
Method Article
Expanding the Toolkit for In Vivo Imaging of Axonal Transport
In This Article
Summary
Using transgenic fluorescent mice, detailed protocols are described to assess in vivo axonal transport of signaling endosomes and mitochondria within motor and sensory axons of the intact sciatic nerve in live animals.
Abstract
Axonal transport maintains neuronal homeostasis by enabling the bidirectional trafficking of diverse organelles and cargoes. Disruptions in axonal transport have devastating consequences for individual neurons and their networks, and contribute to a plethora of neurological disorders. As many of these conditions involve both cell autonomous and non-autonomous mechanisms, and often display a spectrum of pathology across neuronal subtypes, methods to accurately identify and analyze neuronal subsets are imperative.
This paper details protocols to assess in vivo axonal transport of signaling endosomes and mitochondria in sciatic nerves of anesthetized mice. Stepwise instructions are provided to 1) distinguish motor from sensory neurons in vivo, in situ, and ex vivo by using mice that selectively express fluorescent proteins within cholinergic motor neurons; and 2) separately or concurrently assess in vivo axonal transport of signaling endosomes and mitochondria. These complementary intravital approaches facilitate the simultaneous imaging of different cargoes in distinct peripheral nerve axons to quantitatively monitor axonal transport in health and disease.
Introduction
The peripheral nervous system (PNS) connects the central nervous system (CNS) to its distal targets, permitting the relay of efferent signals to exert motor control and afferent signals to provide sensory feedback. Using the multitude of advances in mouse genetics, scientists have developed different mouse models to investigate many diseases/syndromes afflicting the PNS1,2,3. As most neurodegenerative pathologies are multifactorial with cell autonomous and non-autonomous contributions4,5, untangling cell-/neuron-specific pathologies can provide crucial, novel insights into disease mechanisms.
To this end, the development of bacterial artificial chromosome (BAC)-transgenic mice6 has enabled selective endogenous expression of fluorescent proteins in targeted subsets of neurons. For example, BAC-transgenic mice are available, which express green fluorescent protein (GFP) in cholinergic7 or glycinergic neurons8, or a variant red fluorescent protein (tdTomato) in parvalbumin-positive neurons9. Alternatively, selective neuronal expression of fluorescent proteins can be achieved via Cre-loxP technology10. For instance, mouse strains expressing Cre-recombinase in subsets of neurons (e.g., choline acetyltransferase (ChAT)-Cre) can be bred with mice that express a fluorescent protein (e.g., tdTomato or GFP) from a constitutive locus (e.g., Gt(ROSA)26Sor) under the control of a transcriptional repressor flanked by loxP sites11 (e.g., generating mice that express tdTomato only in cholinergic neurons). Indeed, using Cre-loxP recombination, transgenic mice have been generated that express yellow fluorescent protein in axons of the descending corticospinal tract12.
In addition, recent advances in CRISPR/Cas9 gene editing, such as ORANGE, enable the fluorescent tagging of multiple endogenous neuronal proteins, with expression achievable at nanoscale resolution13. Moreover, in combination with Cre-expressing mouse strains, ORANGE-CAKE can be used to tag multiple endogenous proteins in individual neurons13. Alternatively, viral-mediated neuronal tracing also allows the labeling of neuronal subsets and can be achieved with targeted combinations of viral serotypes and/or cell-specific promoters14,15,16,17.
Further to the neuronal labeling methods, mouse lines have also been engineered to express reporter proteins targeting specific organelles, such as mitochondria expressing cyan fluorescent protein (Mito.CFP)18 or autophagosomes expressing GFP (LC3.GFP)19. Moreover, mouse lines have been engineered to assess calcium dynamics specifically in neurons (e.g., Thy1.GCaMP)20,21. Altogether, with the advancement of such models, novel experimental applications enable scientists to ask more precise biological and pathological questions about the CNS and PNS.
The main role of peripheral motor nerves is to transmit electrical signals to skeletal muscle to elicit movement. In addition, and occurring over longer time-scales, neurochemical and physiological messages in the form of diverse organelles (e.g., mitochondria, endolysosomes, signaling endosomes) traverse the cytoskeletal network in a uni- or bi-directional manner to help maintain neuronal homeostasis22,23,24. Impairments in axonal transport have disastrous consequences for neuronal health and are linked to many neurodevelopmental and neurodegenerative diseases25. At the molecular level, impairments in axonal transport can disrupt physiological events regulating synaptic signaling and plasticity, gene transcription, and local translation throughout the axon26,27. While there is a multitude of tools to study these events in cultured cells/neurons28,29, assessing axonal transport dynamics and axonal-linked biological events in vivo are required to confirm key insights into physiological and pathological processes30.
Over the years, the Schiavo Laboratory has optimized protocols to ask diverse questions about axonal transport31,32,33,34,35,36. These experiments have expanded from the discovery that a fluorescently labeled atoxic fragment of tetanus neurotoxin (HCT) is internalized into axon terminals in skeletal muscle through interactions with nidogens and polysialogangliosides37. Once internalized, HCT is retrogradely transported in Rab7-positive, neurotrophin-containing signaling endosomes that are destined for the cell bodies of motor and sensory neurons38,39,40,41. In parallel, advances in imaging technology have enabled the real-time analysis of peripheral nerve bundles and individual axons in live, anesthetized mice30. The first foray into assessing in vivo axonal transport dynamics in pathology revealed presymptomatic impairments in the transport of signaling endosomes and mitochondria in the SOD1G93A mouse model of amyotrophic lateral sclerosis (ALS)35. Importantly, these defects are unlikely to simply represent secondary consequences of neurodegeneration, given the finding that motor neuron loss can occur in the absence of axonal transport perturbations in a mouse model of Kennedy's disease42 and a heterozygous mutant FUS model of ALS43. Such axonal transport deficits can be remedied in ALS mice using inhibitors of specific kinases33 or growth factor receptors34. Moreover, treating neurons with a specific histone deacetylase blocker alters mitochondrial transport in vivo36. Most recently, we report that the BDNF-dependent modulation of axonal transport is dysregulated in distinct motor neuron subtypes in ALS mice44.
By using an ever-expanding toolkit for assessing axonal transport dynamics28,29, this video protocol outlines several applications that will permit further insights into different biological and pathological scenarios. First, transgenic mice that selectively express fluorescent proteins in cholinergic neurons (i.e., motor neurons) are used to discriminate between motor and sensory axons both in vivo and ex vivo. Fluorescently labeled HCT is then loaded into signaling endosomes in three transgenic lines to differentiate axonal transport dynamics in distinct peripheral neurons. The next experimental protocol details a multiplex fluorescence approach to assess mitochondrial transport specifically in motor neurons by breeding ChAT.tdTomato mice with Mito-CFP mice. Finally, instructions are provided on how to concurrently image mitochondria and signaling endosomes within the same axon in vivo.
Protocol
All mouse handling and experiments were performed in accordance with the Animals (Scientific Procedures) Act (1986) and were approved by the University College London - Queen Square Institute of Neurology Ethics Committee.
1. Animals
- House all animals in individually ventilated cages in a temperature- and humidity-controlled environment and maintain them on a 12 h light/dark cycle with ad libitum access to food and water.
- Use both male and female mice of the following transgenic strains: 1) heterozygous Tg(Chat-EGFP) GH293Gsat/Mmucd mice, referred to as ChAT.eGFP mice; 2) heterozygous B6.Cg-Tg(Hlxb9-GFP)1Tmj/J, referred to as HB9.GFP mice; and 3) heterozygous B6.Cg-Tg(Thy1-CFP/COX8A)S2Lich/J, referred to as Mito.CFP mice.
- Generate ChAT.tdTomato mice by crossing homozygous B6;129S6-Chattm2(cre)Lowl/J, referred to as ChAT.Cre mice, with homozygous B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, referred to as Rosa26.tdTomato mice.
- Generate ChAT.tdTomato::Mito.CFP mice by crossing heterozygous ChAT.tdTomato mice with heterozygous Mito.CFP mice.
2. Intramuscular injections of fluorescent HCT
- Pre-surgery preparation
- Express HCT (HCT441, residues 875-1315) fused to an improved cysteine-rich tag in bacteria as a glutathione-S-transferase fusion protein as per 45. Label HCT with AlexaFlour555 C2 maleimide31, dialyze it in ice-cold dialysis buffer (10 mM HEPES-NaOH, 100 mM NaCl, pH 7.4), freeze it in liquid nitrogen, and store it at -80 °C. Before performing in vivo experiments, first test HCT in vitro for successful uptake and transport in primary neurons.
- Dilute fluorescent HCT (e.g., HCT-555) to a final and experimentally consistent concentration ranging from 2.5 to 10 µg/µL in sterile phosphate-buffered saline (PBS) in a 0.2 mL tube. At this step, add more compounds/factors to the HCT solution if required (e.g., brain-derived neurotrophic factor).
NOTE: The final volume must be appropriate for the size of the muscle(s) of interest. For example, prepare an injection volume of 3-4 µL for the tibialis anterior (TA) muscle and ~1 µL for the smaller soleus muscle. Keep the working concentration of HCT between 2.5 and 10 µg/µL regardless of the final volume. - Mix the HCT solution using a pipette or vortex, and briefly spin down at low speed using a desktop centrifuge to collect the liquid and remove large bubbles. Protect the HCT from light and transport on ice.
- Use a pulled glass micropipette for optimal intramuscular injections into smaller muscles (e.g., soleus) or for intrasciatic nerve injections. Pull graded glass micropipettes (as per 46) before surgery.
NOTE: To enable pipetting and restrict flow up and out the back of the micropipette, carefully break off a small piece from the sharp tip using fine forceps under a dissecting microscope. Take care to dispose of the broken end in the appropriate bin. - Sterilize and clean all surgical tools prior to use.
- Surgery-intramuscular injections
- Prepare for surgery by securing a sterile surgical drape on a heat mat set to 37°C. Position and focus the operating microscope. For surgery to commence, unpack onto the surgical drape the presterilized surgical tools, surgical tape, sterile cotton swabs, 70% (v/v) ethanol in water, sterile saline, sutures, and a Hamilton needle or pulled glass micropipettes.
- Ensure the anesthetic machine has sufficient oxygen and isoflurane for the duration of the surgical procedure. Direct the flow of anesthesia to the induction chamber and switch on the anesthetic machine.
- To begin, use an oxygen flow rate of 1-2 L/min and 5% isoflurane. Place the mouse in the induction chamber to initiate anesthesia. When the righting reflex is absent, reduce anesthesia to 2-3% isoflurane, direct the flow of anesthesia to the mouthpiece, and transfer the mouse to the mouthpiece located in a separate area of the surgical space.
- Ensure both the corneal and pedal withdrawal reflexes are absent before shaving the area of fur covering the muscle(s) to be injected. When complete, remove as much shaved fur from the mouse as possible using the sticky side of surgical tape, and place the mouse on weighing scales to record its pre-surgical weight.
- Carefully apply eye lubricant using a cotton swab and transfer the mouse and mouthpiece to the surgical area.
NOTE: Try to limit the amount of shaved fur that is also transferred to the surgical area. Use surgical tape to secure the head to the mouthpiece to prevent the mouse from slipping out. Using a separate cotton swab, apply ethanol to the shaved region to sterilize and reduce fur contamination. - Position the body according to the muscle to be injected. For example, for the TA, place the mouse on its back and stretch out the hindlimb at ~10° from the midline. Alternatively, for soleus injections, place the animal on its side and extend the hindlimb at ~45° from the midline. When the hindlimb is in the correct position, use surgical tape across the foot to prevent unwanted movement during surgery.
NOTE: The injection procedures for TA, gastrocnemius, and soleus muscles have been previously detailed32. - Before making an incision, confirm that the anesthesia is sufficient by testing the pedal withdrawal reflex. Monitor the anesthesia continually and maintain it throughout the surgical procedure with regular assessment of breathing and the withdrawal reflex.
- At this point, draw the working HCT solution into the Hamilton syringe or pulled glass micropipette.
- Make a small incision over the muscle(s) of interest in the area(s) that correspond(s) with the motor end plate regions46,47,48. Pierce the external fascia on the muscle and slowly inject the HCT as per 32. Leave the syringe/micropipette in position for 5-10 s before slowly withdrawing.
- Close the incisions with 1-2 sutures and transfer the mouse to an isolated recovery cage. Monitor the mouse post surgery for a minimum of 30 min, before returning it to the home cage. When the mouse has successfully recovered and post surgical monitoring is complete, return the cage to normal housing conditions.
3. In vivo axonal transport
- Exposing the sciatic nerve
- Set the microscope environmental chamber to 37 °C at least 1 h prior to imaging.
- Prepare to expose the sciatic nerve by arranging the surgical drape, tools, tape, sterile cotton swabs, 70% ethanol, and sterile saline around the surgical area. Ensure the anesthetic machine has sufficient stores of oxygen and isoflurane for up to 2 h per mouse. Create a wedge out of parafilm or invisible tape by cutting it into a narrow rectangle (e.g., ~1 cm width for larger mice) with an angled tip and place it underneath the exposed sciatic nerve to aid the imaging process. Place the induction chamber on top of a heat mat and set it to body temperature.
NOTE: Four hours is ample time for HCT to have been taken up and retrogradely transported from the site of injection to the sciatic nerve; hence, a single mouse can be readied for re-anesthesia after this time. - Direct the flow of anesthesia into the induction chamber, switch on the anesthetic machine with an oxygen flow rate of 1-2 L/min and 3-4% isoflurane, and place the mouse in the induction chamber to initiate anesthesia.
NOTE: As the in vivo axonal transport experiment is a terminal procedure, there is no need to lubricate the eyes. - When the righting reflex is absent, reduce anesthesia to 2-3% isoflurane, direct the flow of anesthesia to the mouthpiece, and transfer the mouse to the mouthpiece. Use surgical tape to secure the head to the mouthpiece, extend the targeted hindlimb at ~45° from the midline, and use surgical tape over the foot to maintain this position.
NOTE: Reduced anesthesia is advantageous at this point as it can limit the impact of breathing artifacts during the imaging process. - Ensure corneal and pedal withdrawal reflexes are absent, and then use scissors to cut away the skin overlying the sciatic nerve32 (i.e., a large area extending from the central spinal cord to mid-lower hindlimb). Remove the overlying biceps femoris muscle, as well as any other musculature and connective tissue that is near the sciatic nerve. Avoid damaging the sciatic nerve and surrounding blood vessels, especially those located near the lateral aspect of the patella/proximal aspect of the lateral gastrocnemius head.
- When the intact sciatic nerve is sufficiently exposed, apply prewarmed sterile saline to the area around the sciatic nerve to prevent desiccation. Use curved forceps to disrupt the deep-lying connective tissue and place the pre-prepared parafilm 'wedge' underneath the nerve. When complete, place saline-soaked cotton wool on the exposed area and move the mouse into the induction chamber positioned on top of the heat mat (set to 37 °C), which should still be filled with isoflurane in O2.
- In vivo axonal imaging
- Place a 22 x 64 mm coverglass on the customized microscope stage and secure its position with tape. Select and apply immersion oil to the objective, and then connect the microscope stage to the inverted microscope. Slowly raise the oil-immersed objective until contact is made between the oil and coverglass.
NOTE: Either the 40x, 1.3 numerical aperture (NA) DIC Plan-Apochromat or 63x, 1.4 NA DIC Plan-Apochromat oil-immersion objectives can be used to image in vivo transport in the sciatic nerve. - Move the anesthetic mouthpiece onto the microscope stage and secure the anesthesia hoses with tape to prevent disturbance to the anesthesia. Remove the cotton wool from the sciatic nerve and transfer the mouse from the induction chamber to the mouthpiece, with the exposed nerve facing the coverglass. Use surgical tape to ensure the mouse's head is fixed to the mouthpiece and maintain the lowest, effective level of anesthesia. Gently lift the mouse by its tail and add sterile saline to the coverslip near the exposed sciatic nerve to restrict desiccation and aid imaging.
NOTE: Close all doors of the environmental chamber to ensure the area remains at body temperature. - Using the oculars, locate the sciatic nerve, determine the optimal focal point, and select an area of interest containing motile axonal organelles.
NOTE: A detailed explanation of this process has been previously described32. - Switch to the computer software by clicking the Acquisition button (or equivalent), and select an area of interest. Use a digital zoom to obtain a total of >80x magnification and rotate the selected area to horizontally visualize the axons (e.g., right-to-left moving retrograde cargo and left-to-right moving anterograde cargo).
NOTE: The directionality parameters are user-dependent, but must remain consistent throughout experiments. - Optimize the signal intensity by adjusting parameters such as laser intensity (0.2 - 1%), pinhole aperture (1 AU - max), gain (Master) (700 - 1000), digital offset (-50 - 0), and digital gain (1.0 - 4.0). To reduce the potential influence of phototoxicity, maintain laser intensity at ≤ 1% where possible, with a maximum laser intensity of 2%. Change all other parameters before adjusting the laser intensity for optimal signal detection.
- Click the Regions box (or equivalent), select a rectangular region of interest, then in the Acquisition Mode (or equivalent), set the frame size to a minimum 1024 x 1024 pixels, and commence time-lapse acquisition of 100-1,000 frames.
NOTE: The desired frame acquisition rate is user-dependent (e.g., transport can be assessed with frame rates between 0.1 and 6 s) and can be adjusted with software parameters, such as region of interest, scan speed time, acquisition averaging, and laser directionality. For example, to obtain a slower frame rate, increase the height/width of the region of interest, acquire slower scan speeds, increase the acquisition averaging, and use single laser directionality, and vice versa for a faster frame rate. The frame acquisition rate must remain consistent across comparable datasets because imaging at different frequencies may cause inconsistencies. Fast cargoes such as signaling endosomes require a faster frame rate (e.g. 0.1-3 s) compared with slower organelles like mitochondria which can be analysed using a slower rate (e.g. 2.5-6 s). - Aim to capture a minimum of 10 motile cargoes from a minimum of three axons per mouse.
NOTE: Based on two-sample, two-sided power calculations (with standard power of 0.8 (1−β) and type I error rate of 5% (α)), sample sizes of 6-8 are sufficient to identify axonal transport differences between wild-type and disease models35,43. - Once imaging is complete, euthanize the mouse immediately while under anesthesia (e.g., cervical dislocation). Post mortem tissue, such as muscles and sciatic nerves, can also be harvested for further analysis.
- Place a 22 x 64 mm coverglass on the customized microscope stage and secure its position with tape. Select and apply immersion oil to the objective, and then connect the microscope stage to the inverted microscope. Slowly raise the oil-immersed objective until contact is made between the oil and coverglass.
Results
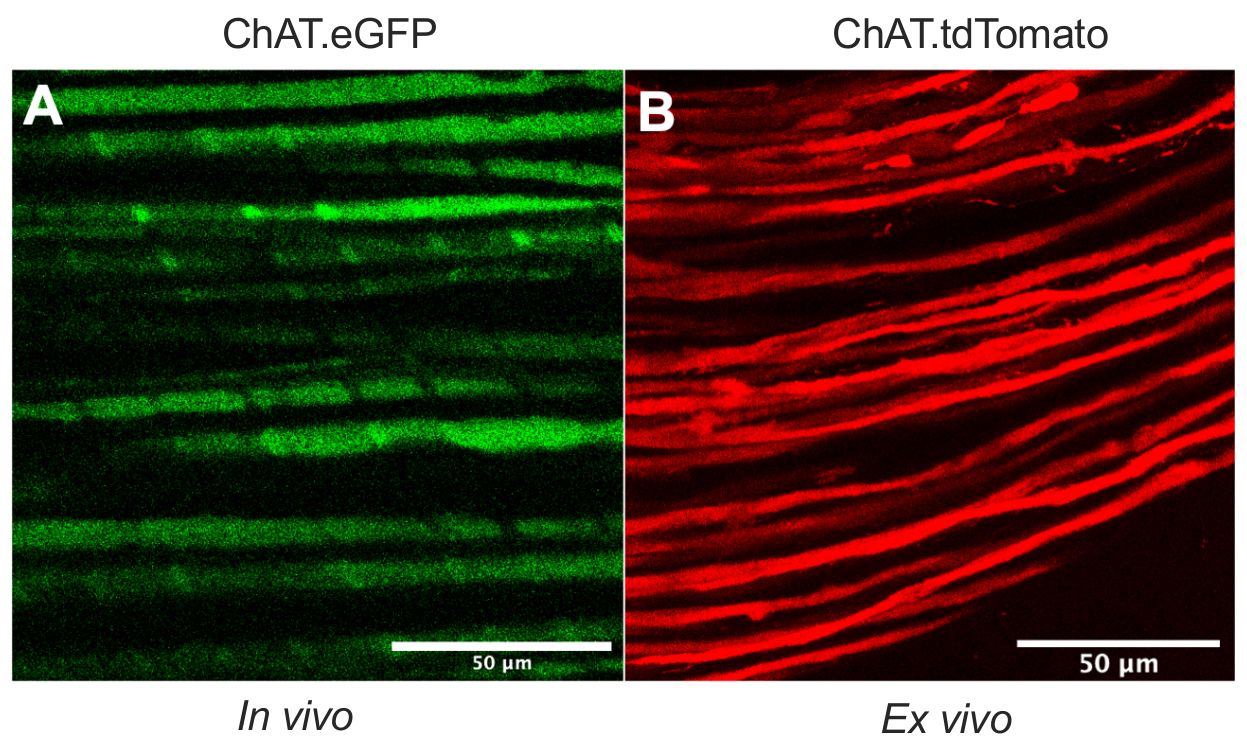
This paper details a versatile protocol that expands the in vivo axonal transport toolkit in rodent models. Figure 1 demonstrates that motor neuron axons can be differentiated from both sensory neuron axons and Schwann cells by using transgenic mice. Figure 1A depicts eGFP expression in cholinergic motor axons from a live, anesthetized ChAT.eGFP mouse. Figure 1B uses an alternative method to attain tdTomato expression in a freshly excised nerve (i.e., no additional tissue processing) from a ChAT.tdTomato mouse. Hence, using transgenic strains such as ChAT.eGFP, ChAT.tdTomato or Hb9.GFP enables motor axon-specific labeling in vivo.
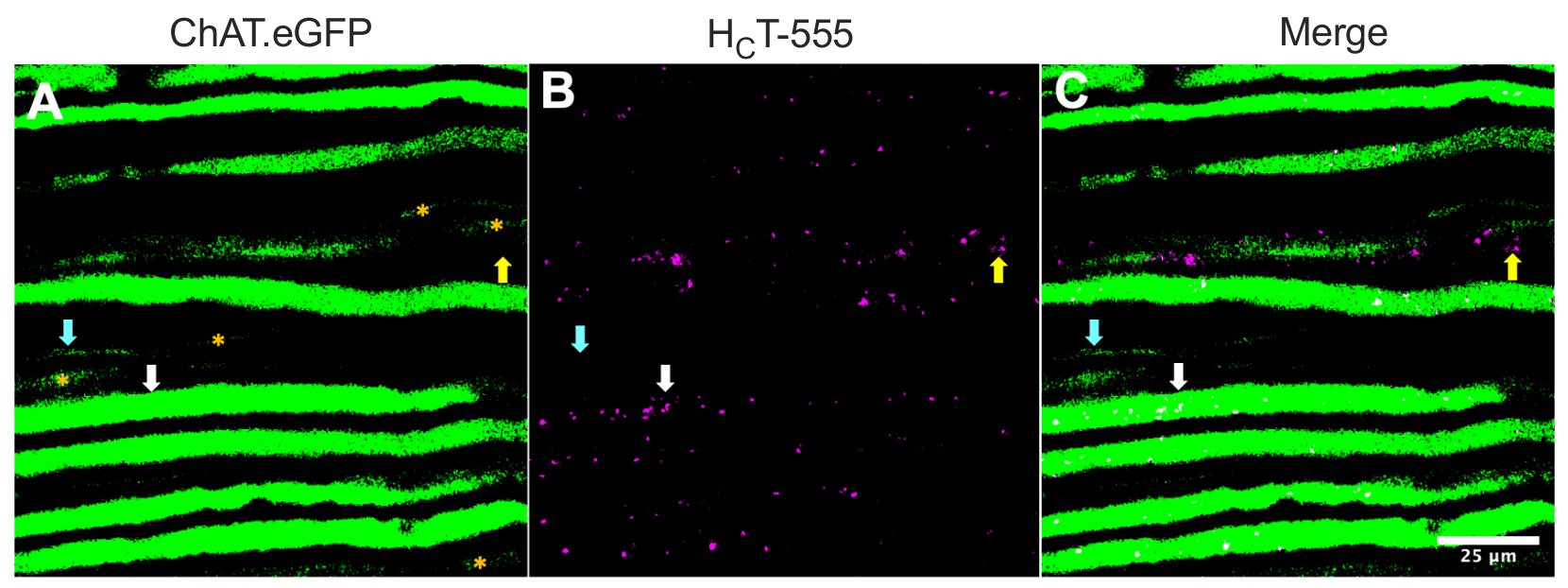
Alternatively, axons can also be identified by injecting tracers/markers (e.g., HCT31,32 or viruses encoding eGFP15) into skeletal muscles. Figure 2 highlights such an application, depicting eight robustly expressing ChAT.eGFP-positive axons that contain HCT-555-positive signaling endosomes (white arrows), ~4 h post probe injection into the TA muscle. Using this experimental design, we could identify TA-innervating α-motor neurons, which are predominantly fast-fatigable44. A further five ChAT.eGFP axons with less robust eGFP expression (Figure 2A, orange asterisks) were partially out of focus and likely to be located slightly deeper within the sciatic nerve.
Moreover, we identified HCT-555-positive signaling endosomes in eGFP-negative sensory axons (yellow arrows). As such, using this experimental paradigm, one can specifically assess and compare the axonal transport of signaling endosomes in motor versus sensory neurons in vivo. Indeed, using this transgenic reporter strain, we discovered that transport of signaling endosomes in ChAT.eGFP-positive motor axons is faster than in ChAT.eGFP-negative sensory axons, which can be reliably differentiated using axon widths43.
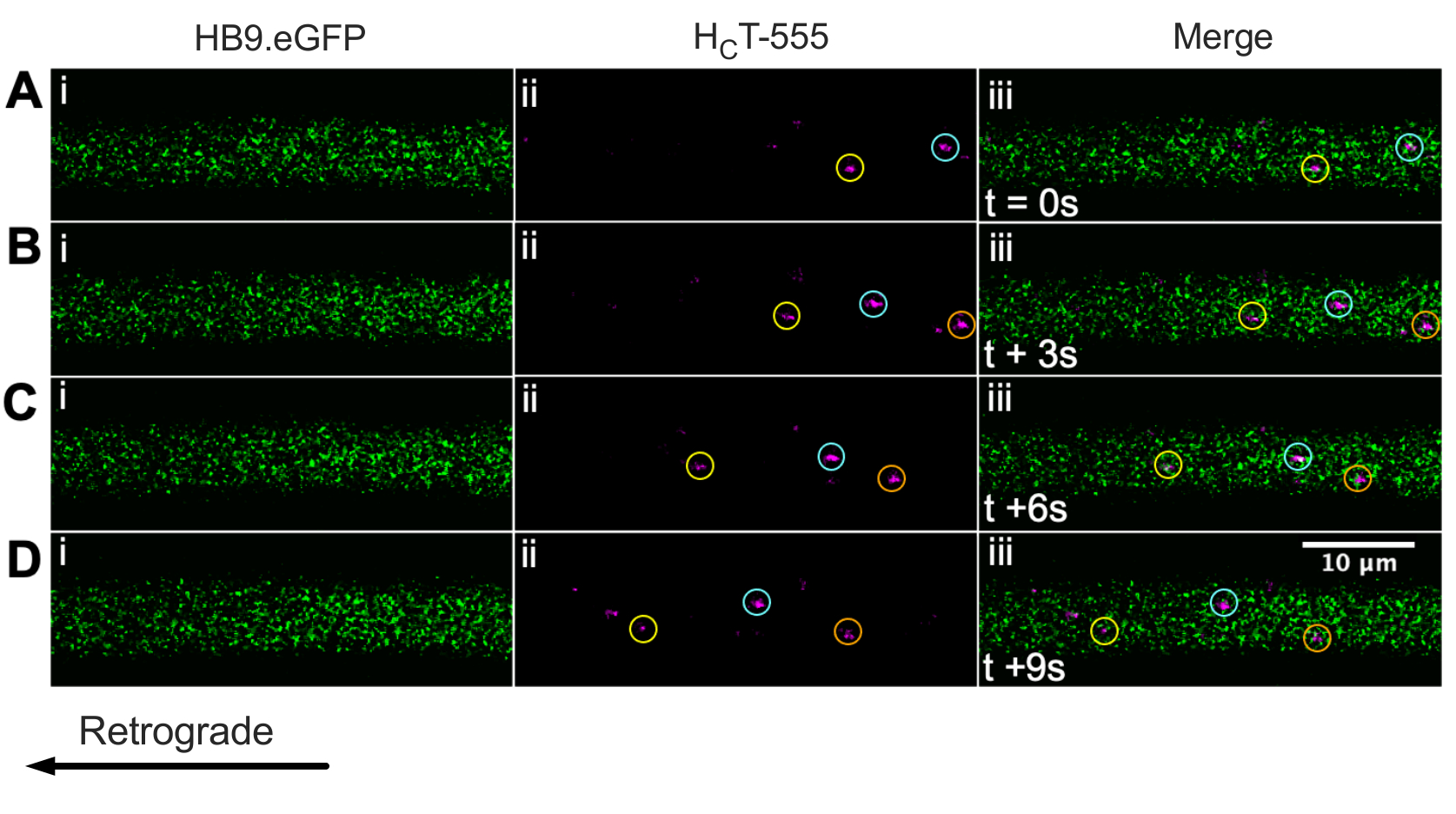
We have previously identified motor neuron axons using the ChAT.eGFP mouse in vivo43. We now report that HB9.GFP mice can also be used to achieve motor neuron axon identification in vivo. Indeed, Figure 3 presents a series of time-lapse images of HB9.GFP axons containing retrogradely moving HCT-555-positive signaling endosomes. Note that, unlike ChAT-driven expression, GFP has a more punctate/granular pattern in HB9.GFP axons; the reason for this is unclear.
We have previously described how to monitor in vivo mitochondrial dynamics in sciatic nerves via intrasciatic nerve injections of the mitochondrial-targeting dye, tetramethylrhodamine, ethyl ester, perchlorate (TMRE)32,36. To reliably differentiate motor versus sensory mitochondria, the Mito.CFP mouse, which expresses CFP under the Thy1 promoter18, can be crossed with transgenic mice that express a fluorescent reporter gene in specific neuronal types. Indeed, by breeding Mito.CFP mice with ChAT.tdTomato mice (referred to as ChAT.tdTomato::Mito.CFP), we could visualize mitochondria specifically in motor axons, as shown in Figure 4. In this live multiplex example, five ChAT.tdTomato axons could be visualized, four of which contain CFP-positive mitochondria. Moreover, the node of Ranvier (white arrow in panel iii) could also be identified. Furthermore, the nodes of Ranvier are clearly detectable in ChAT.eGFP, HB9.GFP and Mito.CFP mice (not shown). These double-transgenic strains enable time-lapse intravital imaging of live, anesthetized mice to monitor motor neuron-specific mitochondrial content and axonal transport dynamics.
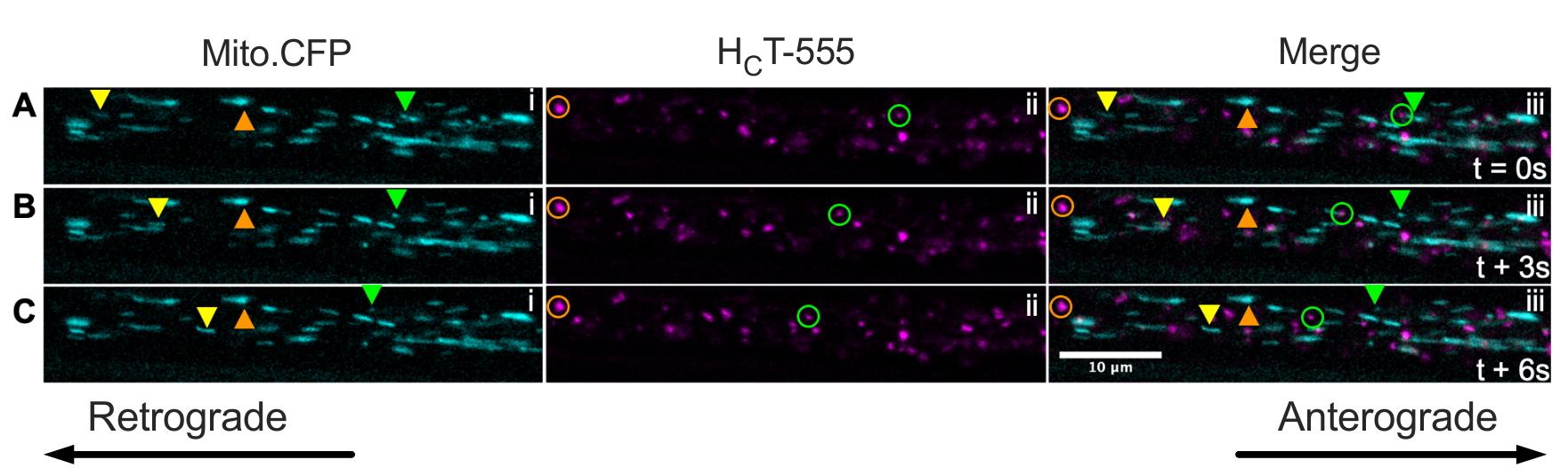
Finally, signaling endosomes and mitochondria can be concurrently visualized within the same axons in vivo by injecting HCT into the muscles of Mito.CFP mice (Figure 5). Intramuscular injections of HCT-555 were performed in TA muscle in a Mito.CFP mouse ~4 h prior to imaging. Both mitochondria (i panels) and signaling endosomes (ii panels) were simultaneously visualized in muscle-specific axons (i.e., axons innervating the TA). Indeed, anterogradely (yellow triangles) and retrogradely (green triangles and circles) moving organelles as well as stalled organelles (orange triangles and circles) can be observed. Using this experimental paradigm, one can assess the complex functional interactions between axonal mitochondria and signaling endosomes in vivo. Overall, we demonstrate several different experimental approaches to assess axonal transport of signaling endosomes and/or mitochondria, specifically in cholinergic motor neurons in vivo.

Figure 1: Sciatic nerve motor axons. (A) Representative single-plane image of eGFP-positive motor axons obtained in vivo from a ChAT.eGFP mouse. (B) Representative single-plane image of tdTomato-positive motor axons in an excised sciatic nerve from a ChAT.tdTomato mouse. Distinctions in axon caliber result from differences in mouse age and size. Scale bars = 50 µm. Abbreviations: eGFP = enhanced green fluorescent protein; ChAT = choline acetyltransferase. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: In vivo axonal transport of signaling endosomes in live sciatic nerve motor and sensory neurons of a ChAT.eGFP mouse. (A-C) Representative images of cholinergic axons expressing eGFP (A) and containing HCT-555-positive signaling endosomes (B), and the merge (C). The white arrows highlight an eGFP-positive motor axon containing HCT-555-positive signaling endosomes, the cyan arrows identify a motor axon lacking HCT-555-positive signaling endosomes, and the yellow arrows highlight an eGFP-negative sensory axon transporting HCT-555-positive signaling endosomes. Orange asterisks identify motor axons with weaker eGFP expression. Scale bar = 25 µm. Abbreviations: eGFP = enhanced green fluorescent protein; ChAT = choline acetyltransferase; HCT-555 = tetanus toxin-binding domain. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Time-lapse image series representing in vivo axonal transport of signaling endosomes in live motor neurons of an HB9.GFP mouse. (A-D) Time-lapse images taken every 3 s depicting motor neuron axons expressing green fluorescent protein (i) and containing HCT-555-positive signaling endosomes (ii), and the merge (iii). Each circle of the same color identifies the same moving endosome across different frames. Retrograde movement is from right to left. Scale bar = 10 µm. Abbreviations: GFP = green fluorescent protein; HCT-555 = tetanus toxin-binding domain. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: In vivo axonal transport of mitochondria in live sciatic nerve motor neurons of a ChAT.tdTomato:: Mito.CFP: mouse. (A-C) Representative images of tdTomato-positive motor axons (A), containing CFP-positive mitochondria (B), and the merge (C). The inset images i-iii contain a higher magnification from each panel. The white arrow represents a suspected node of Ranvier. Scale bars = 25 (A-C) and 10 µm (i-iii). Abbreviations: ChAT = choline acetyltransferase; CFP = cyan fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Time-lapse image series representing concurrent in vivo axonal transport of mitochondria and signaling endosomes in live sciatic nerve motor neurons of a Mito.CFP mouse. (A-C) Time-lapse images taken every 3 s depicting axonal transport of both mitochondria (i) and signaling endosomes (ii) within the same sciatic nerve axon (iii). The yellow triangles identify anterogradely moving cargoes, the green circles/triangles identify retrogradely moving cargoes, and the orange circles/triangles identify stationary cargoes. Anterograde movement is from left to right, whereas retrograde movement is in the opposite direction. Scale bar = 10 µm. Abbreviations: HCT-555 = tetanus toxin-binding domain; CFP = cyan fluorescent protein. Please click here to view a larger version of this figure.
{kind=link}
Discussion
This protocol details steps to assess in vivo axonal transport of signaling endosomes and mitochondria in intact axons of the mouse sciatic nerve. Indeed, an experimental setup is provided that enables users to 1) distinguish motor from sensory neurons in vivo, in situ, and ex vivo by using mice expressing fluorescent reporter proteins selectively expressed in motor neurons; 2) assess in vivo axonal transport of signaling endosomes specifically in motor neuron axons using three different transgenic mice; 3) investigate in vivo axonal transport of mitochondria specifically in motor neuron axons; and 4) concurrently assess in vivo transport dynamics of signaling endosomes and mitochondria within the same axon. This approach has vast potential for investigating axonal transport in basal conditions and can be used to assess pathological perturbations in different diseases affecting peripheral motor and sensory nerves.
Using previous experimental paradigms as a foundation31,32, here we have detailed novel, robust ways to differentiate axonal transport occurring in motor versus sensory neurons using transgenic reporter mice. Using the Mito.CFP mouse, this approach has been further developed to assess in vivo mitochondrial transport by avoiding intrasciatic nerve injections of TMRE36. This circumvents possible neural damage and perturbations in axonal transport caused by the intranerve injection of the probe. Furthermore, this protocol allows the visualization of axonal transport of multiple organelles in motor axons innervating muscles with distinct physiological properties (e.g., fast-twitch fatigable muscles vs. slow-twitch fatigue-resistant muscles). As such, signaling endosome and/or mitochondrial axonal transport dynamics can be assessed in different subsets of α-motor neurons44. Moreover, the axonal transport of those organelles in pathological settings can also be assessed through crossbreeding with mouse models of different neurodegenerative diseases1,2,3.
The axonal transport toolkit is continuously expanding28,29, and ex vivo protocols have been developed to assess transport dynamics using cultured mouse ventral horn explants49 or excised mouse nerve-muscle preparations50. Furthermore, the development of protocols to assess axonal transport in induced human pluripotent stem cell (hiPSC)-derived cortical51 neurons or hiPSC-derived spinal motor neurons52 has enabled investigation of human neurons with disease-causing mutations. Such cutting-edge protocols in mouse tissue and human cells can provide critical insights into neuronal function, facilitate novel pathomechanistic discovery in neurodegenerative disease models, and be used to test therapeutic molecules and strategies.
Several critical steps need to be followed for the successful implementation of these techniques, and some important notes have been provided in the protocol section. The major requirements for intravital imaging are the inverted confocal microscope with customized stage insert and the equipment to maintain anesthesia and optimum temperature. Indeed, a specialized mobile anesthetic system is needed for 1) induction of anesthesia, 2) dissection/tissue processing (i.e., exposing the sciatic nerve), and 3) maintaining anesthesia during intravital imaging (as previously detailed in 31,32). Especially when using higher magnification objectives (e.g., 40x or 63x), the depth of anesthesia can impact the image quality, as deeper anesthesia induces large 'gaspy' breaths that result in frequent shifts in focus. Such large movements will undoubtedly impact post imaging transport analyses (e.g., tracking cargoes using the Fiji plugins TrackMate53 or KymoAnalyzer54) as the breathing movements produce artifacts in time-lapse videos that can render them unsuitable for automated tracking or require more time-consuming assessment. Moreover, we have also observed imaging artifacts caused by pulsating arteries within the sciatic nerve, which can only be resolved by choosing a different imaging region. The microscope must be equipped with an environmental chamber capable of maintaining constant body temperature, as temperature and pH influence axonal transport55. Furthermore, the application of analgesics post surgery should be avoided, as they can alter transport dynamics56. If the experimental design is longitudinal and requires repeated imaging (e.g.,57), the dissection protocols need to be appropriately adjusted to be minimally invasive and may require additional ethical/licence approval.
Certain experimental considerations need to be kept in mind. First, most of the protocols detailed herein involve the use of transgenic mice that possess fluorescent reporter proteins in mitochondria or motor neuron axons. Each of these mouse lines should be bred and imaged as hemi-/heterozygote. The exceptions, however, are the ChAT.Cre and Rosa26.tdTomato mouse lines that can be separately maintained as homozygotes, with the resulting hemizygote offspring enabling tdTomato expression in cholinergic neurons after Cre-loxP recombination. When cross-breeding transgenic hemi-/heterozygote mice (e.g., Mito.CFP) with other transgenic hemi-/heterozygote mice (e.g., ChAT.eGFP), one needs to carefully consider the breeding strategy, as obtaining the desired numbers of double-mutant progeny can be time-consuming. Moreover, when breeding the F1 generation of ChAT.Cre and Rosa26.tdTomato mice (i.e., ChAT.tdTomato) with additional transgenic strains (e.g., Mito.CFP), one should expect even fewer mice carrying the desired triple transgenes. In addition, one must also consider the potential fluorophore overlap when breeding two-reporter mice with nearby wavelength properties (e.g., Mito-CFP-excitation: 435 nm, emission: 485 nm, bred with ChAT.eGFP-excitation: 488 nm, emission: 510 nm), although it may be possible to overcome this problem with spectral unmixing58.
This technique has some limitations to be considered. In this work and our previous protocols31,32, we have shown how several genetically encoded markers and different staining methods can be used to label and track distinct organelles in vivo. However, not all probes are suitable for this experimental approach. We assessed injections into either TA or soleus muscle of cholera toxin beta subunit (CTB)-488 (0.5-1.5 µg/µL ~4 h before imaging), a probe routinely used to label motor neuron cell bodies in in vivo retrograde tracer experiments59,60. However, when injected alone or co-injected with HCT-555, the CTB-488 labeling was poor despite using concentrations similar to those used for successful retrograde motor neuron tracing. Thus, we conclude that, despite CTB being an excellent in vitro marker of signaling endosomes in neuronal cultures61, HCT remains the gold-standard probe to identify signaling endosomes in vivo in sciatic nerve axons.
Using different routes, we also tested probes routinely used for labeling lysosomes, such as LysoTracker green DND-26, and markers of active lysosomal hydrolases, such as BODIPY-FL-pepstatin A for Cathepsin D62 and Magic Red for Cathepsin B, but with no success. We tried intramuscular delivery of BODIPY-FL-pepstatin A (2.5 µg into the TA ~4 h before imaging), as well as intrasciatic nerve injection of 2 µL of LysoTracker (10 µM), BODIPY-FL-pepstatin A (10 µM) or Magic Red (1/10) 30-60 min before imaging. Despite these probes highlighting the nerve, we were unable to find clearly labeled organelles. The probes accumulated around axons, likely being retained by Schwann cells. Hence, the unsuccessful labeling of lysosomes may be due to deficient probe delivery into neurons, although the existence of more suitable concentrations cannot be ruled out. Given that TMRE labeling works under similar conditions (i.e., intrasciatic nerve injections), the labeling intensity may be dye-dependent and must be tested for each marker independently. However, we conclude that targeting lysosomes in vivo with these probes is not feasible at the concentrations stated above.
Methods of anesthesia can alter distinct physiological readouts (e.g., cochlea function63 and cortical electrophysiology64); however, whether anesthesia influences in vivo axonal transport in the sciatic nerve is currently unknown. Given the reduced neuromuscular activity under isoflurane-induced anesthesia, it is possible that transport kinetics differ compared to the wakeful state. However, the only in vivo study that has directly investigated this revealed that transport of dense core vesicles in thalamocortical projections does not differ between anesthetized and awake mice65. Furthermore, because distinctions in transport between wild-type and disease model mice are detectable under anesthesia35,43, it is clear that isoflurane exposure does not prevent the identification of perturbances in signaling endosome or mitochondrial trafficking.
This protocol has other potential applications, which have been described below. Breeding of the transgenic mice outlined in this protocol (e.g., Mito.CFP, ChAT.eGFP) with neurodegenerative disease mouse models1,2,3 will enable neuron subtype- and/or cargo-specific investigations. Moreover, recently developed mouse Cre lines66 would also permit the visualization of fluorescent reporter proteins in distinct sensory axon populations. For example, Rosa26.tdTomato mice can be crossed with a neuropeptide Y receptor-2-expressing (Npy2r).Cre mouse to enable tdTomato fluorescence in myelinated A-fiber nociceptors67. Furthermore, temporal control can also be achieved by using inducible Cre systems (e.g., tamoxifen)68. Another potential application relies on the availability of transgenic mice expressing fluorescent reporter proteins in Schwann cells. Indeed, S100-GFP69 and PLP-GFP70 mice enable in vivo and/or in situ imaging of Schwann cells and have been at the forefront of research involved in Schwann cell migration during peripheral nerve regeneration.
In addition to these applications and complementing the Mito.CFP mouse is the availability of several transgenic mouse lines that express fluorescent proteins in distinct organelles, such as mitochondria and autophagosomes. For example, investigating in vivo mitochondrial transport might be possible with the mito::mKate2 mouse71 or the photoconvertible mitoDendra mouse57. Moreover, in vivo mitophagosome transport may be possible using the pH-sensitive mito-Keima mouse72 and the mito-QC mouse73 for mitophagy analyses. Furthermore, the lysosomal labeling difficulties we encountered may be overcome by using mice expressing LAMP1-GFP, with the caveat that LAMP1 is also present in endocytic organelles distinct from lysosomes74.
In summary, we have provided novel ways to assess in vivo axonal transport of several organelles in specific peripheral nerve axons from diverse transgenic mice. The concurrent imaging of different organelles will be particularly important, given recent findings of axonal interactions and co-trafficking of organelles such as mitochondria and endosomes75,76. We believe that the presented methods will be useful for improving understanding of the basal physiology of axons in vivo and untangling important pathomechanisms driving neurodegeneration of peripheral nerves.
Disclosures
The authors have no conflicts of interest.
Acknowledgements
We would like to thank Robert M. Brownstone (Queen Square Institute of Neurology, University College London) for sharing the ChAT-eGFP, ChAT.Cre and Rosa26.tdTomato mice, and Pietro Fratta (Queen Square Institute of Neurology, University College London) for sharing the HB9.GFP mouse. We would like to thank Elena R. Rhymes, Charlotte J.P. Kremers, and Qiuhan Lang (Queen Square Institute of Neurology, University College London) for critical reading of the manuscript. This work was supported by a Junior Non-Clinical Fellowship from the Motor Neuron Disease Association (UK) (Tosolini/Oct20/973-799) (APT), the Wellcome Trust Senior Investigator Awards (107116/Z/15/Z and 223022/Z/21/Z) (GS), a UK Dementia Research Institute Foundation award (GS); and a Medical Research Council Career Development Award (MR/S006990/1) (JNS).
Materials
| Name | Company | Catalog Number | Comments |
| 0.2 mL PCR tube | |||
| 70% (v/v) ethanol in distilled water | |||
| AlexaFlour555 C2 maleimide | ThermoFisher Scientific | A-20346 | Can also use AlexaFlour-488 or -647 Maleimide |
| B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | 7909 | Rosa26.tdTomato mice |
| B6.Cg-Tg(Hlxb9-GFP)1Tmj/J mice | Jackson Laboratory | 5029 | HB9.GFP mice |
| B6.Cg-Tg(Thy1-CFP/COX8A)S2Lich/J mice | Jackson Laboratory | 7967 | Mito.CFP mice |
| B6;129S6-Chattm2(cre)Lowl/J mice | Jackson Laboratory | 6410 | ChAT.Cre mice |
| Computer with microscope control and image acquisition software | Zeiss | Zen | |
| Cotton swab | |||
| Desktop centrifuge | |||
| Dissecting microscope | |||
| Eye lubricant | |||
| Fine curved forceps | Dumont | ||
| Fine straight forceps | Dumont | ||
| Glass coverslip (22 x 64 mm, thickness no. 1) | |||
| Graduated, glass micropipette with microliter markings and plunger | Drummond Scientific | 5-000-1001-X10 | |
| Hair clippers | |||
| Hamilton microliter syringe (701 N, volume 10 μL, needle size 26 s G, bevel tip, needle L 51 mm) | Merck | 20779 | |
| HcT-441 | N/A | N/A | See Restani et al., 2012 for more details |
| Heating pad | |||
| Immersion oil for fluorescent imaging at 37 °C | |||
| Inverted confocal microscope with environmental chamber | Zeiss | LSM 780 | Most inverted confocals should be adaptable |
| Isoflurane | |||
| Isoflurane vaporizer/anesthesia machine with induction cham-ber and mask stabilizer | |||
| Magic tape | invisible tape | ||
| Micropipette puller | |||
| Parafilm | Parafilm | ||
| Phosphate-buffered saline (PBS): 137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4–HCl, pH 7.4 | |||
| Recombinant human brain-derived neurotrophic factor (BDNF) | Peprotech | 450-02 | BDNF that can be co-injected with HcT-555 |
| Saline | |||
| Scalpel blade | Dumont | ||
| Small spring scissors | Dumont | ||
| Surgery/operating microscope | |||
| Surgical drape | |||
| Surgical suture | |||
| Surgical tape | |||
| Tg(Chat-EGFP) GH293Gsat/Mmucd mice | MMRRC | 000296-UCD | ChAT.eGFP |
| Vortex mixer |
References
- Webster, R. G. Animal models of the neuromuscular junction, vitally informative for understanding function and the molecular mechanisms of congenital myasthenic syndromes. International Journal of Molecular Sciences. 19 (5), 1326 (2018).
- Sleigh, J. N., Gillingwater, T. H., Talbot, K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Disease Models & Mechanisms. 4 (4), 457-467 (2011).
- De Giorgio, F., Maduro, C., Fisher, E. M. C., Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Disease Models & Mechanisms. 12 (1), 037424 (2019).
- Ilieva, H., Polymenidou, M., Cleveland, D. W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. The Journal of Cell Biology. 187 (6), 761-772 (2009).
- Brown, R. H., Al-Chalabi, A. Amyotrophic lateral sclerosis. The New England Journal of Medicine. 377 (2), 162-172 (2017).
- Gong, S., et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 425 (6961), 917-925 (2003).
- Tallini, Y. N., et al. BAC transgenic mice express enhanced green fluorescent protein in central and peripheral cholinergic neurons. Physiological Genomics. 27 (3), 391-397 (2006).
- Zeilhofer, H. U., et al. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. The Journal of Comparative Neurology. 482 (2), 123-141 (2005).
- Kaiser, T., Ting, J. T., Monteiro, P., Feng, G. Transgenic labeling of parvalbumin-expressing neurons with tdTomato. Neuroscience. 321, 236-245 (2016).
- Zheng, B., Sage, M., Sheppeard, E. A., Jurecic, V., Bradley, A. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Molecular and Cellular Biology. 20 (2), 648-655 (2000).
- Madisen, L., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience. 13 (1), 133-140 (2010).
- Bareyre, F. M., Kerschensteiner, M., Misgeld, T., Sanes, J. R. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nature Medicine. 11 (12), 1355-1360 (2005).
- Willems, J., et al. A CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLoS Biology. 18 (4), 3000665 (2020).
- Huh, Y., Oh, M. S., Leblanc, P., Kim, K. -. S. Gene transfer in the nervous system and implications for transsynaptic neuronal tracing. Expert Opinion on Biological Therapy. 10 (5), 763-772 (2010).
- Tosolini, A. P., Morris, R. Targeting motor end plates for delivery of adenoviruses: an approach to maximize uptake and transduction of spinal cord motor neurons. Scientific Reports. 6, 33058 (2016).
- Andrews, M. R. Gene therapy in the CNS-one size does not fit all. Gene Therapy. 28 (7-8), 393-395 (2021).
- Kügler, S. Tissue-specific promoters in the CNS. Methods in Molecular Biology. 1382, 81-91 (2016).
- Misgeld, T., Kerschensteiner, M., Bareyre, F. M., Burgess, R. W., Lichtman, J. W. Imaging axonal transport of mitochondria in vivo. Nature Methods. 4 (7), 559-561 (2007).
- Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular Biology of the Cell. 15 (3), 1101-1111 (2004).
- Dana, H., et al. Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS One. 9 (9), 108697 (2014).
- Chen, Q., et al. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 76 (2), 297-308 (2012).
- Maday, S., Twelvetrees, A. E., Moughamian, A. J., Holzbaur, E. L. F. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 84 (2), 292-309 (2014).
- Terenzio, M., Schiavo, G., Fainzilber, M. Compartmentalized signaling in neurons: from cell biology to neuroscience. Neuron. 96 (3), 667-679 (2017).
- Abouward, R., Schiavo, G. Walking the line: mechanisms underlying directional mRNA transport and localisation in neurons and beyond. Cellular and Molecular Life Sciences. 78 (6), 2665-2681 (2021).
- Sleigh, J. N., Rossor, A. M., Fellows, A. D., Tosolini, A. P., Schiavo, G. Axonal transport and neurological disease. Nature Reviews. Neurology. 15 (12), 691-703 (2019).
- Bronfman, F. C., Moya-Alvarado, G. BDNF/TrkB signaling endosomes mediate long-distance dendritic growth by activating CREB/PI3K-mTOR-dependent translation in neuronal cell bodies. BioRxiv. , (2020).
- Nagano, S., Araki, T. Axonal Transport and Local Translation of mRNA in Neurodegenerative Diseases. Frontiers in Molecular Neuroscience. 14, 697973 (2021).
- Boecker, C. A., Olenick, M. A., Gallagher, E. R., Ward, M. E., Holzbaur, E. L. F. ToolBox: Live Imaging of intracellular organelle transport in induced pluripotent stem cell-derived neurons. Traffic. 21 (1), 138-155 (2020).
- Surana, S., et al. The evolution of the axonal transport toolkit. Traffic. 21 (1), 13-33 (2020).
- Sleigh, J. N., Vagnoni, A., Twelvetrees, A. E., Schiavo, G. Methodological advances in imaging intravital axonal transport. F1000Research. 6, 200 (2017).
- Gibbs, K. L., Kalmar, B., Sleigh, J. N., Greensmith, L., Schiavo, G. In vivo imaging of axonal transport in murine motor and sensory neurons. Journal of Neuroscience Methods. 257, 26-33 (2016).
- Sleigh, J. N., Tosolini, A. P., Schiavo, G. In vivo imaging of anterograde and retrograde axonal transport in rodent peripheral nerves. Methods in Molecular Biology. 2143, 271-292 (2020).
- Gibbs, K. L., et al. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death & Disease. 9 (6), 596 (2018).
- Fellows, A. D., Rhymes, E. R., Gibbs, K. L., Greensmith, L., Schiavo, G. IGF1R regulates retrograde axonal transport of signalling endosomes in motor neurons. EMBO Reports. 21 (3), 49129 (2020).
- Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proceedings of the National Academy of Sciences of the United States of America. 107 (47), 20523-20528 (2010).
- Kalinski, A. L., et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. The Journal of Cell Biology. 218 (6), 1871-1890 (2019).
- Bercsenyi, K., et al. Tetanus toxin entry. Nidogens are therapeutic targets for the prevention of tetanus. Science. 346 (6213), 1118-1123 (2014).
- Deinhardt, K., et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 52 (2), 293-305 (2006).
- Debaisieux, S., Encheva, V., Chakravarty, P., Snijders, A. P., Schiavo, G. Analysis of signaling endosome composition and dynamics using SILAC in embryonic stem cell-derived neurons. Molecular & Cellular Proteomics. 15 (2), 542-557 (2016).
- Surana, S., et al. The travel diaries of tetanus and botulinum neurotoxins. Toxicon. 147, 58-67 (2018).
- Villarroel-Campos, D., Schiavo, G., Lazo, O. M. The many disguises of the signalling endosome. FEBS Letters. 592 (21), 3615-3632 (2018).
- Malik, B., et al. Absence of disturbed axonal transport in spinal and bulbar muscular atrophy. Human Molecular Genetics. 20 (9), 1776-1786 (2011).
- Sleigh, J. N., et al. Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Reports. 30 (11), 3655-3662 (2020).
- Tosolini, A. P., Sleigh, J. N., Surana, S., Rhymes, E. R., Cahalan, S. D., Schiavo, G. modulation of axonal transport is selectively impaired in ALS. BioRxiv. , (2021).
- Restani, L., et al. Botulinum neurotoxins A and E undergo retrograde axonal transport in primary motor neurons. PLoS Pathogens. 8 (12), 1003087 (2012).
- Mohan, R., Tosolini, A. P., Morris, R. Intramuscular injections along the motor end plates: a minimally invasive approach to shuttle tracers directly into motor neurons. Journal of Visualized Experiments: JoVE. (101), e52846 (2015).
- Tosolini, A. P., Mohan, R., Morris, R. Targeting the full length of the motor end plate regions in the mouse forelimb increases the uptake of fluoro-gold into corresponding spinal cord motor neurons. Frontiers in Neurology. 4, 58 (2013).
- Mohan, R., Tosolini, A. P., Morris, R. Targeting the motor end plates in the mouse hindlimb gives access to a greater number of spinal cord motor neurons: an approach to maximize retrograde transport. Neuroscience. 274, 318-330 (2014).
- Altman, T., Maimon, R., Ionescu, A., Pery, T. G., Perlson, E. Axonal transport of organelles in motor neuron cultures using microfluidic chambers system. Journal of Visualized Experiments: JoVE. (159), e60993 (2020).
- Boyer, N. P., Azcorra, M., Jung, P., Brown, A. Imaging and analysis of neurofilament transport in excised mouse tibial nerve. Journal of Visualized Experiments: JoVE. (162), e61264 (2020).
- Mou, Y., Mukte, S., Chai, E., Dein, J., Li, X. -. J. Analyzing mitochondrial transport and morphology in human induced pluripotent stem cell-derived neurons in hereditary spastic paraplegia. Journal of Visualized Experiments: JoVE. (156), e60548 (2020).
- Stoklund Dittlau, K., et al. Generation of human motor units with functional neuromuscular junctions in microfluidic devices. Journal of Visualized Experiments: JoVE. (175), e62959 (2021).
- Tinevez, J. -. Y., et al. TrackMate: An open and extensible platform for single-particle tracking. Methods. 115, 80-90 (2017).
- Neumann, S., Chassefeyre, R., Campbell, G. E., Encalada, S. E. KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic. 18 (1), 71-88 (2017).
- Bohnert, S., Schiavo, G. Tetanus toxin is transported in a novel neuronal compartment characterized by a specialized pH regulation. The Journal of Biological Chemistry. 280 (51), 42336-42344 (2005).
- Kanai, A., et al. Low-concentration lidocaine rapidly inhibits axonal transport in cultured mouse dorsal root ganglion neurons. Anesthesiology. 95 (3), 675-680 (2001).
- Bolea, I., Gan, W. -. B., Manfedi, G., Magrané, J. Imaging of mitochondrial dynamics in motor and sensory axons of living mice. Methods in Enzymology. , 97-110 (2014).
- Cohen, S., Valm, A. M., Lippincott-Schwartz, J. Multispectral live-cell imaging. Current Protocols in Cell Biology. 79 (1), 46 (2018).
- Blum, J. A., et al. Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons. Nature Neuroscience. 24 (4), 572-583 (2021).
- Xu, J., et al. An approach to maximize retrograde transport based on the spatial distribution of motor endplates in mouse hindlimb muscles. Frontiers in Cellular Neuroscience. 15, 707982 (2021).
- Wang, T., et al. Flux of signalling endosomes undergoing axonal retrograde transport is encoded by presynaptic activity and TrkB. Nature Communications. 7, 12976 (2016).
- Chen, C. S., Chen, W. N., Zhou, M., Arttamangkul, S., Haugland, R. P. Probing the cathepsin D using a BODIPY FL-pepstatin A: applications in fluorescence polarization and microscopy. Journal of Biochemical and Biophysical Methods. 42 (3), 137-151 (2000).
- Cederholm, J. M. E., et al. Differential actions of isoflurane and ketamine-based anaesthetics on cochlear function in the mouse. Hearing Research. 292 (1-2), 71-79 (2012).
- Michelson, N. J., Kozai, T. D. Y. Isoflurane and ketamine differentially influence spontaneous and evoked laminar electrophysiology in mouse V1. Journal of Neurophysiology. 120 (5), 2232-2245 (2018).
- Knabbe, J., Nassal, J. P., Verhage, M., Kuner, T. Secretory vesicle trafficking in awake and anaesthetized mice: differential speeds in axons versus synapses. The Journal of Physiology. 596 (16), 3759-3773 (2018).
- Gong, S., et al. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. The Journal of Neuroscience. 27 (37), 9817-9823 (2007).
- Arcourt, A., et al. Touch receptor-derived sensory information alleviates acute pain signaling and fine-tunes nociceptive reflex coordination. Neuron. 93 (1), 179-193 (2017).
- Valny, M., Honsa, P., Kirdajova, D., Kamenik, Z., Anderova, M. Tamoxifen in the mouse brain: implications for fate-mapping studies using the tamoxifen-inducible Cre-loxP system. Frontiers in Cellular Neuroscience. 10, 243 (2016).
- Hayashi, A., et al. A double-transgenic mouse used to track migrating Schwann cells and regenerating axons following engraftment of injured nerves. Experimental Neurology. 207 (1), 128-138 (2007).
- Chen, B., Chen, Q., Parkinson, D. B., Dun, X. -. P. Analysis of schwann cell migration and axon regeneration following nerve injury in the sciatic nerve bridge. Frontiers in Molecular Neuroscience. 12, 308 (2019).
- Barrasso, A. P., Tong, X., Poché, R. A. The mito::mKate2 mouse: A far-red fluorescent reporter mouse line for tracking mitochondrial dynamics in vivo. Genesis. 56 (2), (2018).
- Sun, N., et al. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nature Protocols. 12 (8), 1576-1587 (2017).
- McWilliams, T. G., et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. The Journal of Cell Biology. 214 (3), 333-345 (2016).
- Cheng, X. -. T., et al. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. The Journal of Cell Biology. 217 (9), 3127-3139 (2018).
- Cioni, J. -. M., Lin, J. Q., et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell. 176 (12), 56 (2019).
- Liao, Y. -. C., Fernandopulle, M. S., et al. RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell. 179 (1), (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved