Method Article
Sample Preparation to Bioinformatics Analysis of DNA Methylation: Association Strategy for Obesity and Related Trait Studies
* These authors contributed equally
In This Article
Summary
The present study describes the workflow to manage DNA methylation data obtained by microarray technologies. The protocol demonstrates steps from sample preparation to data analysis. All procedures are described in detail, and the video shows the significant steps.
Abstract
Obesity is directly connected to lifestyle and has been associated with DNA methylation changes that may cause alterations in the adipogenesis and lipid storage processes contributing to the development of the disease. We demonstrate a complete protocol from selection to epigenetic data analysis of patients with and without obesity. All steps from the protocol were tested and validated in a pilot study. 32 women participated in the study, in which 15 individuals were classified with obesity according to Body Mass Index (BMI) (45.1 ± 5.4 kg/m2); and 17 individuals were classified without obesity according to BMI (22.6 ± 1.8 kg/m2). In the group with obesity, 564 CpG sites related to fat mass were identified by linear regression analysis. The CpG sites were in the promoter regions. The differential analysis found 470 CpGs hypomethylated and 94 hypermethylated sites in individuals with obesity. The most hypomethylated enriched pathwayswere in the RUNX, WNT signaling, and response to hypoxia. The hypermethylated pathways were related to insulin secretion, glucagon signaling, and Ca2+. We conclude that the protocol effectively identified DNA methylation patterns and trait-related DNA methylation. These patterns could be associated with altered gene expression, affecting adipogenesis and lipid storage. Our results confirmed that an obesogenic lifestyle could promote epigenetic changes in human DNA.
Introduction
Large-scale omics technologies have been increasingly used in studies of chronic diseases. An interesting feature of these methods is the availability of a large amount of generated data to the scientific community. Therefore, a demand to standardize the protocols has arisen to allow technical comparison between studies. The present study suggests the standardization of a protocol to obtain and analyze DNA methylation data, using a pilot study as an applicated example.
Negative energy expenditure predominates in modern human lifestyles, leading to an excessive accumulation of adipose tissue and, consequently, the development of obesity¹. Many factors have increased obesity rates, such as sedentarism, high-calorie diets, and stressful routines. The World Health Organization (WHO) estimated that 1.9 billion adults were obese in 2016, which means that more than 20% of the world's population has over 30 kg/m2 BMI2. The most recent update of 2018 revealed that the prevalence of obesity in the United States of America (USA) was higher than 42%3.
Epigenetics is the structural adaptation of chromosomal regions to register, signal, or perpetuate altered activity states4. DNA methylation is a reversible chemical alteration in the cytosine-guanosine dinucleotides sites (CpG sites), forming 5-methylcytosine-pG (5mCpG). It can modulate gene expression by regulating the access of the transcription machinery to the DNA5,6,7,8. In this context, it is essential to understand which CpG sites are associated with obesity-related traits9. Many factors can support or prevent site-specific DNA methylation. Necessary enzymes for this process, such as DNA methyltransferases10 (DNTMs) and ten-eleven translocations (TETs), can promote DNA methylation or demethylation under environmental exposures11.
Considering the growing interest in DNA methylation studies over the last years, choosing the most appropriate analysis strategy to precisely answer each question has been an essential concern of researchers12,13,14. The 450K DNA methylation array is the most popular method, used in more than 360 publications14 for determining the DNA methylation profile. It can determine the methylation of up to 485,000 CpGs located in 99% of known genes15. However, this array has been discontinued and replaced with the EPIC, covering 850,000 CpG sites. The present protocol can be applied for both 450K and EPIC16,17,18.
The protocol is presented step-by-step in Figure 1 and comprises the following steps: population selection, sampling, experiment preparation, DNA methylation pipeline, and bioinformatics analysis. A pilot study performed in our laboratory is demonstrated here to illustrate the steps of the proposed protocol.

Figure 1: Schematic of the presented protocol. Please click here to view a larger version of this figure.
{kind=link}
Protocol
The ethics committee of Ribeirão Preto Medical School University Hospital of the University of São Paulo (HCRP-USP) approved the study (CAAE:14275319.7.0000.5440). The participants signed the consent form, and all procedures were conducted following the Declaration of Helsinki.
1. Population and sampling
- Recruit participants with BMI >30 kg/m2 for the study. For the present study, 32 women from an admixture population19 between 18 to 60 years old were recruited.
NOTE: In the HCRP-USP, higher rates of obesity were observed mainly in women20. Participants with obesity, with BMI ≥30 kg/m2 (n = 15), were recruited from the Metabolic Diseases outpatient clinic in HCRP-USP. The participants without obesity, with BMI between 18.5 kg/m2 and 24.9 kg/m2 (n = 17), were recruited from other clinics and did not have severe diseases; clinical data are shown in Table 1. - Exclude women who are pregnant, nursing mothers, smokers, and consume alcohol.
2. Anthropometry and body composition
- Measure the weight of the participants using a 200 kg digital anthropometric scale. Ensure to remove shoes and excess clothing.
- Assess height using a stadiometer with a 0.5 cm graduation. Participants were instructed to stand barefoot, feet together, and arms by their sides21.
- Collect the fat mass (FM) information using an electric bioimpedance. After 12 h of fasting, assess with an empty urinary bladder. Place the participants in the supine position, with legs apart and arms parallel without touching the body. Instruct participants to remove all metal accessories22.
- Obtain the waist circumference (WC) using an inextensible measuring tape with a 0.1 mm graduation. The tape was placed horizontally around the middle waist, just above the hip bones. The measurement occurred just after a breath out. According to the Centers for Diseases Control and Prevention (CDC), the WC cut-off for women is 88 cm23.
Table 1: Population characteristics. All variables were parametric (p > 0.05, Shapiro Wilk), and differences were evaluated using an Independent t-Test, in which p < 0.05 was considered significant. *p < 0.0001. Please click here to download this Table.
3. Collection of biological material
- Collect peripheral blood after 12 h of fasting, as described previously24.
- Collect 4 mL of the whole blood samples in collection tubes with anticoagulant (e.g., EDTA) and store them on ice for transportation.
4. DNA extraction
- Centrifuge each tube containing the blood at 2,000 x g for 10 min. Collect around 300 µL of the buffy coat (white interphase).
- Extract DNA from the peripheral blood using a commercially available instrument (see Table of Materials), as described previously25. Elute the DNA in 480 µL of ultrapure H2O.
- Assess DNA concentration and quality using a fluorometer. The integrity was evaluated using a 1% agarose gel analysis25. Store in a freezer at -80 ˚C.
5. Preparation before methylation analysis
- Ensure that the samples have the minimum DNA concentration (500 ng is the ideal initial input to start), but dilution may vary between samples in this step.
- Some reagents are not included in the microarray kit26, so purchase them separately27.
- Check if the kit contains all reagents used during the procedure; they are irreplaceable. Check the expiration date of all the reagents. Ensure other reagents and solutions not provided in the kit are also available (95% formamide/1 mM EDTA, 0.1 N NaOH, absolute ethanol, 2-propanol).
CAUTION: Formamide is a volatile reagent; the quality of the results can be improved if the product is fresh. The integrity and quality of formamide and absolute ethanol are considered critical because they can affect the staining process proposed by the manufacturer. The chosen alcohols must be ultrapure products specific for molecular biology analysis. - Once the microarray kit is acquired, download the Chips Decode Maps (DMAPs). Those files are available on the manufacturer's website 24-48 h after the kit is shipped and are excluded on the expiration date28. To download the mentioned files, perform the steps below.
- Ensure a computer with internet access is available. Perform the download using the Software Decode File Client.
- Before installing the program, ensure that the computer has outbound and inbound access granted by the institution; the Decode File Client does not have security restrictions.
- Log in to the Decode File Client software using the Access by BeadChip option. This login is performed using MyIllumina user ID and password. Registration on this page is required28.
- After logging in to the Decode File Client, enter the information listed below in their respective fields on the software home page28: List of barcodes; List of Box Ids; Purchase order number (PO); and Sales order number (remember not to include the letters that follow the numbers).
- Select the DMAPs to download according to the barcode number in the purchase box and specify the destination in the dialog box to save the DMAPs.
- Click the button to start the download.
NOTE: At the end of the download, make sure that the downloaded folders are not empty and store the files safely, as they will be necessary for the final step to convert the experiments to Intensity Data Files (IDAT files). After the expiry date, Illumina may remove the DMAPs data from its online database.
6. DNA methylation pipeline
NOTE: The DNA methylation experiment is divided into 4 days (Figure 1). Follow the manufacturer's recommendations and specifications to obtain accurate results.
- Day 1: Bisulfite treatment
- Start by denaturing 500 ng of genomic DNA, then add the conversion reagents. To perform this step, follow the recommendations of the bisulfite and microarray kits29.
- Treat the denatured DNA with 130 µL of the bisulfite conversion reagent at the standard concentration provided by the manufacturer.
- Incubate the tube in a thermocycler for 16 cycles at 95 °C for 30 s and 50 °C for 1 h. Ramp down the thermocycler to 4 °C for 10 min.
- Perform the washing step by adding 100 µL of the washing buffer and then centrifuging at full speed for 30 s. The concentration is not provided in the user guide, and the manufacturer standardizes the volumes.
NOTE: This reagent comes with the kit and does not need to be diluted or prepared.
- Day 2: Amplification - Methylation Assay, Manual Protocol
- Preheat the hybridization oven to 37 °C and allow the temperature to equilibrate.
- Apply a barcode label to a 0.8 mL microplate.
- Thaw the amplification reagents (Multi-Sample Amplification Mix 1, Random Primer Mix, and Multi-Sample Amp Master Mix) at room temperature (those reagents come with the kit and do not need to be diluted or prepared). Then, perform a gentle inversion at least 10x until the tubes' entire contents are mixed.
- Dispense 20 µL of the Multi-Sample Amplification Mix 1 (MA1) into the wells of the plate.
- Transfer 4 µL of the DNA sample from the bisulfite converted plate to the corresponding wells on the 0.8 mL plate. At this point, record the original DNA sample's ID on the laboratory tracking form corresponding to the plate wells.
- Dispense 4 µL of 0.1 N NaOH into each well of the plate containing the Multi-Sample Amplification Mix 1 (MA1) and the DNA sample.
- Seal the plate with a plastic seal.
- Vortex the plate to mix the reagents with the samples at 1,600 rpm for 1 min. Incubate for 10 min at room temperature and carefully remove the seal.
- Pipette 68 µL of Random Primer Mix (RPM) into each well of the plate. Pipette 75 µL of Multi-Sample Amp Master Mix (MSM) into each well of the plate (without removing the previous solution). Use a new seal to cover the plate.
NOTE: Take care to combine the wells with the plate and orient it correctly. - Vortex the sealed plate at 1,600 rpm for 1 min. Incubate in the hybridization oven for 20-24 h at 37 °C.
- Day 3: Hybridization - Methylation Assay, Manual Protocol
- Preheat the block to 37 °C.
- Thaw the contents of the tube at room temperature. Carefully and gently invert the Fragmentation Solution (FMS) tubes at least 10x to thoroughly mix the contents.
- Carefully remove the plate from the hybridization oven. Pulse-centrifuge the plate at 280 x g.
- Now remove the seal from the plate. In each well of the plate containing a sample, add 50 µL of Fragmentation Solution (FMS) and seal the plate again.Vortex the plate at 1,600 rpm for 1 min.
- Centrifuge the plate at 280 x g for a few seconds (perform pulse centrifugation). Then, place the sealed plate on the heating block at 37 °C for 1 h.
- If it was decided to pause the experiment and freeze the plate, thaw it at room temperature, and centrifuge it at 280 x g. If it is not frozen, skip this step and move to Step 6.3.7.
- Leave the heating block to preheat to 37 °C.
- Leave the Wash Buffer to thaw at room temperature. When defrosted, invert it at least 10x to mix the contents.
- Remove the seal from the plate.
- Add 100 µL of Precipitation Solution (PM1) to each well of the plate containing samples.
- Seal the plate again with the seal.
- Vortex the plate at 1,600 rpm for 1 min.
- Incubate at 37 °C for 5 min.
- Then, place in the pulse centrifuge at 280 x g for 1 min.
NOTE: Set the centrifuge at 4 °C for the next step. - Carefully remove the seal from the plate and discard it.
- Add 300 µL of 2-propanol (100%) to each well containing the sample.
- Seal the plate with extreme care using a new seal. Take care not to shake the plate.
- Invert the plate at least 10x, mixing the entire contents.
- Incubate at 4 °C for 30 min.
- Centrifuge at 3,000 x g at 4 °C for 20 min.
NOTE: At the end of Step 6.3.20, immediately remove the centrifuge plate. - Remove the seal from the plate and discard it.
- Quickly invert the plate and drain the liquid on a pad to discard the supernatant. Then, hit the plate in a dry area on a paper towel to drain the liquid.
- Tap firmly several times for 1 min or until all the wells are free of any liquid.
- Leave the plate inverted and uncovered for 1 h at room temperature.
NOTE: It is expected to see blue pellets at the bottom of the wells. - Preheat the hybridization oven to 48 °C.
- Turn on the heat block to preheat. Allow 20 min for this process.
- Thaw Resuspension, Hybridization, and Wash Solution at room temperature. Invert at least 10x. Check that there is no salt precipitated in the solution.
- Add 46 µL of Resuspension, Hybridization, and Wash Solution (RA1) to each well of the plate.
NOTE: Any remaining reagents must be reserved for the staining and hybridization steps. - Apply the seal to the plate. Use an aluminum seal to cover the plate. Hold tight to perform the sealing evenly.
- Check if all wells are safely closed to avoid evaporation.
- Carefully place the newly sealed plate in the hybridization oven and incubate at 48 °C for 1 h.
- Vortex the plate at 1,800 rpm for 1 min.
- Pulse centrifuge at 280 x g.
- Transfer the plate to the heat block at 95 °C for 1 h.
- Prepare the Hybridization (Hyb) Chambers by placing them together. Dispense 400 µL of Humidifying Buffer (PB2) into the reservoirs of the Hyb Chambers. Put the lid on immediately.
- Load each sample from the plate into the sample entry opening of the microarray.
- Load the Hyb Chamber inserts containing the microarrays into the Hyb Chamber. Close the Hyb Chamber crosswise to avoid possible shifting of the Hyb Chamber.
NOTE: Hyb Chambers need to be incubated at 48 °C for at least 16 h, but not more than 24 h.
- Day 4: Staining - Methylation Assay, Manual Protocol
- Resuspend the Staining Solution 4 with 330 mL of 100% EtOH, obtaining a final volume of 350 mL. Shake the Staining Solution 4 bottle vigorously to ensure complete resuspension. Keep at room temperature.
NOTE: Staining Solution 4 can be stored for up to 2 weeks at a temperature between 2 °C and 8 °C. - Remove the chambers from the hybridization oven and allow them to cool on the bench for 30 min before opening.
- While the Hyb Chamber is cooling, fill two washing containers with Wash Buffer (200 mL per washing container). Remember to label each container as "Wash Buffer" (PB1 and PB2).
- Fill the microarray alignment accessories with 150 mL of Wash Buffer.
- Separate the plastic spacers and, if necessary, clean the glass.
- To wash the microarray slides, attach the wire loop to the grid and immerse the dripping equipment in the containers with 200 mL of Wash Buffer for a total of 10x.
- Remove all inserts from the Hybridization Chamber and remove each array from the inserts. Remove the seal.
NOTE: During removal, do not touch the exposed matrices. - While removing the microarray slides from the Hybridization Chamber, be careful not to spill anything.
- Wash the microarrays in the wash buffer (PB1) solution slowly and lightly, moving up and down 10x. Move to a fresh PB1 solution and perform the wash by slowly moving the microarrays 10x up and down in the solution.
- Remove the microarray from the container and confirm the absence of residue.
- Use a transparent spacer at the top of each microarray and guide the spacers to the corresponding positions.
NOTE: Be careful to use only the transparent spacers, not the white ones. - Position the alignment bar on the alignment accessory.
- Place a glass backplate on the top of the clear spacer; be careful to cover the entire microarray. Then, attach metal clamps to the flow chambers. If necessary, use scissors to trim the ends of the flow chamber spacers.
- Immediately wash the Hybridization Chamber reservoirs using ddH2O and scrub with a small cleaning brush. Do not allow any Humidifying Buffer to remain in the reservoir.
- Move to the next step that is responsible for the Extending and Staining Microarray.
NOTE: Leave all the flow chambers horizontally mounted on the laboratory bench. Only re-handle them when the entire staining step is ready. Heat the Resuspension, Hybridization, and Wash Solution to a temperature between 20 °C and 25 °C. Mix gently until no crystal is seen in the solution. - Place the reagents in a rack in sequence. If any are frozen, leave them at room temperature and then invert them at least 10x to mix the solutions.
- Fill the water circulator to the appropriate level.
- Turn on the pump and set the temperature to 44 °C.
- Remove any bubbles that are attached to the chamber rack.
- Perform tests on the chamber rack using a temperature probe. All tested locations must be between 44 °C ± 0.5 °C.
NOTE: The following steps must be performed without interruption. - When the rack reaches a temperature of 44 °C, quickly place each assembly in the flow chamber.
NOTE: For the following steps, pipette all reagents into the back glass plate. - Pipette 150 µL of Resuspension, Hybridization, and Wash Solution (RA1). Then, incubate for 30 min. Repeat this step a total of 5x.
- Pipette 450 µL of Staining Solution 1 (XC1) and incubate for 10 min.
- Pipette 450 µL of Staining Solution 2 (XC2) and incubate for 10 min.
- Pipette 200 µL of Two-Color Extension Master Mix (TEM) and incubate for 15 min.
- Pipette 450 µL of 95% formamide/1 mM EDTA and incubate for 1 min. Repeat twice.
- Incubate for 5 min.
- Decrease the temperature of the chamber rack to 32 °C (indicated on Superior Two-Color Master Mix).
- Pipette 450 µL of Stain Solution 3 (XC3) and incubate for 1 min. Repeat 1x.
- Wait for the rack in the chamber to reach the correct temperature.
NOTE: To read the microarray image immediately after the coloring process, turn on the scanner at this stage. - Now dispense 250 µL of Superior Two-Color Master Mix (STM), followed by a 10 min incubation.
- Then, dispense 450 µL of Stain Solution 3 (XC3), followed by 1 min incubation. Repeat 1x.
- Wait for 5 min and then add 250 µL of Anti-Stain 2-Color Master Mix (ATM) and incubate for 10 min.
- Now dispense 250 µL of Superior Two-Color Master Mix (STM), followed by a 10 min incubation.
- Then, dispense 450 µL of Stain Solution 3 (XC3), followed by 1 min incubation. Repeat 1x.
- Wait for 5 min.
- Add 250 µL of Anti-Stain 2-Color Master Mix (ATM) and incubate for 10 min.
- Add 450 µL of Stain Microarray Solution 3 (XC3) and incubate for 1 min, then repeat again.
- Wait for 5 min.
NOTE: Do the following repetition scheme: Step 6.4.34., Step 6.4.35. and Step 6.4.36.; Step 6.4.37., 6.4.38., and Step 6.4.39.; Step 6.4.34., Step 6.4.35., and Step 6.4.36. - Now, immediately remove the flow chambers from the rack and carefully place them horizontally on the laboratory bench. Make sure the temperature is ambient.
- 6.4.41.Place 310 mL of Wash Solution (PB1) for each microarray in a wash container.
- Remove the metal clamps and the glass, and finally, the microarray.
- Place the microarrays on a staining rack and place them in the wash container containing 10x PB1 solution. Ensure that the barcodes are facing away from whoever is handling them and that all the microarrays are submerged.
- Move slowly and lightly with up and down movements 10x. The microarray should be removed entirely from the solution before being submerged again. Then, let it remain submerged for 5 min.
- Vigorously shake the Stain Solution 4 (XC4) reagent to ensure total resuspension. Place 310 mL of XC4 into a wash container.
NOTE: Do not let the solution sit in the wash container for more than 10 min. - Lightly move the staining stand to the other container with XC4.
- Move slowly and lightly with up and down movements 10x. Ensure that the microarray is removed entirely from the solution before being submerged again.
- Then, let the microarray remain submerged for 5 min.
- Move the staining stand out of the solution and place it in a tube holder with the barcodes facing up.
- Carefully remove the microarray using locking forceps and place them on a shelf to dry.
- Dry them using the vacuum desiccator for 50-55 min at 675 mm Hg (0.9 bar).
- Clean the back of each microarray using EtOH.
CAUTION: Avoid touching the stripes, and do not allow EtOH to drip on the stripes in any way. - Proceed to image the microarray.
- Carefully store the microarray in a storage box at room temperature.
NOTE: Microarray images must be taken within 72 h.
- Resuspend the Staining Solution 4 with 330 mL of 100% EtOH, obtaining a final volume of 350 mL. Shake the Staining Solution 4 bottle vigorously to ensure complete resuspension. Keep at room temperature.
7. Bioinformatics analysis
NOTE: It is necessary to change some attributes of the kind of microarray (e.g., arraytype = "450k" should be replaced by arraytype = "EPIC"). The author of the ChAMP pipeline describes in detail all fields that should be modified to analyze arrays on the Bioconductor page of the package30.
- Sample sheet
- Transfer the IDAT files to the computer to proceed with bioinformatics analysis31. Create a sample sheet to carry out the data analysis31.
NOTE: Three columns are essential for this spreadsheet file. The first is Sample_name. This column must include the names or codes used to identify the samples. The second is the Sentrix_position. This column places the corresponding R01C01 of the samples, with R01 being responsible for the chip row and the column being referred to as C01. The third essential column is the Sentrix_ID, and it is responsible for receiving the number that identifies the microarray. Make sure to put the correct numbers. It is possible to add other information on the sample sheet as variable values. In the present experiment, data from the metal analysis were included.
- Transfer the IDAT files to the computer to proceed with bioinformatics analysis31. Create a sample sheet to carry out the data analysis31.
- ChAMP pipeline
- Install RStudio (V.4.0.2) on the computer to perform the analysis30.
NOTE: Ensure that the computer has a minimum of 8 G of RAM. Data analysis will struggle or crash with a lower amount of RAM. - After installation, open RStudio and install the ChAMP Bioconductor package30 using the respective command line (see Supplementary Material 1).
NOTE: If there is any error at the end of the installation, install the required ChAMP libraries manually, one by one26. - Now, import the raw data and perform the analysis. See Supplementary Material 1.
champ.load() imports the raw data and does the filtering process, excluding probes with low detection pval, multihit sites, non-CG probes, CpG sites near to SNPs, and CpGs located in sexual chromosomes.
champ.impute() performs KNN imputation as default.
champ.QC retrieves data quality graphs (e.g., density plot graph).
champ.SVD evaluates batch effects based on provided metadata in the sample sheet.
champ.refbase() performs cell type estimation and correction.
champ.DMP( ) retrieves differentially methylated positions and trait associations such as fat mass to check the DNA methylation between groups and change the parameters, as shown in Supplementary Material 1.
- Install RStudio (V.4.0.2) on the computer to perform the analysis30.
- Enrichment (String DB)
NOTE: Functional enrichment is used to infer the biological functions of a set of genes.- To enrich data in a string, select the list of targets to enrich and use as input from: https://string-db.org/cgi/input?sessionId=bywt5Gl8Bawy&input_page_active_form
=multiple_identifiers - For organism, select homo sapiens and press the Search button.
- To navigate through the enrichment, click on the Analysis button.
- To enrich data in a string, select the list of targets to enrich and use as input from: https://string-db.org/cgi/input?sessionId=bywt5Gl8Bawy&input_page_active_form
- GEO submission
- Deposit the data to a public repository like NCBI's GEO. For this, register for the submitter account.
NOTE: Complete the GEO submission (https://submit.ncbi.nlm.nih.gov/geo/submission/) by downloading the metadata sheet with descriptive information and protocols for the overall experiment and individual samples. GEO archive submissions can be created in any spreadsheet software. Second, add the raw data file generated from the cell-ranger count script for all libraries into the directory. IDAT and associated files or a matrix worksheet contains non-normalized data. The third folder is the processed data files. The matrix table is a spreadsheet containing the final, normalized values comparable across rows and samples and preferably processed as described in any accompanying manuscript. - Place processed data files generated from the cell-ranger count script for all libraries into the directory. Use the GEO submitter's FTP server credentials to transfer the directory containing all three components.
- Deposit the data to a public repository like NCBI's GEO. For this, register for the submitter account.
Results
After using the ChAMP pipeline, 409,887 probes were considered in the analysis after all filters (unqualified CpGs, non-CG probes, CpG near SNPs, multihit sites, and CpGs related to XY); a scheme is represented in Figure 2.

Figure 2: Pipeline of the bioinformatics analysis. Please click here to view a larger version of this figure.
{kind=link}
Furthermore, the density plot revealed that all samples had similar densities of the beta distribution related to the quality control steps. This analysis evaluates the distribution of beta values and points out if there are any samples that need to be excluded. In this batch, no samples were excluded based on this analysis.

Figure 3: Beta values density plot obtained with the ChAMP package. Please click here to view a larger version of this figure.
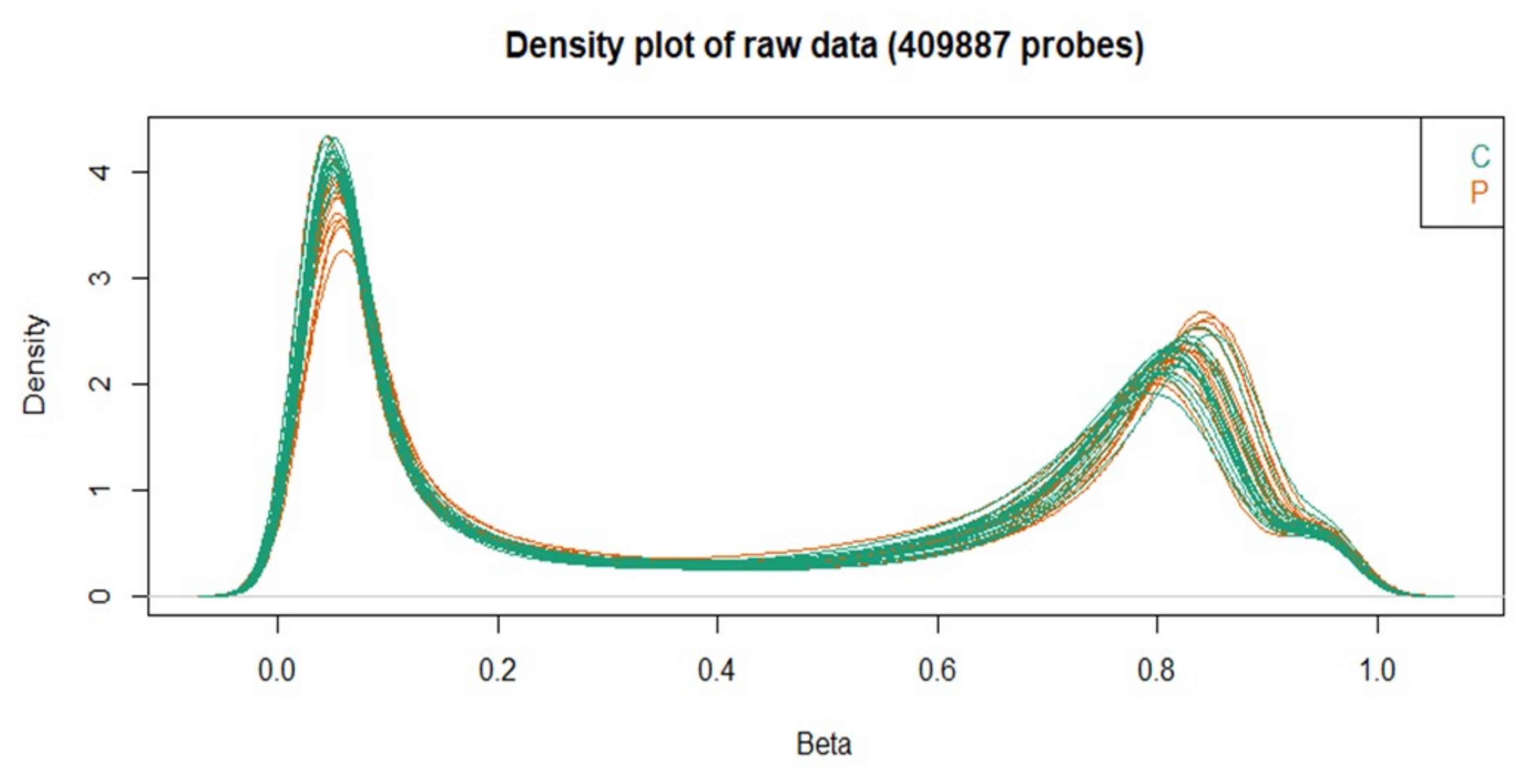{kind=link}
The singular value decomposition (SVD) analysis was used to verify the principal components that significantly influenced DNA methylation data variability. The data revealed that BMI, WC (p < 1e10-5), and FM (p < 0.05) had a significant effect on the data variability (Figure 4).

Figure 4: Singular value decomposition analysis. Please click here to view a larger version of this figure.
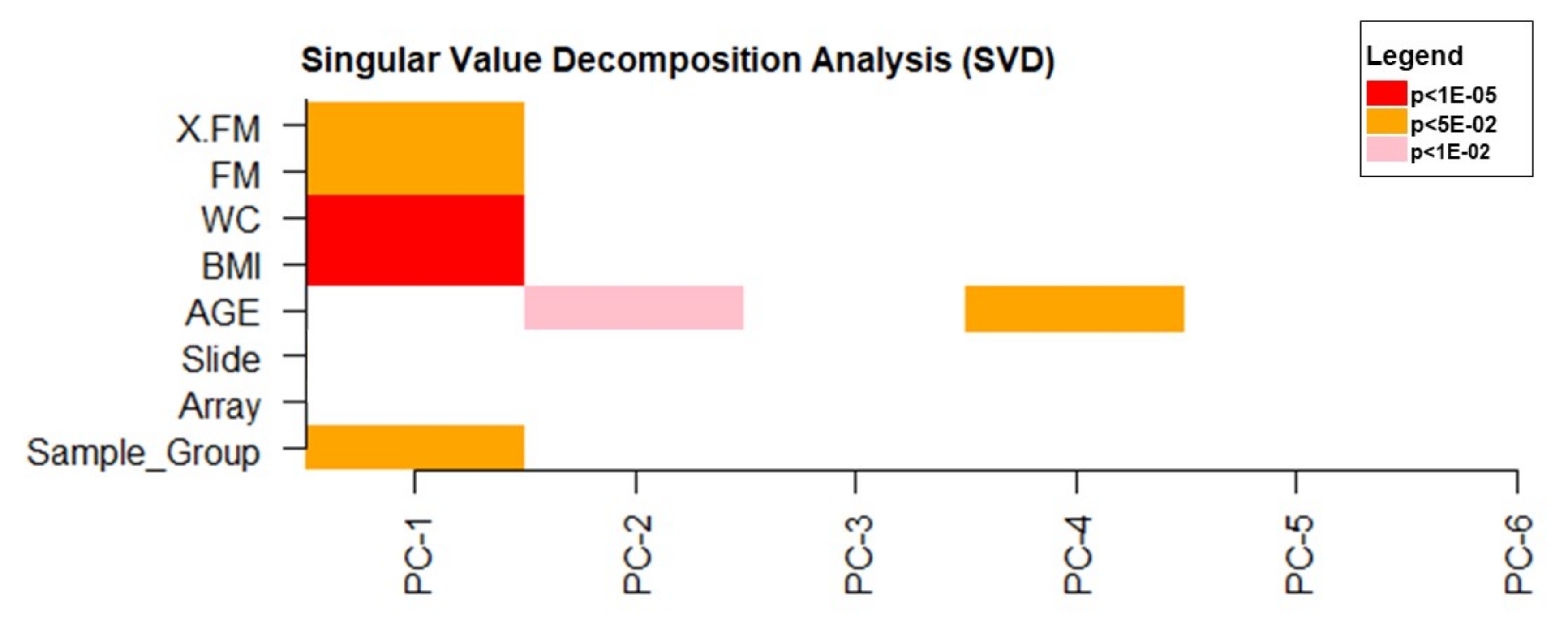{kind=link}
Cell type estimation revealed that both natural killer cells (NK) and B cells were higher in obese women (Figure 5).

Figure 5: Cell fractions estimated by Houseman's method. Gran: granulocyte. CD4T: helper T cell, lymphocyte. CD8T: cytotoxic T cell, lymphocyte. Mono: monocyte. B Cell: B lymphocyte. NK: natural killer cells, lymphocytes. *p < 0.05. Please click here to view a larger version of this figure.
{kind=link}
DNA methylation levels between obese and non-obese women differed before and after cell-type correction. Before DNA methylation data correction for cell types, 43,463 differentially methylated positions (DMPs) were observed, and 3,329 CpGs remained significant after. 445 CpG sites were in intergenic regions (IGR), and 2,884 were in genic regions, with most in the promoter region (n = 1,438). The distribution along all regions were TSS1500 (n = 612), TSS200 (n = 826), 5'UTR (n = 390), first exon (n = 273), body (n = 724), and 3'UTR (n = 59). Considering Δβ values <-0.05 and >0.05, 162 CpGs were hypomethylated and 576 were hypermethylated in obese compared to non-obese individuals (Figure 6). The data are available in the GEO database under the register code GSE166611.

Figure 6: Differentially methylated positions. Please click here to view a larger version of this figure.
{kind=link}
It is possible to evaluate the different methylation between groups and find CpG sites associated with specific traits in methylation studies. For fat mass studies, 13,222 CpG sites were found. 6,159 CpGs in the promoter region were related to fat mass, with 470 hypermethylated and 94 hypomethylated (Figure 7), and the respective genes were enriched (Table 2).

Figure 7: Promoter regions of hypomethylated and hypermethylated genes, independently related to fat mass. Please click here to view a larger version of this figure.
{kind=link}
Table 2: Functional enrichment of the genes from the differentially methylated CpG sites. Please click here to download this Table.
Supplemental Material 1: Please click here to download this File.
Discussion
DNA methylation arrays are the most used methods to access DNA methylation due to their cost-benefit ratio14. The present study described a detailed protocol using a commercially available microarray platform to evaluate DNA methylation in a pilot study performed in a Brazilian cohort. The obtained results from the pilot study confirmed the effectiveness of the protocol. Figure 3 shows the sample comparability and the complete bisulfite conversion32.
As a quality control step, the ChAMP algorithm recommended the exclusion of CpGs sites during the filtering process. The aim of excluding probes is to improve data analysis and eliminate bias. The low-quality CpGs (p-values lower than 0.05) were removed to eliminate experimental noise in the dataset. The targets remained passed in the density plot analysis. Zhou33 described the importance of filtering CpGs near SNPs to avoid mismatches, misinterpretation of polymorphic cytosines' methylation, and causing switch color of type I probe design34. Also, as XY chromosomes are differentially impacted by imprinting, Heiss and Just35 reinforced the importance of filtering those probes because, in females, the problems with hybridization may be confounding factors35.
The DMAPs expiration date, the formamide opening date, the analytical quality of the absolute ethanol, and total leucocyte counts are considered critical steps in the protocol.
Furthermore, according to our observations, the cell-type estimation is essential in performing the bioinformatics analysis. The Houseman method performs the cell type estimation as described in Tian's study30. This method is based on 473 specific CpG sites that can predict the percentages of the most important cell types, such as granulocytes, monocytes, B cells, and T cells36. We used the recommended function "myRefbase" from the ChAMP package. After the estimation, the ChAMP algorithm adjusts the beta values and eliminates this bias from the dataset. This step is crucial in studies focused on obesity because this population has a considerable difference in white blood cells due to their chronic inflammatory state.
We only changed the original cap map for the common PCR seal regarding method modification and troubleshooting. After each centrifugation process, the seal was changed for a new one. We could not use the standard heat sealing and adapted it using aluminum foil around the plate.
Although commercial assays have been considered a gold standard for epigenetic studies, one limitation of the protocol could be the specificity of the reagents and equipment from a unique brand37,38,39,40. Another limitation is the lack of indicators that allow identification of the correct progress of the experiment41.
The standardization of the present protocol represents a great guide for epigenetic research, reducing human errors during the process and allowing successful data analysis and comparability between different studies.
According to our results, DNA methylation experiments are suitable for studies comparing individuals with and without obesity43. Also, the proposed bioinformatics analysis provided high-quality data and could be considered in large-scale studies.
Using the SVD analysis, we identified that the obesity-related traits (BMI, WC, and FM) influenced the variability in DNA methylation data. As a significant result, the cell-type estimation indicates that both natural killer cells (NK) and B cells were higher in women with obesity than in women without obesity (Figure 5). The higher counts of those cells could be explained by the low-grade inflammatory state of these individuals44. We observed that patients with obesity have hypo- and hypermethylated CpGs in promoter regions of genes associated with fat mass. Most of the sites were hypomethylated, which could be related to the natural increase in reactive oxygen species (ROS) levels in these individuals. This oxidative stress condition may promote guanine perturbance at the dinucleotide site, forming 8-hydroxy-2'-deoxyguanosine (8-OHdG), resulting in a 5mCp-8-OHdG dinucleotide site, and causing TET enzymes recruitment. All these events could be responsible for promoting DNA hypomethylation and hypermethylation by different mechanisms of action45.
In addition, the rate of adipogenesis seems to increase in individuals with obesity, with approximately 10% of new cells to old cells46,47. Epigenetic contributions, emphasizing the obesogenic environment, can alter the cells' proliferation and differentiation rates, favoring the development of fat mass48. Epigenetic changes can also affect adipogenic programs, facilitating or restricting their development. Primary transcription factors (PPARγ or C / EBPα) or the assembly of multiprotein complexes, positioned in downstream promoter regions operated by including or excluding epigenetic modifying enzymes, regulate gene expression through hyper- or hypomethylation45. The PPARγ pathway has been previously described to alter the WNT pathway, which had genes enriched in this study. Although it is still unknown how WNT signaling occurs during adipogenesis, recent studies have reported that it might have essential roles in adipocyte metabolism, particularly under obesogenic conditions49.
Disclosures
The authors of this article have no conflicts of interest and have no financial disclosure to declare.
Acknowledgements
We would like to thank Yuan Tian, Ph.D. (tian.yuan@ucl.ac.uk) for being available to answer all doubts about the ChAMP package. We also thank Guilherme Telles, Msc. for his contribution both to the technical and scientific issues from this paper; he made important considerations regarding epigenetics and video capturing and formatting techniques (guilherme.telles@usp.br). Consumables Funding: São Paulo Research Foundation (FAPESP) (#2018/24069-3) and National Council for Scientific and Technological Development (CNPq: #408292/2018-0). Personal funding: (FAPESP: #2014/16740-6) and Academic Excellence Program from Coordination for Higher Education Staff Development (CAPES:88882.180020/2018-01). The data will be made publicly and freely available without restriction. Address correspondence to NYN (e-mail: nataliayumi@usp.br) or CBN (e-mail: carla@fmrp.usp.br).
Materials
| Name | Company | Catalog Number | Comments |
| Absolute ethanol | J.T. Baker | B5924-03 | |
| Agarose gel | Kasvi | K9-9100 | |
| Electric bioimpedance | Quantum BIA 450 Q - RJL System | ||
| Ethylenediaminetetraacetic acid (EDTA) | Corning | 46-000-CI | |
| EZ DNA Methylation-Gold kit | ZymoResearch, Irvine, CA, USA | D5001 | |
| Formamide | Sigma | F9037 | |
| FMS—Fragmentation solution | Illumina | 11203428 | Supplied Reagents |
| HumanMethylation450 BeadChip | Illumina | ||
| Maxwell Instrument | Promega, Brazil | AS4500 | |
| MA1—Multi-Sample Amplification 1 Mix | Illumina | 11202880 | Supplied Reagents |
| MicroAmp Optical Adhesive Film | Thermo Fisher Scientific | 201703982 | |
| MSM—Multi-Sample Amplification Master Mix | Illumina | 11203410 | Supplied Reagents |
| NaOH | F. MAIA | 114700 | |
| PB1—Reagent used to prepare BeadChips for hybridization | Illumina | 11291245 | Supplied Reagents |
| PB2—Humidifying buffer used during hybridization | Illumina | 11191130 | Supplied Reagents |
| 2-propanol | Emsure | 10,96,34,01,000 | |
| RA1—Resuspension, hybridization, and wash solution | Illumina | 11292441 | Supplied Reagents |
| RPM—Random Primer Mix | Illumina | 15010230 | Supplied Reagents |
| STM—Superior Two-Color Master Mix | Illumina | 11288046 | Supplied Reagents |
| TEM—Two-Color Extension Master Mix | Illumina | 11208309 | Supplied Reagents |
| Ultrapure EDTA | Invitrogen | 155576-028 | |
| 96-Well Reaction Plate with Barcode (0.1mL) | ByoSystems | 4346906 | |
| 96-Well Reaction Plate with Barcode (0.8mL) | Thermo Fisher Scientific | AB-0859 | |
| XC1—XStain BeadChip solution 1 | Illumina | 11208288 | Supplied Reagents |
| XC2—XStain BeadChip solution 2 | Illumina | 11208296 | Supplied Reagents |
| XC3—XStain BeadChip solution 3 | Illumina | 11208392 | Supplied Reagents |
| XC4—XStain BeadChip solution 4 | Illumina | 11208430 | Supplied Reagents |
References
- Manna, P., Jain, S. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metabolic Syndrome and Related Disorders. 13 (10), 423-444 (2015).
- . Obesity Available from: https://www.who.int/health-topics/obesity#tab=tab_1 (2017)
- Adult Obesity Facts. Center for Disease Control and Prevention Available from: https://www.cdc.gov/obesity/data/adult.html (2021)
- Bird, A. Perceptions of epigenetics. Nature. 396, (2007).
- Kouzarides, T. Chromatin modifications, and their function. Cell. 128 (4), 693-705 (2007).
- Berger, F. The strictest usage of the term epigenetic. Seminars in Cell & Developmental Biology. 19 (6), 525-526 (2008).
- Henikoff, S., Greally, J. M. Epigenetics, cellular memory, and gene regulation. Current Biology. 26 (14), 644-648 (2016).
- Laker, R. C., et al. Transcriptomic and epigenetic responses to short-term nutrient-exercise stress in humans. Scientific Reports. 7 (1), 1-12 (2017).
- Wahl, S., et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 541, 81-86 (2017).
- Jin, B., Robertson, K. D. DNA methyltransferases, DNA damage repair, and cancer. Epigenetic Alterations in Oncogenesis. 754, 3-29 (2013).
- Jang, H. S., Shin, W. J., Lee, J. E., Do, J. T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes. 8 (6), 148 (2017).
- Shen, L., Waterland, R. A. Methods of DNA methylation analysis. Current Opinion in Clinical Nutrition & Metabolic Care. 10 (5), 576-581 (2007).
- Olkhov-Mitsel, E., Bapat, B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer Medicine. 1, 237-260 (2012).
- Kurdyukov, S., Bullock, M. DNA methylation analysis: choosing the right method. Biology. 5 (1), 3 (2016).
- Yong, W. -. S., Hsu, F. -. M., Chen, P. -. Y. Profiling genome-wide DNA methylation. Epigenetics & Chromatin. 9 (1), 1-16 (2016).
- Dugué, P. A., et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addiction Biology. 1, 12855 (2021).
- Colicino, E., et al. Blood DNA methylation sites predict death risk in a longitudinal study of 12, 300 individuals. Aging. 14, 14092-14124 (2020).
- Karlsson, L., Barbaro, M., Ewing, E., Gomez-Cabrero, D., Lajic, S. Genome-wide investigation of DNA methylation in congenital adrenal hyperplasia. The Journal of Steroid Biochemistry and Molecular Biology. 201, 105699 (2020).
- Ribeiro, R. R., Guerra-Junior, G., de Azevedo Barros-Filho, A. Bone mass in schoolchildren in Brazil: the effect of racial miscegenation, pubertal stage, and socioeconomic differences. Journal of Bone and Mineral Metabolism. 27 (4), 494-501 (2009).
- Filozof, C., et al. Obesity prevalence and trends in Latin-American countries. Obesity Reviews. 2 (2), 99-106 (2001).
- Chadid, S., Kreger, B. E., Singer, M. R., Bradlee, M. L., Moore, L. L. Anthropometric measures of body fat and obesity-related cancer risk: sex-specific differences in Framingham Offspring Study adults. International Journal of Obesity. 44 (3), 601-608 (2020).
- Nicoletti, C. F., et al. DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. European Journal of Clinical Nutrition. 74 (9), 1345-1353 (2020).
- Assessing Your Weight. Centers for Disease Control and Prevention Available from: https://www.cdc.gov/healthyweight/assessing/index.html (2020)
- Giavarina, D., Lippi, G. Blood venous sample collection: Recommendations overview and a checklist to improve quality. Clinical Biochemistry. 50 (10-11), 568-573 (2017).
- Duijs, F. E., Sijen, T. A rapid and efficient method for DNA extraction from bone powder. Forensic Science International: Reports. 2, 100099 (2020).
- . Infinium HD Methylation Assay, Manual Protocol Available from: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium_hd_methylation/infinium-hd-methylation-guide-15019519-01.pdf (2015)
- Serrano, J., Snuderl, M. Whole genome DNA methylation analysis of human glioblastoma using Illumina BeadArrays. Glioblastoma. , 31-51 (2018).
- Leti, F., Llaci, L., Malenica, I., DiStefano, J. K. Methods for CpG methylation array profiling via bisulfite conversion. Disease Gene Identification. 1706, 233-254 (2018).
- Noble, A. J., et al. A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing. PeerJ. 9, 10762 (2021).
- Tian, Y., et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 33, 3982-3984 (2017).
- Turinsky, A. L., et al. EpigenCentral: Portal for DNA methylation data analysis and classification in rare diseases. Human Mutation. 41 (10), 1722-1733 (2020).
- . The Chip Analysis Methylation Pipeline Available from: https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html (2020)
- Zhou, W., Laird, P. W., Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Research. 45 (4), 22 (2017).
- Wanding, Z., Triche, T. J., Laird, P. W., Hui, S. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Research. 46 (20), 123 (2018).
- Heiss, J. A., Just, A. C. Improved filtering of DNA methylation microarray data by detection p values and its impact on downstream analyses. Clinical Epigenetics. 11, 15 (2019).
- Houseman, E. A., et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 13, 86 (2012).
- Valavanis, I., Sifakis, E. G., Georgiadis, P., Kyrtopoulos, S., Chatziioannou, A. A. A composite framework for the statistical analysis of epidemiological DNA methylation data with the Infinium human Methylation 450K BeadChip. IEEE Journal of Biomedical and Health Informatics. 18 (3), 817-823 (2014).
- Sun, N., Zhang, J., Zhang, C., Shi, Y., Zhao, B., Jiao, A., Chen, B. Using Illumina Infinium HumanMethylation 450K BeadChip to explore genomewide DNA methylation profiles in a human hepatocellular carcinoma cell line. Molecular Medicine Reports. 18 (5), 4446-4456 (2018).
- Moran, S., Arribas, C., Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 8 (3), 389-399 (2016).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Lehne, B., et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biology. 16 (1), 1-12 (2015).
- Wang, J., Zhang, H., Rezwan, F. I., Relton, C., Arshad, S. H., Holloway, J. W. Pre-adolescence DNA methylation is associated with BMI status change from pre- to post-adolescence. Clinical Epigenetics. 25, 64 (2021).
- Maugeri, A. The effects of dietary interventions on DNA methylation: implications for obesity management. International Journal of Molecular Sciences. 21, 8670 (2020).
- DeFuria, J., et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proceedings of the National Academy of Sciences. 110 (13), 5133-5138 (2013).
- Kietzmann, T., Petry, A., Shvetsova, A., Gerhold, J. M., Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. British Journal of Pharmacology. 174, 1533-1554 (2017).
- Chase, K., Sharma, R. P. Epigenetic developmental programs and adipogenesis: implications for psychotropic induced obesity. Epigenetics. 8 (11), 1133-1140 (2013).
- Spalding, K. L., et al. Dynamics of fat cell turnover in humans. Nature. 453, 783-787 (2008).
- Ross, S. E., et al. Inhibition of adipogenesis by Wnt signaling. Science. 289, 950-953 (2000).
- Bagchi, D. P., et al. Wnt/β-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Molecular Metabolism. 42, 101078 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved