Milling is carried out at multiple points at one region along this gradient where the ice thickness generates lamellae that are a suitable size for tomography. This approach works well for smaller cells as you can produce lamellae containing a slice through multiple cells. For larger cells or clumps of cells, it might be the case that one cell or cell clump may produce one lamella.
Grid handling and sample transfer remain one of the main challenges of this workflow. Lamellae can be lost due to breakages or cracks, curtaining artefacts obscuring the biology, excessive surface contamination, as well as grid orientation problems within the TEM, requiring an extra handling step to reposition the grid. The degree of losses varies from day-to-day and grid-to-grid, but it is improved through practice and handling experience over time. In this study, it was found that the grids can be carefully reorientated in the autoloader multiple times without destroying lamellae and this sometimes has the benefit of washing off surface ice. Another main limitation of this workflow is the time it takes to produce lamellae. As production is slow, it is critical to have a properly optimized sample, making the milling as efficient as possible.
A number of adaptations to FIB-milling of vitreous biological samples have recently been introduced. A game-changer is the implementation of cryogenically cooled lift-out tools within the cryoFIB-SEM chamber that enable larger blocks of material to be milled from high-pressure frozen samples. The blocks can be attached to a metal rod or picked up in a gripper and moved to a second sample position, containing a specially modified EM grid. The blocks can then be coated with organoplatinum and milled to generate lamellae. The ability to mill lamellae from high-pressure frozen material means that much larger cells and tissues can be processed, specifically targeting areas by correlative fluorescence microscopy12. Other recent adaptations to the FIB-milling method include reducing curtaining artifacts by wedge pre-milling the sample16, microfluidic cryo-fixation49 and photo-micropatterning of electron microscopy grids to improve cell distribution50. Also, it has been demonstrated that milling micro-expansion gaps either side of a lamella can alleviate compression by the surrounding sample as it reaches its final thinness51. This may be particularly useful when milling continuous cell layers, such as the sample in this study, where bending of lamellae is sometimes seen during the final polishing step.
The future of electron microscopy will likely be the determination of in situ molecular structures by sub-tomogram averaging and FIB-milling is an important tool that will facilitate the production of vitreous biological samples for these types of workflows. While FIB-milling is still in its infancy for biological applications, methods development is happening at a rapid pace thanks to the hard work of researchers, both academic and at national facilities, plus commercial investment in developing cryoFIB-SEM technology to support research.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was funded in whole, or in part, by the Wellcome Trust (212916/Z/18/Z). For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
This project for which this method was developed was funded by a Medical Research Council grant MR/P010288/1 awarded to Helen R. Saibil, Roland A. Fleck, and Michael J. Blackman. Cultures of P. falciparum were grown at The Francis Crick Institute, with the support of members of Michael J. Blackman's group. The authors would like to thank Dr. Ser Ying (Michele) Tan for providing the images of compound 2 and E64-treated schizonts in thin blood smears. Most of the lamellae were produced with support from staff at eBIC and we are grateful for access to the Scios dual-beam cryoFIB-SEM on research proposal NT21004. The authors also acknowledge the support of the Royal Society Industry Fellowship scheme (INF\R2\202061) in continuing the development of the FIB-milling technique within the CUI. The authors would also like to thank Helen R. Saibil for useful discussions in relation to this methods paper and supervising the project.
Materials
| Name | Company | Catalog Number | Comments |
| c-clips | Thermo Fisher Scientific | 1036171 | |
| Clipping station | Thermo Fisher Scientific | n/a | Direct quote from Thermo |
| Clipping station | Sub-angstrom | CSA-01 | |
| Compound 2 | n/a | n/a | Synthesised by Dr Simon A. Osborne, LifeArc |
| Cryo-FIB specific autogrid rims | Thermo Fisher Scientific | 1205101 | |
| E64 | Sigma | E3132 | |
| Emitech K100X glow discharge unit | Quorum | n/a | |
| Gibco RPMI 1640 media | Thermo Fisher Scientific | 12633012 | Formulation used for culturing is custom made (REF 041-91762 A) and includes Albumax, glutamine, HEPES and hypoxanthine supplements |
| Giemsa Stain | VWR International | 350864X | |
| Glass slides | Thermo Fisher Scientific | 11562203 | |
| Grid boxes | Sub-angstrom | PB | For clipped grids |
| Grid boxes | Thermo Fisher Scientific | n/a | For clipped grids - direct quote from Thermo Fisher Scientific |
| Grid boxes | Agar Scientific | AGG3727 | |
| Home-made manual plunge freezing rig | n/a | n/a | With an insulated ethane pot (high-density foam) and liquid nitrogen bath (polystyrene) on a bench top in a containment facility. |
| Human blood | n/a | n/a | UK National Blood and Transplant service |
| JEOL 4700F Z JSM with a Leica VCT500 stage cooling system | JEOL | n/a | FIB-SEM |
| Leica EM ACE600 with a VCT500 cryostage | Leica | n/a | sputter coater |
| Leica EM ACE900 | Leica | n/a | e-beam rotary coater. In a humidity controlled room. |
| Linkam cryo-stage for light microscope | Linkam | Model No. CMS196 | Cassettes for clipped and un-clipped grids. In a humidity controlled room. |
| Methanol | Sigma | 179957 | |
| Nikon Eclipse E200 light microscope | Nikon | n/a | with Linkam cryo-stage in a humidity controlled room. |
| Percoll | VWR International | 17-0891-01 | Solution for percoll cushion is 35 ml 10x PBS, 150 ml RPMI 1640 media and 315 ml Percoll |
| Quantifoil copper 200 mesh 2/4 holey carbon EM Finder grids | Quantifoil | N1-C17nCuH2-01 | 100 pack |
| Quantifoil copper 200 mesh 2/4 holey carbon EM grids | Quantifoil | N1-C17nCu20-01 | 100 pack |
| Quorum PP3010 prep-chamber | Quorum | n/a | sputter coater |
| Scios dual beam equipped with a Quorum PP3010 transfer stage. | FEI | n/a | FIB-SEM |
| Staedtler Lumocolor fine black permanent marker pen | Viking Direct | ND538522 | |
| TFS Titan Krios 300 kV TEM | Thermo Fisher Scientific | n/a | TEM equipped with a K2 or K3 camera. |
| Whatmann grade 1 filter paper | Sigma | WHA1001150 | |
| Zeiss Axio Scope A1 light microscope | Zeiss | n/a | with Linkam cryo-stage in a humidity controlled room. |
References
- Baumeister, W. From proteomic inventory to architecture. FEBS Letters. 579 (4), 933-937 (2005).
- Rigort, A., Villa, E., Bäuerlein, F. J. B., Engel, B. D., Plitzko, J. M. Integrative approaches for cellular cryo-electron tomography: Correlative imaging and focused ion beam micromachining. Methods in Cell Biology. 111, 259-281 (2012).
- Oikonomou, C. M., Chang, Y. W., Jensen, G. J. A new view into prokaryotic cell biology from electron cryotomography. Nature Reviews Microbiology. 14, 205-220 (2016).
- Wagner, F. R., et al. Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nature Protocols. 15 (6), 2041-2070 (2020).
- Giannuzzi, L. A., Stevie, F. A. A review of focused ion beam milling techniques for TEM specimen preparation. Micron. 30 (3), 197-204 (1999).
- Narayan, K., Subramaniam, S. Focused ion beams in biology. Nature Methods. 12 (11), 1021-1031 (2015).
- Rigort, A., et al. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proceedings of the National Acadamy of Science of the United States of America. 109 (12), 4449-4454 (2012).
- Marko, M., Hsieh, C., Schalek, R., Frank, J., Mannella, C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nature Methods. 4 (3), 215-217 (2007).
- Villa, E., Schaffer, M., Plitzko, J. M., Baumeister, W. Opening windows into the cell: Focused-ion-beam milling for cryo-electron tomography. Current Opinion in Structural Biology. 23 (5), 771-777 (2013).
- Wang, K., Strunk, K., Zhao, G., Gray, J. L., Zhang, P. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. Journal of Structural Biology. 180 (2), 318-326 (2015).
- de Winter, D. A. M., et al. In-situ integrity control of frozen-hydrated, vitreous lamellas prepared by the cryo-focused ion beam-scanning electron microscope. Journal of Structural Biology. 183 (1), 11-18 (2013).
- Schaffer, M., et al. A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue. Nature Methods. 16 (8), 757-762 (2019).
- Mahamid, J., et al. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 351 (6276), 969-972 (2016).
- Albert, S., et al. Proteasomes tether to two distinct sites at the nuclear pore complex. Proceedings of the National Acadamy of Science of the United States of America. 114 (52), 13726-13731 (2017).
- Guo, Q., et al. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell. 172 (4), 696-705 (2018).
- Schaffer, M., et al. Optimized cryo-focused ion beam sample preparation aimed at in situ structural studies of membrane proteins. Journal of Structural Biology. 197 (2), 73-82 (2017).
- Szwedziak, P., Wang, Q., Bharat, T. A. M., Tsim, M., Löwe, J. Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. eLife. 3, 04601 (2014).
- Carlson, L. A., et al. Cryo electron tomography of native HIV-1 budding sites. PLOS Pathogens. 6 (11), 1001173 (2010).
- Klein, S., et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nature Communications. 11 (1), 5885 (2020).
- Studer, D., Humbel, B. M., Chiquet, M. Electron microscopy of high pressure frozen samples: Bridging the gap between cellular ultrastructure and atomic resolution. Histochemistry and Cell Biology. 130 (5), 877-889 (2008).
- Schertel, A., et al. Cryo FIB-SEM: Volume imaging of cellular ultrastructure in native frozen specimens. Journal of Structural Biology. 184 (2), 355-360 (2013).
- Murphy, G. E., et al. Correlative 3D imaging of whole mammalian cells with light and electron microscopy. Journal of Structural Biology. 176 (3), 268-278 (2012).
- Heymann, J. A. W., et al. Site-specific 3D imaging of cells and tissues with a dual beam microscope. Journal of Structural Biology. 155 (1), 63-73 (2012).
- Spehner, D., et al. Cryo-FIB-SEM as a promising tool for localizing proteins in 3D. Journal of Structural Biology. 211 (1), 107528 (2020).
- Kamino, T., Yaguchi, T., Ohnishi, T., Ishitani, T., Osumi, M. Application of a FIB-STEM system for 3D observation of a resin-embedded yeast cell. Journal of Electron Microscopy. 53 (5), 563-566 (2004).
- Han, H., Zuber, B., Dubochet, J. Compression and crevasses in vitreous sections under different cutting conditions. Journal of Microscopy. 230, 167-171 (2008).
- Matias, V. R. F., Al-amoudi, A., Dubochet, J., Beveridge, T. J. Cryo-transmission electron microscopy of frozen-hydrated sections of Escherichia coli and Pseudomonas aeruginosa. Journal of Bacteriology. 185 (20), 6112-6118 (2003).
- Al-Amoudi, A., Studer, D., Dubochet, J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. Journal of Structural Biology. 150 (1), 109-121 (2005).
- Al-Amoudi, A., et al. Cryo-electron microscopy of vitreous sections. The EMBO Journal. 23 (18), 3583-3588 (2004).
- Bouchet-Marquis, C., Dubochet, J., Fakan, S. Cryoelectron microscopy of vitrified sections: A new challenge for the analysis of functional nuclear architecture. Histochemistry and Cell Biology. 125 (1-2), 43-51 (2006).
- Hayles, M. F., et al. The making of frozen-hydrated, vitreous lamellas from cells for cryo-electron microscopy. Journal of Structural Biology. 172 (2), 180-190 (2010).
- Dubochet, J., Adrian, M., Chang, J. -. J., Lepault, J., McDowall, A. W. Cryoelectron microscopy of vitrified specimens. Cryotechniques in Biological Electron Microscopy. , 114-131 (1987).
- Hsieh, C., Schmelzer, T., Kishchenko, G., Wagenknecht, T., Marko, M. Practical workflow for cryo focused-ion-beam milling of tissues and cells for cryo-TEM tomography. Journal of Structural Biology. 185 (1), 32-41 (2014).
- Harapin, J., et al. Structural analysis of multicellular organisms with cryo-electron tomography. Nature Methods. 12 (7), 634-636 (2015).
- Rubino, S., et al. A site-specific focused-ion-beam lift-out method for cryo Transmission Electron Microscopy. Journal of Structural Biology. 180 (3), 572-576 (2012).
- Duyvesteyn, H. M. E., et al. Machining protein microcrystals for structure determination by electron diffraction. Proceedings of the National Acadamy of Science of the United States of America. 115 (38), 9569-9573 (2018).
- Blackman, M. J. Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Microbes as Tools for Cell Biology. 45, 213-220 (1995).
- Hagen, W. J. H., Wan, W., Briggs, J. A. G. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. Journal of Structural Biology. 197 (2), 191-198 (2017).
- Zheng, S. Q., et al. MotionCor2 - anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods. 14 (4), 331-332 (2017).
- Grant, T., Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 4, 06980 (2015).
- Rohou, A., Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology. 192 (2), 216-221 (2015).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Turoňová, B., Schur, F. K. M., Wan, W., Briggs, J. A. G. Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4 Å. Journal of Structural Biology. 199 (3), 187-195 (2017).
- Arnold, J., et al. Site-specific cryo-focused ion beam sample preparation guided by 3d correlative microscopy. Biophysical Journal. 110 (4), 860-869 (2016).
- Riglar, D. T., et al. Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host & Microbe. 9 (1), 9-20 (2011).
- Hanssen, E., et al. Electron tomography of Plasmodium falciparum merozoites reveals core cellular events that underpin erythrocyte invasion. Cellular Microbiology. 15 (9), 1457-1472 (2013).
- Buckley, G., et al. Automated cryo-lamella preparation for high-throughput in-situ structural biology. Journal of Structural Biology. 210 (2), 107488 (2020).
- Zachs, T., et al. Fully automated, sequential focused ion beam milling for cryo-electron tomography. eLife. 9, 52286 (2020).
- Fuest, M., et al. In situ microfluidic cryofixation for cryo focused ion beam milling and cryo electron tomography. Scientific Reports. 9 (1), 19133 (2019).
- Toro-Nahuelpan, M., et al. Tailoring cryo-electron microscopy grids by photo-micropatterning for in-cell structural studies. Nature Methods. 17 (1), 50-54 (2020).
- Wolff, G., et al. Mind the gap: Micro-expansion joints drastically decrease the bending of FIB-milled cryo-lamellae. Journal of Structural Biology. 208 (3), 107389 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved