
Method Article
Simple In-House Ultra-High Performance Capillary Column Manufacturing with the FlashPack Approach
In This Article
Summary
Here we present a protocol for the optimized FlashPack capillary column packing procedure. Application of an optimized protocol to a common 100-bar pressure bomb setup allows 10-times faster packing and manufacturing of long ultra-high performance capillary columns.
Abstract
Capillary ultra-high performance liquid chromatography (UHPLC) is currently a method of choice for the sample separation step in LC-MS-based proteomics. However, capillary columns are much less robust in comparison to their higher flow countertypes. Because of easy contamination and blocking, they often need replacement. That makes them a markedly expensive part of the total LC-MS analysis cost. In-house packing of UHPLC capillary columns saves a lot of money and allows customization. However, the standard packing procedure in the 100-bar pressure bomb works well only for HPLC columns but is too slow for UHPLC sorbents. Here we provide a description of an optimized FlashPack protocol applied to the same 100-bar pressure bomb setup. The method is based on packing from ultra-high sorbent concentration slurry and is developed for in-house manufacturing of UHPLC capillary columns of unlimited length in reasonable time.
Introduction
Modern proteomics is based on liquid chromatography-coupled mass spectrometry with the ultra-high performance nano-flow chromatography (50-150 µm column internal diameter (ID)) separation providing the best analysis speed and sensitivity1. While numerous commercial UHPLC capillary columns are available, their price makes up a major part of the consumables cost, especially when multiple diverse projects are run in the laboratory and project-specific column contamination is a frequent issue. Besides, packing of columns in-house allows the use of custom experiment-specific sorbents (such as, e.g., polyCAT-A sorbent2) and column characteristics not available for buying as a ready-made column.
To cope with that, many laboratories pack capillary columns in-house. However, the common packing procedure with a 100 bar pressure bomb (pressure injection cell)3 is ill-suited to the UHPLC column packing due to high backpressure of sub-2 µm UHPLC sorbents resulting in a dramatic packing rate reduction in comparison to larger-sized HPLC sorbents. While short UHPLC columns can still be very slowly packed, manufacturing of long UHPLC columns is physically impossible4.
Standard capillary column packing is done at relatively low pressures-up to 100 bars, and with a very low sorbent slurry concentration. Hence, two possible directions of speeding-up the process are available. It is possible to increase the packing pressure5. However, this requires special equipment and, practically, installation of a new method in the laboratory. Another way is to increase the sorbent slurry concentration6. High sorbent slurry concentration packing is described in combination with ultra-high packing pressure in a previous publication7. However, at 100 bar pressure, which is used in most of the existing packing bombs, higher sorbent concentration results in either packing rate slow-down or outright packing cessation. The effect was recently demonstrated to be due to sorbent clustering at the column entrance, and a simple trick of sorbent cupola destabilization by hammering the column entrance with a magnet bar inside a sorbent vial was suggested4. The resulting method, named FlashPack, uses the same 100-bar pressure bomb packing setup. At the same time, minor but critical changes in the packing procedure allow packing from very high sorbent slurry concentration and production of very long UHPLC columns (50 to 70 cm, and longer) in less than an hour, while a short column can be produced in minutes with the separation quality equal to commercial columns of the same parameters4. The FlashPack approach was already successfully used in multiple proteomics projects for the preparation of both reverse phase (RP)8,9,10,11,12,13,14 and hydrophilic interaction (HILIC)2 capillary columns.
Here we describe in detail, the modifications needed for adaptation of the FlashPack approach to the standard 100-bar pressure bomb packing procedure.
Protocol
The packing protocol consists of five steps (Figure 1): 1) Packing station preparation, 2) capillary preparation, 3) sorbent slurry preparation, 4) capillary packing in the pressure bomb, and 5) column packing-up in the HPLC system, cutting up to the size and UHPLC connection installation. The FlashPack optimization requires adjustments to be made in sections 3 and 4 as compared to the common protocol.
1. Packing station assembly
- Prepare a gas tank filled with either nitrogen, helium or argon furnished with a single stage gas regulator with the outlet pressure > 50 bars. Maximum pressure is limited by the pressure bomb compatibility.
- Connect the regulator to the vent valve of the pressure bomb.
- If the pressure bomb is not equipped with an integrated magnetic stirrer, place the bomb on a magnetic stirrer.
- Connect a narrow ID plastic tubing (e.g., 0.13 mm) to the vent outlet of the pressure bomb and put it into a vessel with water.
2. Capillary preparation
- Prepare a fritted capillary with an integrated glass frit formed from Kasil and formamide15 or a pulled emitter capillary prepared by a laser puller16. The capillary is made 10-15 cm longer than the intended column length.
NOTE: See Table 1 for the discussion of possible issues associated with different capillary sizes and frit types. Table 2 contains an example of a P2000 laser puller program for making pulled-emitter capillaries. - Protect a pulled emitter end with a cut gel-loading pipette tip.
- Cut the tip so it fits tightly around the 360 μm OD capillary (it can be moved along the capillary but requires some effort to do that).
- Slide the cut pipette tip onto the capillary from the direction of the capillary front end and move it up to the emitter end.
- Slide back the protecting tip when the column is spraying. Slide the tip forward to have the emitter end inside the tip when the column is not spraying (even when the column is under the flow, but still not spraying).
3. Sorbent slurry preparation
- Prepare a stock sorbent vial: Place ~50 mg of dry sorbent into a 1.5 mL centrifuge tube. Here, Reprosil Pur C18 is used as an example.
- Add 1 mL of methanol to the sorbent tube.
- To mix it completely, vortex the tube for 10 s using a vortex mixer.
- Sonicate in a sonication bath for 10 s.
- Let the sorbent get soaked thoroughly for 20-30 min. Then, vortex and sonicate it once more.
- Prepare a working sorbent vial. Use a conical bottom vial which fits into the bomb.
NOTE: It can be either another 1.5 mL centrifuge tube or any other vial depending on the particular pressure bomb design. For this experiment, conical bottom screw cap tube cut to the height of the pressure bomb is used. - Resuspend the sorbent in the stock sorbent vial and transfer 500 µL into the working sorbent vial with a magnet bar of 2 x 3 mm size.
- Add methanol up to ~1 mL to the working vial.
- Let the working vial stand on the table for 10 min for the sorbent to settle by gravity.
- If, after settling, the sorbent layer is below 4 mm, add more of the stock sorbent slurry and wait for the sorbent to settle for another 10 min.
NOTE: The prepared working vial is intended for preparation of multiple columns over months. If the working sorbent vial stays without stirring for more than 2 h, it must be vortexed for 10 s, sonicated for 10 s and settled by gravity. Routinely, the sorbent is resuspended in the morning before packing. Then, it is good for packing for the whole day if there are no long pauses between sequential column packing. If the sorbent in the working vial dries out, add methanol, and run the full sorbent preparation procedure as for the stock sorbent vial (steps 3.2-3.5).
4. Capillary packing in a pressure bomb
CAUTION: Always wear protective glasses when working with the pressure bomb. Do not wear gloves. These severely reduce the sense of touch required for proper handling of small diameter capillaries and lead to mistakes.
- Place the sorbent vial into the pressure bomb and fix all the nuts tightly.
- Start the rotation at 60-100 rpm.
- Insert the fritted or pulled emitter capillary into the bomb: push it to the very bottom of the vial, and then lift it up 2-3 mm and fix the nut.
NOTE: Apply a minimum required force to fix the capillary to avoid capillary and ferrule damage. The best is hand-tightening. If a hex-wrench is used, apply minimum sufficient effort for tightening. - Check whether the capillary is properly fixed-it must be impossible to move the capillary by pulling it out by hand.
- Very slowly open the pressure bomb valve while keeping the open end of the capillary pointed away from your face.
- Watch the initial steps of the packing process.
NOTE: Immediately upon pressurization, the sorbent fills the capillary and it becomes non-transparent for the whole length. As soon as the sorbent starts to pack inside the distal end, the backpressure increases, the flow slows down and the even sorbent slurry inside the capillary reforms into several sorbent packets separated by sorbent-free gaps. Already packed sorbent is visible as a densely colored continuously growing region. - Keep the sorbent filled regions to be at least 70% of the capillary length with small sorbent free gaps for the whole duration of the packing process.
- There are several common issues to watch during the packing process, that require on-the-flight setup adjustment to keep efficient sorbent delivery into the capillary.
NOTE: More details on the sorbent-delivery efficiency adjustment are described in Table 3.- Issue 1: When new sorbent stops entering the capillary while the sorbent already inside keeps moving, follow the steps below.
NOTE: This is the most frequent issue. In most cases the capillary entrance gets blocked by self-aggregating sorbent clusters. Apply the following steps one by one until the sorbent flow is restored, and then skip the rest of the issue-related steps.- Increase the rotation speed to 500 rpm and immediately reduce it back to 60-100 rpm. Usually, it restores the sorbent flow. Check the rotation speed to be at least 60 rpm for the rest of the packing process.
- If it does not help, briefly vent the packing bomb and immediately pressurize it back.
- If it does not help or the blocking happens again, reposition the capillary inside the sorbent layer. The absence of the sorbent can be due to the capillary open end being either too high above the magnet bar, so the column end does not touch it, or the capillary sticking into the vial bottom. First, vent the bomb completely, loosen the nut, push the capillary to the bottom, and then pull it 2 mm back. Fix the nut.
- If the blocking persists, vent the system, take out the sorbent vial and vortex and sonicate it once more. Check the capillary frontal end for damage under the microscope and cut ~5 mm of the front end if needed.
- Issue 2: When the sorbent fills only a small part of the capillary with long empty regions, follow the steps below.
- Check the rotation speed. If the rotation is too slow, the cupola breaking is not efficient enough. Increase the rotation speed to ~150 rpm.
- If the rotation is too fast, the sorbent gets resuspended into the larger vial volume and the local sorbent concentration around the column entrance is low. Slow the rotation speed down to 60-100 RPM.
- Check the sorbent level. The same issue with little sorbent inside the capillary is observed when there is not enough sorbent in the vial. When the sorbent gets used up, fill the vial with the new sorbent to keep the sorbent layer no less than 4 mm high after gravity-induced settling.
- Issue 1: When new sorbent stops entering the capillary while the sorbent already inside keeps moving, follow the steps below.
- Keep on packing the column till the target column length plus 5-7 cm is achieved.
- Stop the rotation and very slowly depressurize the bomb.
- Open the bomb valve a little and wait for the bubble burst inside the water bottle to subside. Then, open the valve a little wider and again wait for the bubble burst to slow down.
- Release the pressure in increments until no gas comes out of the valve.
NOTE: Do not open the valve all the way at once - it will lead to bubbling inside the capillary and the sorbent going back into the vial. If that happens, pressurize the bomb back and wait for the column to get packed again.
- When the gas stops coming out of the vent valve, take the packed capillary out of the pressure bomb.
NOTE: Do not let the column dry out. If not connected to the HPLC system immediately for further packing, put the packed capillary into storage by submerging it whole into 10% EtOH solution. A liquid-tight polypropylene food storage container can be used for capillary storage. Disconnected HPLC columns are stored in the same manner. - If no further packing is planned, take out the sorbent vial from the bomb and close it tightly. Keep it for further column packing.
5. Packing in the HPLC column
- Connect the packed capillary to the HPLC system via an HPLC connection.
- Start the flow at 95% solvent B (80 or 100% acetonitrile, 0.1% formic acid (FA)) targeting 250-300 bar pressure. For 40 cm packed capillary, use the flow rate of 200-300 nL/min.
NOTE: The packing flow rate is 200 nL/min for 40-50 cm column with 100 µm ID packed with 2 µm sorbent. Some other column sizes are listed in Table 4. Flow rates for other column lengths and IDs are estimated from the direct proportionality between the backpressure and the column length and cross-section. The exact flow-rate is adjusted to the actual packed length, which is by default longer than the targeted column length. Also note, that the 300 bar pressure targets the physical pressure limit of the HPLC connection. For higher-pressure connections, higher flow-rates up to the connection pressure limit are to be used for faster packing. - Watch for the loose sorbent inside the capillary getting packed and getting added to the total packed length.
- Without stopping the flow, dip the column body two times into the sonication bath.
NOTE: Do not immerse column ends and capillary connections-only a part of the column body. Sonication step helps to improve column reproducibility, especially for extremely long columns >50 cm long (unpublished data); however, it adds a random chance of breaking the self-assembling sorbent frit inside the emitter end of the pulled emitter capillary and blocking the column completely. While sonication can be universally applied to any glass-fritted columns, we suggest sonicating pulled emitter columns only for the column length > 50 cm. - When the sorbent bed stops shrinking, dip the column body into the sonication bath two times more without stopping the flow.
- Run the column for an additional 10 min at 300 bars.
- Stop the flow, wait for the pressure to drop to below three bars, and disconnect the column.
- Visually inspect the column for the lack of gaps and discolorations. If any are found, sonication under the flow can be repeated. For critical experiments, consider making a new column.
- Cut the column to the desired length.
NOTE: Properly done cutting is a prerequisite for column efficiency. Make a notch in polyimide coating with the scribe, partially crack the capillary and pull two pieces apart. - Polish the column front end on a ceramic wafer or with lapping film.
- Reconnect the column to the LC system using a UHPLC connection.
- Start the working flow rate at 2% B depending on the column ID according to Table 4. Wait for the pressure to equilibrate and check the column backpressure.
NOTE: The working flow rate is adjusted according to column parameters. For instance, a 30 cm long 100 µm ID column is run at 500 nL/min. - Ensure that the backpressure is within 5% of the expected value (see Table 5), This confirms that the column is packed properly and is ready to use.
NOTE: The column backpressure is the total pressure in the gradient channel of the HPLC system with the column connected minus the backpressure of the capillaries before the column. At the same time, the values in Table 5 are arbitrary (they give an arbitrary scale of what to expect). The intra-laboratory similarity of the backpressure from column to column is a more important indicator that everything works properly. The actual absolute backpressure depends on many parameters, such as the sorbent size and characteristics, the capillary ID, manufacturer and batch, the shape of the pulled emitter end or the density and length of the glass frit, the solvent characteristics and the ambient temperature in the room, etc. If the backpressure is too high, see Table 1 for possible issues.
Results
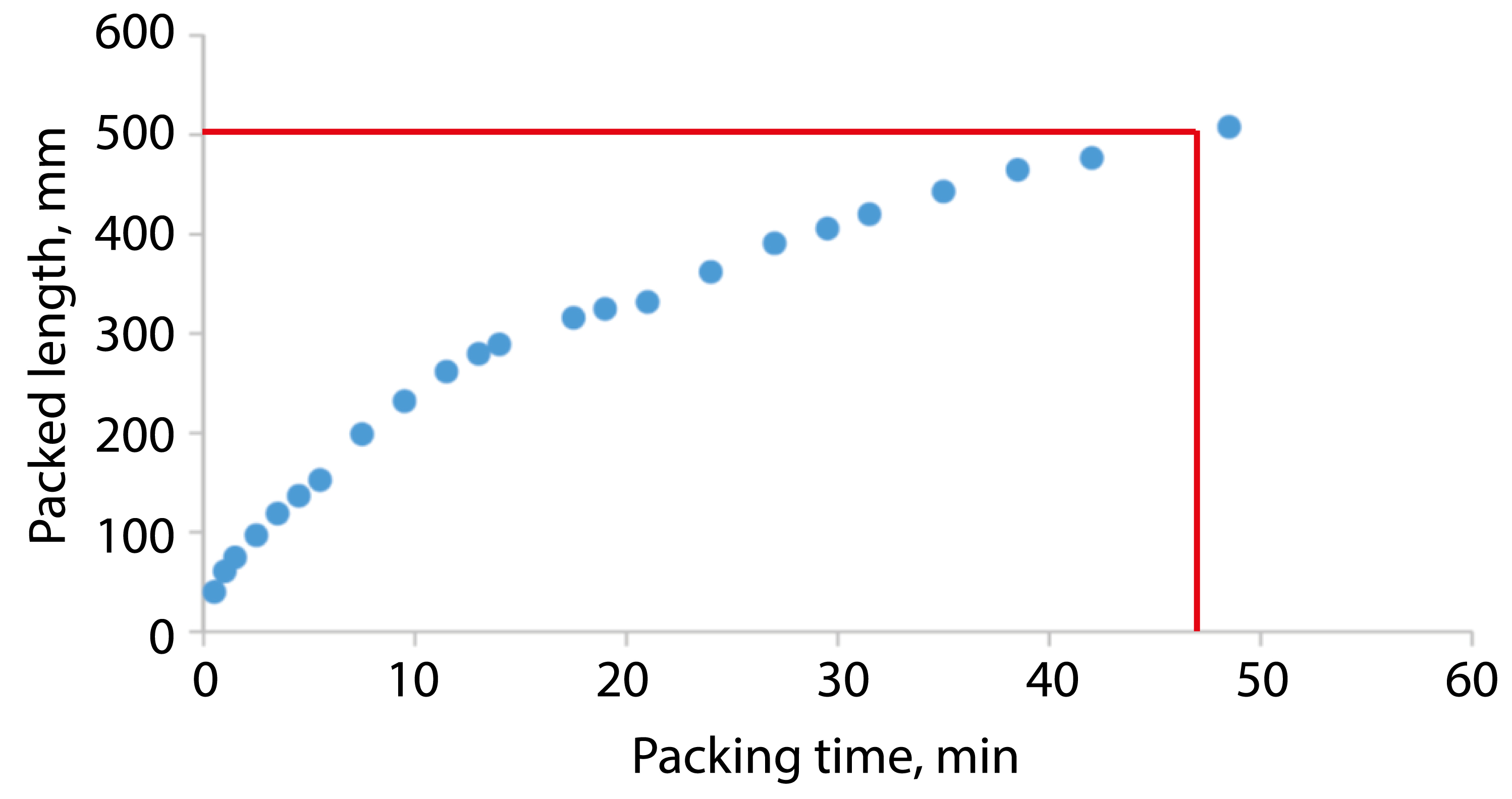
The FlashPack approach is based on the standard packing setup and follows the same packing pipeline. Packing is done into standard fritted or pulled emitter capillaries. The principal optimization lies in the sorbent slurry concentration: the standard method is incompatible with a high-concentrated sorbent suspension used in FlashPack. The result is a fast production method for long UHPLC columns, for example, a column packed for 50 cm length with 1.9 µm sorbent in less than 1 h (Figure 2).
To demonstrate the application of the FlashPack approach, a 30 cm 100 µm ID capillary column was prepared (Table 6). Packing of ReprosilPur C18 1.9 µm sorbent was done at 60 bars into a 50 cm long 100 µm ID pulled emitter capillary, prepared by a P2000 laser puller. The capillary was packed to ~40 cm in 40 min with some more loose sorbent left inside the capillary. The packed capillary was connected to an HPLC system and run at 300 nL/min with solvent B (80% acetonitrile, 0.1% FA). After two rounds of 5 s sonication, the final packed length was 43 cm. The column was disconnected, cut to 30 cm and connected to the HPLC system using a UHPLC connection. We routinely use 360 µm sleeveless PEEK nut-ferrule and 360 µm stainless steel union. This combination holds at least up to 700 bars if tightened strongly. The manufactured column has a backpressure of 520 bars at 2% solvent B at 500 nL/min, which is consistent with the expected value range (Table 5).
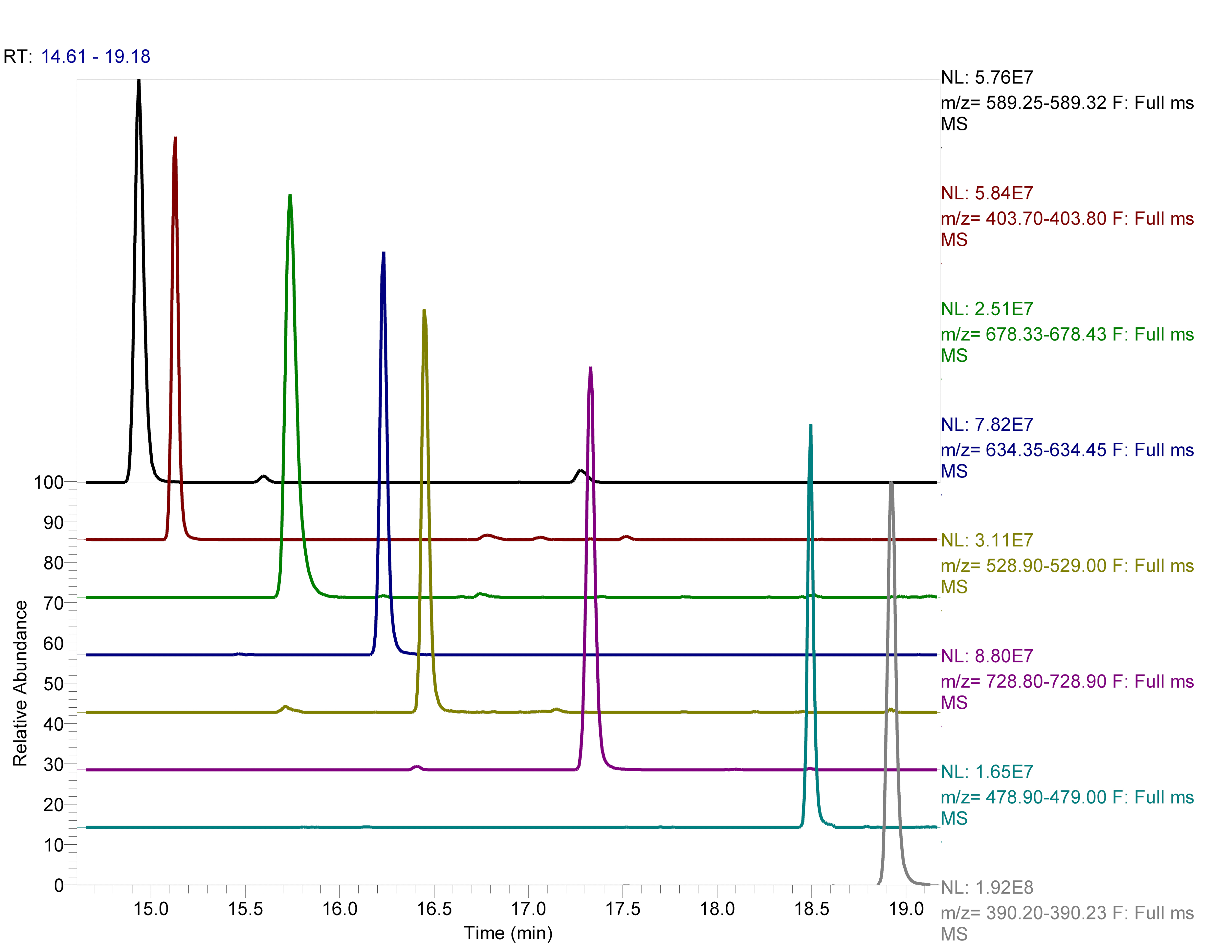
As a demonstration of the column efficiency, we used the manufactured 30 cm column to separate 50 fmol of a tryptic digest of cytochrome C protein in a 15 min gradient from 2% to 50% B. Extracted ion chromatograms showed the peaks to be highly symmetrical with minimum tailing. Average FWHM was around 3 s (Figure 3).
Table 1: Troubleshooting for high working backpressure of the column. Please click here to download this Table.
Table 2: P2000/F laser puller program. P2000/F laser puller program for the preparation of pulled emitter capillaries from 360 µm OD 100 µm ID fused-silica polyimide coated capillaries without internal coating at room temperatures 23-25 °C. Please click here to download this Table.
Table 3: FlashPack-specific packing issues and checkpoints to control during the packing process. Please click here to download this Table.
Table 4: Exemplary packing and working flow rates for different column IDs and length. Please click here to download this Table.
Table 5: Expected backpressure for a column packed with 2 µm spherical sorbents and run at working flow rate (according to the column ID) in RP solvent system at RT. Please click here to download this Table.
Table 6: Exemplary packing of 30 cm UHPLC column. Please click here to download this Table.

Figure 1: Capillary column packing scheme. Stages 1 to 3 are preparatory, followed by pressure bomb packing and finished by HPLC packing-up. Stages 3 and 4 are modified for the ultra-efficient FlashPack protocol. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Packing rate for a fritted capillary 100 µm ID with ReprosilPur C18 AQ 1.9 µm at 100 bars in methanol. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Extracted ion chromatograms of tryptic peptides of cytochrome C. Extracted ion chromatograms of tryptic peptides of cytochrome C after separation of 50 fmol in 30 cm long 100 mm ID pulled emitter capillary column packed with ReprosilPur C18 AQ 1.9 µm in a gradient of buffer B (80% acetonitrile, 0.1% FA) and in buffer A (2% acetonitrile, 0.1% FA) from 2% to 40% B in 15 min at 500 nL/min at RT. Detection was performed using a mass-spectrometer. Absolute intensities and extracted m/z ranges for each peptide are shown to the right of the spectra. Please click here to view a larger version of this figure.
{kind=link}
Discussion
In-house capillary column packing is highly popular in large laboratories working on multiple independent projects. However, a common packing method from a low concentration sorbent suspension has major limitations in the speed and is unable to produce long UHPLC columns.
FlashPack is a modification of the standard packing procedure which makes packing from a very high sorbent concentration possible. The theoretical basis of the method lies in the continuous sorbent cupola destabilization at the column entrance for the whole packing duration. The latter is technically achieved by column entrance being continuously hit with a magnet bar. The method of cupola destabilization is intentionally developed to have the packing setup completely similar to the common packing process, but the trick of FlashPack lies in the details of the sorbent slurry preparation, capillary positioning, and magnet bar usage during the packing process.
The sorbent slurry is prepared as a sediment sorbent layer in a large solvent volume. It is interesting that the pressure bomb-based packing does not require the same packing conditions for column to column. In FlashPack, we never know the exact sorbent slurry concentration around the column entrance. It is impossible to measure and control exactly, as it also changes during the packing process. However, the final columns are still very reproducible4 irrespective of how the packing was achieved.
The basis for the fast packing lies in the efficient sorbent cupola destabilization. For this reason, it is important to control sorbent entering the capillary and to maintain the optimal cupola destabilization conditions throughout the whole packing duration. There are several possible issues that might prevent efficient sorbent delivery. Some examples of these are sorbent layer resuspension by fast magnetic bar rotation, inefficient cupola destabilization due to either wrong relative capillary to the magnet bar positioning or too slow magnet bar rotation. The issues themselves and how they are to be addressed are discussed in detail in the protocol section.
After the column is packed, the major column parameter to check is the column backpressure. The pressure values listed in Table 5 provides a reference point to what is expected for one of the popular sub 2 µm bead size sorbent-ReproSil PUR C18 AQ (1.9 µm). At the same time, additional backpressure might be added by the frit or a too narrowly pulled emitter and one should constantly monitor for that. If packing is done into a pulled emitter, we still suggest measuring the expected column backpressure for the particular sorbent in use by packing fritted capillaries first, and then to see whether the self-assembling frit adds too much. For any high-pressure issues, use the guidelines provided in Table 1 to pinpoint the problem.
In our experience, a packed column without discolorations, gaps, and with the proper backpressure works in 100% of the cases and gives the separation quality close to what can be expected from the column length and sorbent characteristics. A column with discolorations is not guaranteed to work properly but can still give satisfactory results.
Most of the time, if there are any problems with the separation quality, they do not come from the column itself, but rather from other parts of the separation system, namely, pumps, solvents, or connections. Especially potentially harmful is any post-column connections. Bad connection with a dead volume between the emitter and the fritted column leads to major peak broadening and tailing due to very low flow rates in capillary chromatography.
One more important issue specific to the FlashPack approach is that it uses a lot of expensive sorbents in a working sorbent slurry vial. Please remember, that the sorbent slurry in FlashPack is intended for multiple use. Take care of the sorbent. Avoid unnecessary magnet bar stirring to reduce sorbent grinding-remember to stop the rotation as soon as the packing is finished. And do not leave the open sorbent vial in the pressure bomb to avoid sorbent drying. Though the sorbent can still be used after that, it takes time to remake the sorbent slurry.
The method works equally well for both fritted capillaries and pulled-emitter capillaries. The FlashPack principle increases the packing rate for capillary IDs from 20 to 250 µm (smaller and larger were not tested). It is also applicable to all the sorbents, both fully and superficially porous, we could test (reflecting that the sorbent cupola formation in high sorbent slurry concentration is not limited specifically to RP sorbents). Besides, solvent parameters clearly affect the packing according to their physical and chemical characteristics. For example, less viscous acetone gives even higher packing rate than methanol at the same packing pressure. However, it is also less polar than methanol and reduces sorbent particles sticking to each other. The effect by itself prevents sorbent cupola formation in the beginning of the packing when the flowrate is still high. However, reduction in sorbent particle interaction also leads to less reliable self-assembling frit formation and more frequent pulled-end blocking during the packing. So, while acetone is better for packing of fritted capillaries, it is less suitable for pulled-emitter capillaries, with the methanol as a slurry solvent being slower but suitable for both types of ending. Packing from hexane or dichloromethane (DCM) are extreme cases of switching to acetone from methanol: they are even less polar, so they prevent sorbent cupola formation completely, however they are not fit for pulled-emitter packing at all. Besides, it was noted that extremely low DCM polarity leads to sorbent particles sticking to the internal capillary wall and making a thick layer on it. The layer thickness gradually grows and random local blocks form resulting in the column packed in several parts separated by regions without sorbent. Such effect was observed for C18 Peptide Aeris sorbent.
Another observed issue was YMC Triart C18 sorbent not being suspended in methanol properly, but to form some sort of flakes. However, that does not prevent it from getting packed with the FlashPack and giving very decent separation efficiency (unpublished data). Thus, while not being optimal for some cases, methanol was the most universal solvent to work for all the tested sorbents and columns. It is necessary to mention that we did not yet analyze how different slurry solvents affect column separation efficiency. At the same time, the efficiency of columns packed from methanol is already completely equal to commercial columns for the same sorbents4.
FlashPack is not the only existing approach to improve the packing rate of UHPLC columns. Fast packing from high sorbent slurry concentration is also possible with the use of ultra-high pressure packing7. The advantage of FlashPack is that it is much simpler as it does not require special ultra-high pressure pumps and pressure bombs for sorbent delivery and capillary connections. At the same time, it was demonstrated that the columns packed at extreme pressures can have separation efficiency higher than lower pressure packed columns17. And while FlashPack produces columns identical to commercial ones used in the comparison4, for which we do not know the packing method, it was not yet tested how FlashPack columns stand against ultra-high pressure packed columns.
In summary, the described FlashPack method can be easily adapted to the existing packing protocol in the laboratory with some adjustments made to the protocol, while the setup stays completely the same. It speeds up the HPLC capillary column packing to minutes' time and allows production of long UHP capillary columns, which is plainly impossible with the standard packing procedure. The overall economy in the time and money for the laboratory by application of the FlashPack approach can be counted in tens of thousands of Euros per year. Additionally, ability to produce UHP capillary columns locally opens the possibilities for experiment customization impossible with the available commercial products.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
The work was supported by RSF grant 20-14-00121. The authors thank P. V. Shliaha (Memorial Sloan Kettering Cancer Center) for fruitful discussions.
Materials
| Name | Company | Catalog Number | Comments |
| Acetonitrile with 0.1% (v/v) Formic acid | Merck | 1.59002 | |
| centrifuge tube 1.5 mL | Eppendorf | ||
| Ceramic Scoring Wafer | Restek | 20116 | any ceramic wafer is suitable for capillary polishing |
| Diamond-chip bladed scribe | NewObjective | Diamond-chip bladed scribe | recommended for capillary cutting |
| fused silica capillary 100 mm ID 375 mm OD | CM Scientific | TSP100375 | |
| GELoader tips | Eppendorf | 30001222 | |
| HPLC system | ThermoScientific | Ultimate3000 RSLCnano | |
| laser puller | Sutter | P2000/F | |
| magnet bar 2x5 mm | Merck | Z283819 | |
| MeOH | Merck | 1.06018 | |
| microspatula | Merck | Z193216 | |
| PEEK ferrule 360 mm | VICI | JR-C360NFPK | use to connect the column to UPLC union |
| pipette tip, 1000 uL | Merck | Z740095 | |
| pipette, 1000 uL | Gilson | Pipetman L P1000L | |
| pressure bomb | NextAdvance | PC-77 MAG | |
| regulator | GCE | Jetcontrol 600 200/103 | |
| Reprosil Pur C18 AQ 120 1.9 mm | Dr. Maisch | r13.aq.0001 | |
| Screw cap tubes without caps, conical bottom, self-standing, 0.5 mL | Merck | AXYST050SS | |
| Screw cap tubes without caps, conical bottom, self-standing, 1.5 mL | Merck | AXYST150SS | |
| Screw caps with O-rings | Merck | AXYSCOC | |
| sonication bath | Elma | Elmasonic S30 H | |
| union HPLC | VICI | JR-C360RU1PK6 | HPLC connection from 1/16 OD HPLC capillary to 360 um capillary column |
| union UPLC | VICI | JR-C360RU1FS6 | UPLC connection from 1/16 OD HPLC capillary to 360 um capillary column |
| vortex | BioSan | V-1plus | |
| Water with 0.1% (v/v) Formic acid | Merck | 1.59013 |
References
- Shishkova, E., Hebert, A. S., Now Coon, J. J. Now, more than ever, proteomics needs better chromatography. Cell Systems. 3 (4), 321-324 (2016).
- Shliaha, P. V., et al. Middle-down proteomic analyses with ion mobility separations of endogenous isomeric proteoforms. Analytical Chemistry. 92 (3), 2364-2368 (2020).
- . Pressure injection cells - next advance - laboratory instruments Available from: https://www.nextadvance.com/pressure-injection-cells-lc-ms-capillary-column-packing-loader/?target=Overview (2021)
- Kovalchuk, S. I., Jensen, O. N., Rogowska-Wrzesinska, A. FlashPack: Fast and simple preparation of ultrahigh-performance capillary columns for LC-MS. Molecular & Cellular Proteomics: MCP. 18 (2), 383-390 (2019).
- MacNair, J. E., Lewis, K. C., Jorgenson, J. W. Ultrahigh-pressure reversed-phase liquid chromatography in packed capillary columns. Analytical Chemistry. 69 (6), 983-989 (1997).
- Bruns, S., et al. Slurry concentration effects on the bed morphology and separation efficiency of capillaries packed with sub-2 µm particles. Journal of Chromatography. A. 1318, 189-197 (2013).
- Godinho, J. M., Reising, A. E., Tallarek, U., Jorgenson, J. W. Implementation of high slurry concentration and sonication to pack high-efficiency, meter-long capillary ultrahigh pressure liquid chromatography columns. Journal of Chromatography. A. 1462, 165-169 (2016).
- Andrzejczak, O. A. The effect of phytoglobin overexpression on the plant proteome during nonhost response of barley (Hordeum vulgare) to wheat powdery mildew (Blumeria graminis f. sp. tritici). Scientific Reports. 10 (1), 9192 (2020).
- Elchaninov, A., et al. Comparative analysis of the transcriptome, proteome, and miRNA profile of kupffer cells and monocytes. Biomedicines. 8 (12), 627 (2020).
- Babenko, V. V., et al. Draft genome sequences of Hirudo medicinalis and salivary transcriptome of three closely related medicinal leeches. BMC Genomics. 21 (1), 331 (2020).
- Babenko, V. V., et al. Identification of unusual peptides with new Cys frameworks in the venom of the cold-water sea anemone Cnidopus japonicus. Scientific Reports. 7 (1), 14534 (2017).
- Loughran, G., et al. Unusually efficient CUG initiation of an overlapping reading frame in POLG mRNA yields novel protein POLGARF. Proceedings of the National Academy of Sciences of the United States of America. 117 (40), 24936-24946 (2020).
- Radzisheuskaya, A., et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nature Structural & Molecular Biology. 26 (11), 999-1012 (2019).
- Rubtsova, M., et al. Protein encoded in human telomerase RNA is involved in cell protective pathways. Nucleic Acids Research. 46 (17), 8966-8977 (2018).
- Maiolica, A., Borsotti, D., Rappsilber, J. Self-made frits for nanoscale columns in proteomics. PROTEOMICS. 5 (15), 3847-3850 (2005).
- Ishihama, Y., Rappsilber, J., Andersen, J. S., Mann, M. Microcolumns with self-assembled particle frits for proteomics. Journal of Chromatography. A. 979 (1-2), 233-239 (2002).
- Shishkova, E., Hebert, A. S., Westphall, M. S., Coon, J. J. Ultra-high pressure (>30,000 psi) packing of capillary columns enhancing depth of shotgun proteomic analyses. Analytical Chemistry. 90 (19), 11503-11508 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved