
Method Article
A Novel Method to Determine the Longitudinal Antibacterial Activity of Drug-Eluting Materials
In This Article
Summary
Here, we present a protocol to evaluate the antibacterial efficacy of an antibiotic-eluting polymer to simulate prophylactic clinical application by using a commercially available real-time ATP-based luminescent microbial viability assay. This method enables the monitoring of the longitudinal activity of drug-eluting materials and can be widely adapted to test anti-microbial drug delivery platforms.
Abstract
Ultrahigh molecular weight polyethylene (UHMWPE) is widely used in total joint arthroplasties as a load-bearing surface. Periprosthetic joint infections, the majority of which occur shortly after joint replacement, constitute almost 25% of total knee revision surgeries, and the complete eradication of bacterial infection poses a major challenge. A promising way to tackle this problem is to ensure the local sustained delivery of antibiotic concentrations sufficient to inhibit the bacteria to support routine systemic antibiotic prophylaxis. There is increased research into the development of efficient local drug delivery devices. Although established antibacterial testing methods for drugs can be used to test the antibacterial efficacy of drug-eluting materials, they are lacking in terms of providing real-time and longitudinal antibacterial efficacy data that can be correlated to the elution profiles of antibiotics from these devices. Here, we report a direct and versatile methodology to determine the antibacterial efficacy of antibiotic-eluting UHMWPE implants. This methodology can be used as a platform to avoid bacterial culture at each time point of a lengthy experiment and can also be adapted to other local drug delivery devices.
Introduction
Osteoarthritis is a significant degenerative condition that afflicts 500 million adults worldwide1. The gold standard in the treatment of end-stage arthritis is total joint replacement, which is projected to be performed in over 2 million patients annually in the United States alone by 20302, with global demand also increasing tremendously3,4. The procedure involves the replacement of the articulating cartilaginous surfaces of the joints with load-bearing synthetic materials made of metal/ceramic and highly crosslinked ultrahigh molecular weight polyethylene (UHMWPE). Total joint replacements significantly improve the quality of life for patients suffering from arthritis5, but a significant complication that endangers the performance of the implants and the satisfaction of the patients is periprosthetic joint infection (PJI), which accounts for 15%-25% of revision surgeries6. The cause of infection in most cases is believed to be the microbial contamination of the implant site during surgery7. The contaminating population then expands on the implant and tissue surfaces8. The host immune system is triggered in response, but the growth rate and biofilm formation of the contaminating organisms can enable them to evade the immune cells, which can lead to a heightened cytokine and chemokine storm without the eradication of the bacteria9,10,11. The resulting deep infection of the bone jeopardizes the fixation and performance of the implant as well as the health of the patient12.
Staphylococcus aureus and coagulase-negative staphylococci are the predominant causative organisms of PJI13. The severity of staphylococcal infections is high due to their increased resistance to antibiotics, biofilm formation, and ability to persist as small colony variants14,15,16. Implant-associated infections occur due to the adhesion of microorganisms onto the surface of the implant and the establishment of a complex biofilm matrix that enables the bacteria to evade deleterious host immune factors and effective concentrations of antibiotics14,15,16. Conventional treatment methods include intravenous and oral antibiotic combinations for a prolonged period17. A major drawback of this approach is the low bioavailability of the drug at the infection site and the difficulty of achieving sufficient concentrations of antibiotics to eradicate the bacteria consistently over the treatment period, which often results in poor outcomes18. To address these shortcomings, a fully functional localized drug-eluting UHMWPE implant was designed to ensure a sustained release of effective concentrations of antibiotics into the joint space19. Local elution from the drug-eluting implant is used as a complementary tool to prevent the growth and colonization of any bacteria remaining after the implant removal and debridement of the tissue. In vitro antibacterial testing of this antibiotic-eluting UHMWPE can be performed by incubating the material directly with the bacteria and quantifying the bacteria by the spread-plate method20,21. Alternatively, aliquots of eluent media can be tested against bacteria using methods such as the agar disk-diffusion method, broth dilution methods, and time-kill testing22. All these methods rely upon growth observation about 16-18 h after the collection of the aliquots by means of colony counting and turbidity measurements. The elution of antibiotics from such devices can be longitudinally quantified using these in vitro methods; however, to determine the translational value of these concentration profiles, a robust real-time in vitro method to assess antibacterial activity is needed.
The microbial viability assay used in this study was developed for the quantification of viable bacteria by measuring the luminescence corresponding to adenosine triphosphate (ATP) released from a live bacterial cell using luciferin-luciferase-based detection technology. ATP, as the energy currency of living cells, serves as a direct indicator of physiologically viable cells. The reagent performs cell lysis, which releases ATP molecules from viable cells that are then detected in the form of luminescence. This luminescence has a direct correlation to the proportion of viable bacterial cells within the sample23. This is a real-time, simple, versatile, fast, single reagent-based assay that can be adapted into a variety of assay designs and conditions with both gram-positive and gram-negative microorganisms. Here we report a method to determine the real-time antibacterial activity of antibiotic-eluting UHMWPE incubated with laboratory and clinical strains of S. aureus using a modified time-kill assay based on the microbial viability assay (e.g., BacTiter Glo). While the methodology is described with a specific implant material and a specific orthopedic application, the method can provide a platform for testing other antibiotic-eluting delivery devices with clinical applications.
Protocol
1. Preparation of reagents
- Mueller-Hinton broth (MHB) preparation: Dissolve 22 g of cation-adjusted MHB in 1 L of deionized water. Autoclave the media at 121 °C and 15 lb pressure for 20 min, making sure the bottle is loosely capped. Store the prepared sterile broth at 4 °C until use.
- Tryptic soy agar (TSA) preparation: Dissolve 20 g of TSA powder in 500 mL of deionized water. Autoclave the agar mixture at 121 °C and 15 lb for 20 min. Transfer the sterile media to a 55 °C water bath to bring down the temperature of the molten agar. Pour the cooled prepared agar onto sterile Petri dishes (~15 mL), and allow it to solidify. Store the solidified agar plates inverted to avoid condensation.
- Tryptic soy broth (TSB) preparation: Dissolve 15 g of TSB powder in 500 mL of deionized water. Autoclave the broth mixture at 121 °C and 15 lb for 20 min. Store the prepared sterile broth at 4 °C until use.
- Phosphate-buffered saline (PBS): Autoclave commercially purchased 1x PBS at 121 °C and 15 lb for 20 min before sterile use.
- Microbial viability assay reagent preparation: Equilibrate the contents of the assay to room temperature. Mix 10 mL of buffer with the lyophilized luminescent reagent powder, and store the prepared reagent at −20 °C as 1 mL aliquots. Before each time point, thaw the light-sensitive reagent, and bring it to room temperature.
- Gentamicin sulfate stock solution: Dissolve 80 mg of gentamicin sulfate (GS) in 10 mL of 1x PBS. Vortex the drug solution thoroughly to obtain an 8 mg/mL GS stock solution.
- Vancomycin hydrochloride stock solution: Dissolve 10 mg of vancomycin hydrochloride powder thoroughly in 10 mL of deionized water to obtain a 1 mg/mL stock solution.
- Boric acid buffer solution: Dissolve 24.7 g (0.4 M) of boric acid in 900 mL of deionized water. Adjust the pH of the solution to 10.4 with a 50% (w/v) sodium hydroxide solution. Further to this step, add deionized water to make it to 1 L.
- O-pthaldialdehyde reagent (OPA): Dissolve 0.2 g of OPA in 1 mL of methanol. Add 19 mL of 0.4 M boric acid buffer (pH 10.4) to the OPA solution. Add 0.4 mL of 2-mercaptoethanol to the OPA-boric acid mixture. Cover the reagent vial in aluminum foil, and store at 4 °C for use up to 2-3 days post preparation.
CAUTION: Avoid contact with the skin, eyes, and clothing. Handling of the OPA and 2 mercaptoethanol reagents should be performed only under a fume hood with appropriate exhaust ventilation while wearing proper personal protective equipment. Avoid breathing in the vapors, dust, or aerosols.
2. Preparation of virgin and drug-loaded UHMWPE
- Sift 2 g each of gentamicin sulfate and vancomycin hydrochloride powders with a sieve (75 µm pore size), and dehydrate at 45 °C in a vacuum oven (<0.1 atm) for 18-24 h.
- Blend the dehydrated drug with UHMWPE powder at 7% w/w (1.12 g of antibiotic + 14.88 g of UHMWPE) in a container using a mechanical mixer for 5 min.
- Preheat an aluminum bronze custom mold (85 mm x 50 mm) with stainless steel insert plates to 180 °C in a convection oven for 1 h. Preheat the platens of the press to 170 °C.
- Add the blended powder to the mold, and compress at 10 MPa, 170 °C for 10 min, followed by a 45 min water cooling cycle at 10 MPa.
- Remove the molded UHMWPE block (~3.5 mm) from the press using a hydraulic press.
- Mill the top and bottom surfaces of the block using a computerized numerical control (CNC) to remove any surface irregularities and contaminants.
- Cut the block into 3 mm x 5 mm x 20 mm strips using the CNC.
- Prepare virgin UHMWPE (without the addition of antibiotics) using the same methodology as described as a control for the study.
Supplementary Figure 1: Parts of the mold used for molding the UHMWPE samples. (A) Stainless steel insert plate; (B) mold cavity; (C) plunger; (D) backplate Please click here to download this File.
3. Determining the elution kinetics of drug-eluting UHMWPE
- Place a 3 mm x 5 mm x 20 mm strip in a 3 mL syringe with a Luer lock and a 25 G needle.
- Wash the strip by filling the syringe with deionized water and inverting the syringe multiple times.
- Fill the syringe with deionized water up to the 2 mL graduation.
- Place the syringe setup on a shaker at 100 rpm at room temperature.
- At each time point (6 h, 1 day, 2 days, 3 days, 1 week), collect the eluent in a 2 mL vial.
- At each time point, wash the strip by filling the syringe with deionized water and inverting the syringe. Discard the solution, and fill the syringe with de-ionized water up to the 2 mL graduation to continue the experiment until the next time point.
4. Determination of the vancomycin concentration
- Detect vancomycin concentration spectrophotometrically in deionized water at a peak absorption wavelength of 280 nm.
- Prepare known concentration range standards by adding 100 µL of 1 mg/mL stock solution to the UV 96-well plate and serially diluting using 100 µL of deionized water to obtain six levels of known concentrations (1 mg/mL to 0.03125 mg/mL).
- Transfer 100 µL of the eluents to UV-clear 96-well plates, and determine the concentration at 280 nm using a microplate reader.
- Generate a calibration curve by plotting the known drug concentrations against the corresponding absorbance values. Calculate the unknown drug concentration in the eluent using the linear equation generated by the calibration curve.
- Determine the drug mass by multiplying the concentration with the eluent volume (~1.7 mL).
- Calculate the cumulative drug release (mg) at a time point by summing the drug release from all the preceding time points.
- Normalize the drug release to the surface area of the strip (3.5 cm2) to determine the cumulative drug release per centimeter squared (cm2) of the implant.
5. Determination of the gentamicin concentration by the OPA tagging method 27
- Prepare GS standard solutions to perform the OPA assay.
- Transfer 1 mL of the 80 µg/mL GS solution to a centrifuge tube.
- Add 500 µL of PBS to five more centrifuge tubes.
- Perform a serial dilution with these tubes to obtain GS concentrations of 80 µg/mL, 40 µg/mL, 20 µg/mL, 10 µg/mL, 5 µg/mL, and 2.5 µg/mL. These serve as the calibration solutions for the assay.
- In a clear 96-well plate, add 80 µL of PBS to well A-1 and 80 µL of the calibration solutions to wells A-2 to A-7 in ascending concentration. This serves as an internal calibration for the assay.
- Add 80 µL of each sample to the empty wells in triplicate to be analyzed in the same 96-well plate. Limit the number of samples per well plate to <50 such that the timing of adding the OPA solution to the samples is approximately the same for all samples and to avoid evaporation.
- Add OPA reagent to all the standards and sample solutions.
- In a fume hood, fill a 25 mL reagent reservoir with methanol.
- Add 8 mL of methanol and 1 mL of the prepared OPA solution to a second reagent reservoir (section 1.8). Mix the solution thoroughly by pipetting up and down.
- With a multichannel 100 µL micropipette, add 48 µL of methanol from the first reservoir to all the sample-containing wells in the 96-well plate.
- Further to this step, add 72 µL of diluted OPA solution from the second reservoir to all the sample-containing wells in the 96-well plate. Incubate the well plate for 10 min at room temperature.
- Measure the fluorescence intensity (excitation of 340 nm, emission of 455 nm) using a microplate reader immediately after the 10 min incubation.
- Plot the calibration curve using the intensity readings for the solutions with known concentrations of GS. Add a linear line to determine the best fit and generate the corresponding equation.
- Calculate the drug concentration in the eluent from the calibration curves generated by plotting the known drug concentrations against the corresponding absorbance values.
- Calculate the drug mass by multiplying the concentration with the eluent volume (~1.7 mL).
- Determine the cumulative drug release (mg) at a time point by summing the drug release from all the preceding time points.
- Normalize the drug release to the surface area of the strip (3.5 cm2) to determine the cumulative drug release per centimeter squared (cm2) of the implant.
6. Bacteria preparation
NOTE: The following S. aureus strains were used in this study: type strain 12600, clinical strains L1101 and L1163 (obtained from Dr. Kerry LaPlante at the University of Rhode Island). The susceptibility profiles of these strains are presented in Table 1.
| Bacterial strains | Gentamicin MIC | Vancomycin MIC | |
| ATCC 12600 | ≤1 µg/mL (Sensitive) | ≤0.5 µg/mL (Sensitive) | |
| L1101 (Clinical strain) | ≥16 µg/mL (Resistant) | 8 µg/mL (Intermediate) | |
| L1163 (Clinical strain) | ≤1 µg/mL (Sensitive) | 8 µg/mL (Intermediate) | |
Table 1: Antibiotic susceptibility profiles of control and clinical S. aureus strains.
CAUTION: All the steps involving handling bacterial cultures and suspensions were performed in a BSL-2 lab space within a Class II, Type A2 Biosafety Cabinet.
- Thaw the bacterial stocks from −80 °C onto TSA plates to facilitate the growth of S. aureus. Statically incubate the plates overnight at 35 °C.
- Transfer two to three colonies from the overnight-grown agar plate cultures into 1 mL of sterile TSB, and statically incubate overnight at 35 °C.
- Transfer 100 µL of bacteria to a clear 96-well plate to spectrophotometrically measure the turbidity by determining the absorbance at 600 nm.
- Dilute the bacteria 10,000x using sterile MHB before determining the viable bacteria count using the luminescent assay.
7. Performing the real-time microbial viability assay
- Perform the luminescence-based assay using white opaque-bottom 96-well plates.
- Mix 100 µL of diluted bacterial suspensions with 100 µL of assay reagent prepared in section 1.5.
- Place a lid covered with aluminum foil over the prepared 96-well plate, and incubate for 5 mins with 100 rpm shaking.
- Measure the luminescence immediately with a microplate reader using the following settings in Gen5 3.11 software.
- Click New to set up a protocol in the Task Manager window.
- Select Costar 96 white opaque in the drop-down menu for Plate Type.
- Click on Read under the Select Steps > Actions menu.
- Click on Luminescence within the Read Method window. Keep the Endpoint/kinetic and Luminescence fiber options selected under read type and optics type, respectively. Click on OK.
- Keep the <default> settings in the Read Step window (gain = 135; integration time = 1 sec; read height = 4.5 mm). Click on OK.
- The plate layout window appears ready for the run. Check the Use Lid box in the load plate window, and click on OK.
- Place the 96-well plate into the machine to read the luminescence values.
8. Generating a luminescence versus viable count standard curve for each bacterial strain
NOTE: Culture and enumerate all the S. aureus strains according to the methodology described in section 6.
- Prepare serially diluted bacterial suspensions to serve as standards.
- Enumerate the overnight-grown bacterial suspension by spectrophotometrically measuring the turbidity.
- Centrifuge at 6,000 × g for 5 mins to pellet the bacteria, and resuspend the pellet in sterile MHB to adjust the concentration to 1 x 109 CFU/mL.
- Dilute 100 µL of neat suspension using 900 µL of MHB, and perform a 10-fold serial dilution to reach 1 x 101 CFU/mL.
- Perform the luminescence assay on the prepared bacterial suspensions as described in section 7.
- Use sterile MHB as a blank control for the experiment, and subtract the blank luminescence value from the test sample luminescence values.
- Plot the log (CFU) against the log (luminescence) to generate a standard curve. Record the equation and R2 value.
- Determine the CFU/mL corresponding to the luminescence values using the strain-specific equation throughout the study.
9. Time-dependent antibacterial activity study setup

Figure 1: A schematic representation of the experimental setup. Please click here to view a larger version of this figure.
{kind=link}
- Culture the bacteria overnight in 1 mL of TSB broth.
- Dilute the overnight grown bacterial suspension to 105 CFU/mL in sterile MHB (section 6), and verify the viable bacterial count using the luminescence units before the start of the experiment (section 7).
- Place the virgin UHMWPE and drug-loaded UHMWPE strips (3 mm x 5 mm x 10 mm, half the dimension of strips prepared for the elution studies in section 2) within a 3 mL syringe.
- Draw MHB containing 105 CFU/mL into the syringe through the attached needle up to the 1.5 mL mark, which is equivalent to 1.35 mL of MHB (Figure 1: Step 1).
- Place the syringe setup on a shaking incubator (100 rpm) at 37 °C until the indicated time points of 6 h, Day 1, Day 2, Day 3, Day 4, Day 5, Day 6, and Day 7 (Figure 1: Step 2).
- At each indicated time point, take out the syringe setup, and perform a real-time microbial viability assay according to section 7 on 100 µL of bacterial suspension (Figure 1: Step 3-4).
- Determine the viable bacteria count for the 6 h, Day 1, Day 2, Day 3, and Day 7 time points. Determine the CFU/mL from the luminescence units using the corresponding standard curve.
- Verify the absence of viable bacteria in the samples that showed luminescence values below the limit of detection by performing the spread-plate method on TSA plates and incubating at 35 °C overnight. Check for the presence of colonies the next day.
- Centrifuge the remaining bacterial suspension at 10,000 × g for 10 min, and gently aspirate the spent media. For tubes in which pellets are not visually observed due to the antibacterial activity of the drugs, leave 100 µL in the tube to ensure the unseen pellet is not disturbed (Figure 1: Step 5).
- Resuspend the pelleted bacteria in fresh MHB, and draw back the bacterial suspension into the same syringe setup through the attached needle (Figure 1: Step 6).
10. Quantification of adherent bacteria on the UHMWPE surface
- Retrieve virgin UHMWPE and drug-loaded UHMWPE surfaces from the syringe setup after the completion of the study on Day 7.
- Transfer the surfaces to 1.5 mL tubes, and rinse with 1 mL of sterile PBS (three times).
- Sonicate the surface for 40 min in 1 mL of sterile PBS. Determine adherent bacteria viability by performing a luminescence assay on 100 µL of the sonicated sample, as describedin section 7.
Results
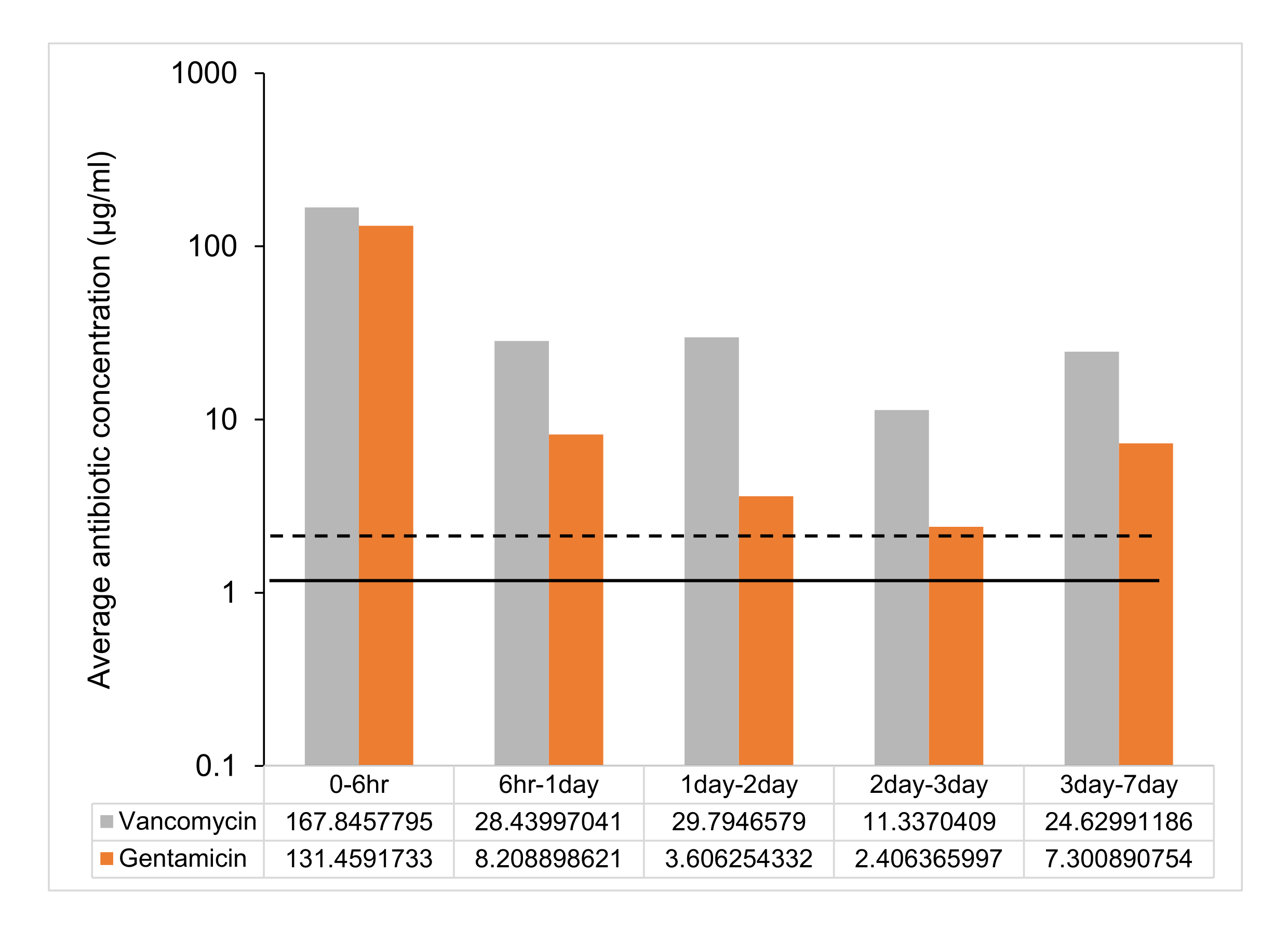
Following the protocol, UHMWPE was molded with gentamicin and vancomycin at 7% w/w and eluted into deionized water. The drug concentration in the eluent from the material was determined at 6 h, 1 day, 2 days, 3 days, and 1 week. The drug release from vancomycin and gentamicin-loaded UHMWPE demonstrated a burst release at 6 h followed by a steady release rate with a release concentration greater than the minimum inhibitory concentration (MIC) until 7 days (Figure 2, Table 2).
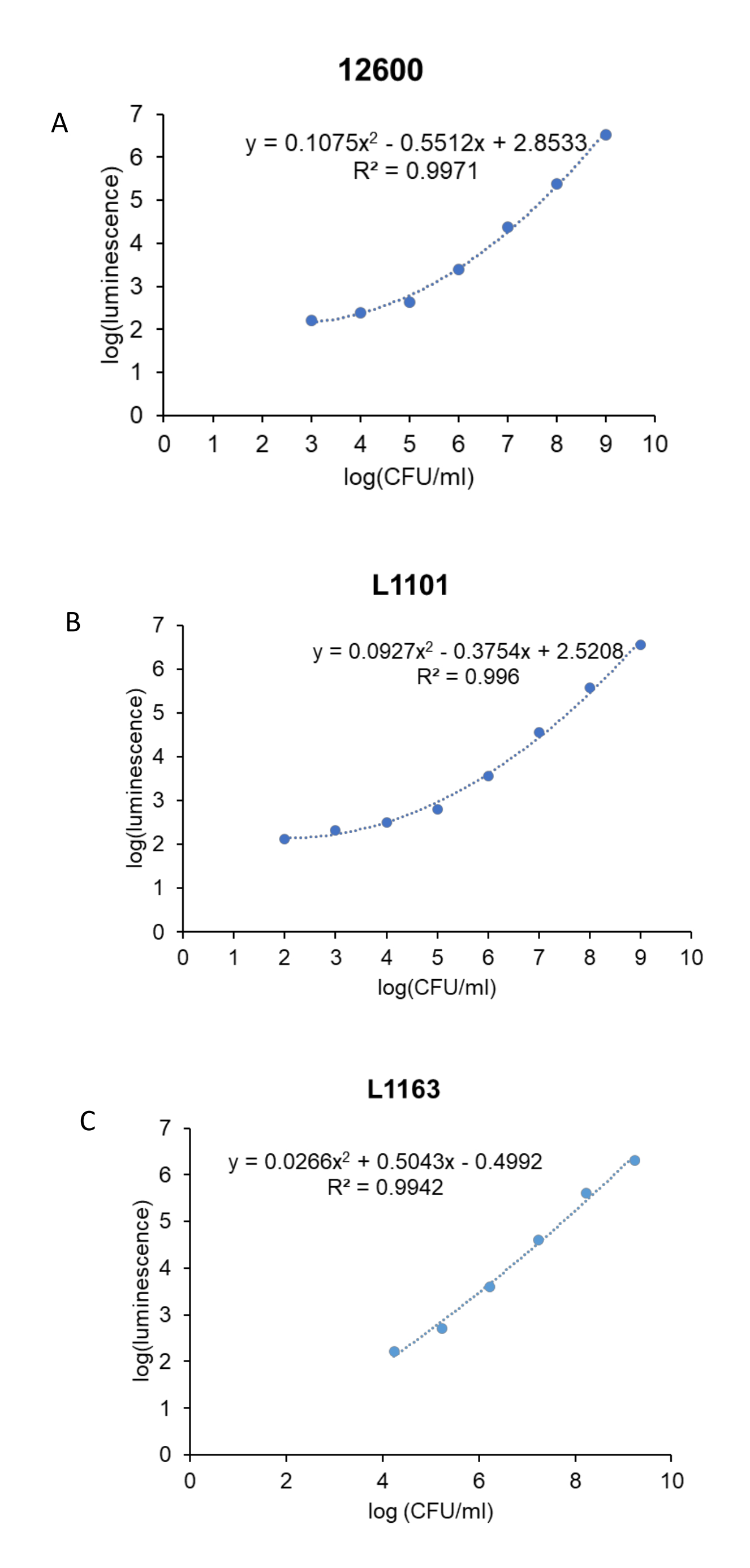
Prior to the antibacterial study, a standard curve was generated to correlate the luminescence units to the CFU/mL of the bacteria for each S. aureus strain (ATCC 12600, L1101, and L1163). The corresponding log (luminescence) values were plotted against the log (CFU/mL) to generate a standard curve (Figure 3). The R2 values calculated were 0.997, 0.996, and 0.994 for ATCC 12600, L1101, and L1163, respectively, indicating a strong fit for the concentration range. The equation derived was subsequently used to calculate the bacterial viability across all the experiments. ATCC 12600 and L1101 demonstrated a limit of detection within a range of 1 x 102-1 x 103 CFU/mL. On the other hand, the limit of detection for the L1163 strain was shown to be 1 x 104 CFU/mL.
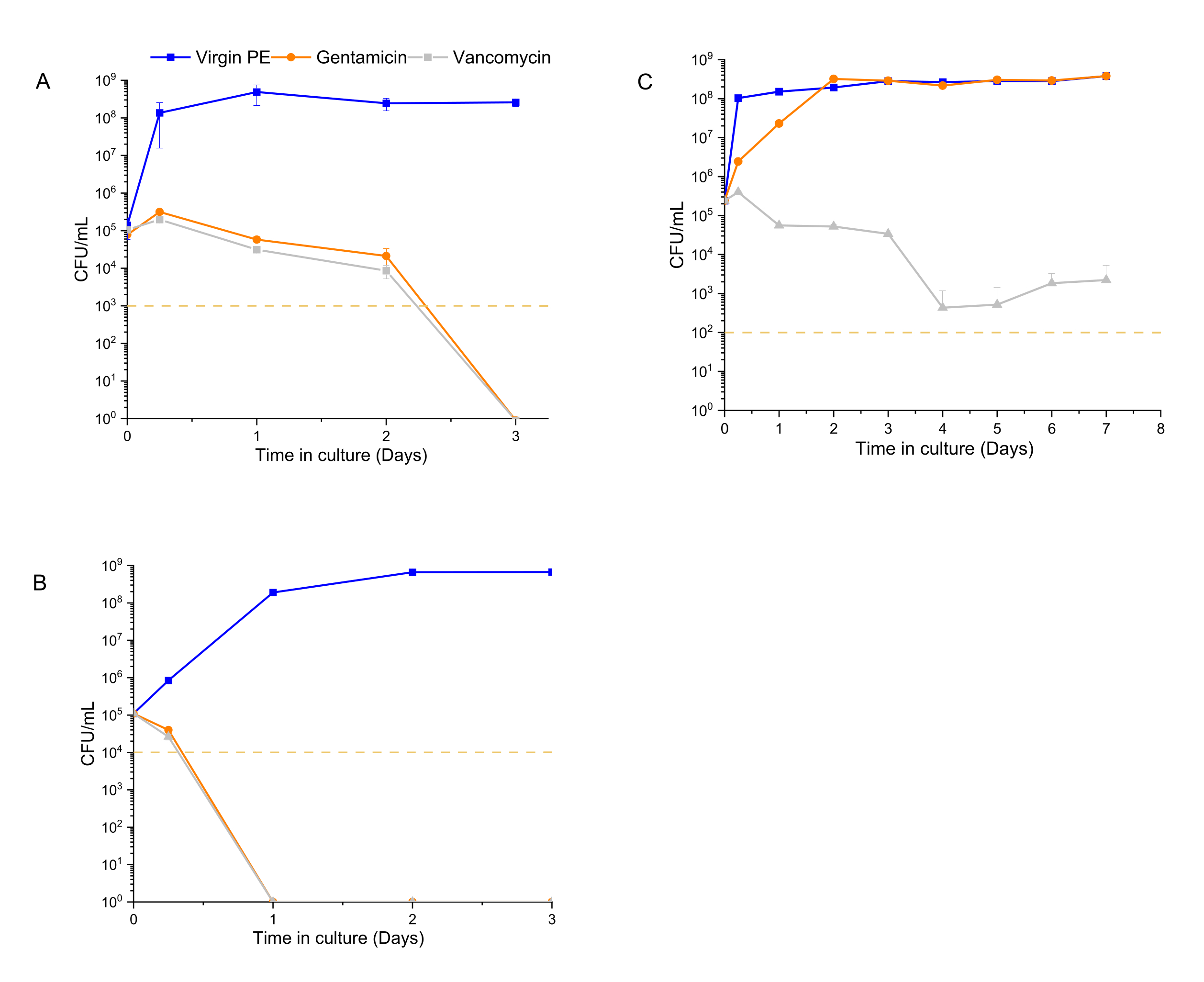
The time-dependent antibacterial activity assay was performed using 1 x 105 CFU/mL as the starting inoculum for 12600, L1163, and L1101, which were exposed to 7% w/w drug-eluting materials for a period of 1 week. At each time point (6 h, 1 day, 2 days, 3 days, 1 week), the medium was refreshed, and the bacterial population was re-suspended. The exposure of the bacteria to the subsequent release of drugs from the material was continued until the next time point. UHMWPE with 7% w/w vancomycin and UHMWPE with 7% w/w gentamycin demonstrated >3log reduction for susceptible ATCC 12600 starting at 6 h, and complete eradication (no colony growth) was observed at the end of 3 days (Figure 4A). For the gentamicin-susceptible and vancomycin-intermediate strain L1163, both drug-eluting materials caused >3log reduction at 6 h, and complete eradication (no colony growth) was observed on day 1 of the experiment (Figure 4B). For the gentamicin-resistant and vancomycin-intermediate strain L1101, gentamicin elution from UHMWPE did not affect the bacterial viability of L1101 (Figure 4C). The bacteria proliferated, and the population stabilized within 6 h in the presence of virgin UHMWPE without antibiotic elution. In the presence of gentamicin-eluting UHMWPE, the population reached a similar growth level on day 2. On the contrary, vancomycin elution from UHMWPE significantly reduced the bacterial viability (>3log) at 6 h, and complete viability loss (no colony growth) was demonstrated by day 4.
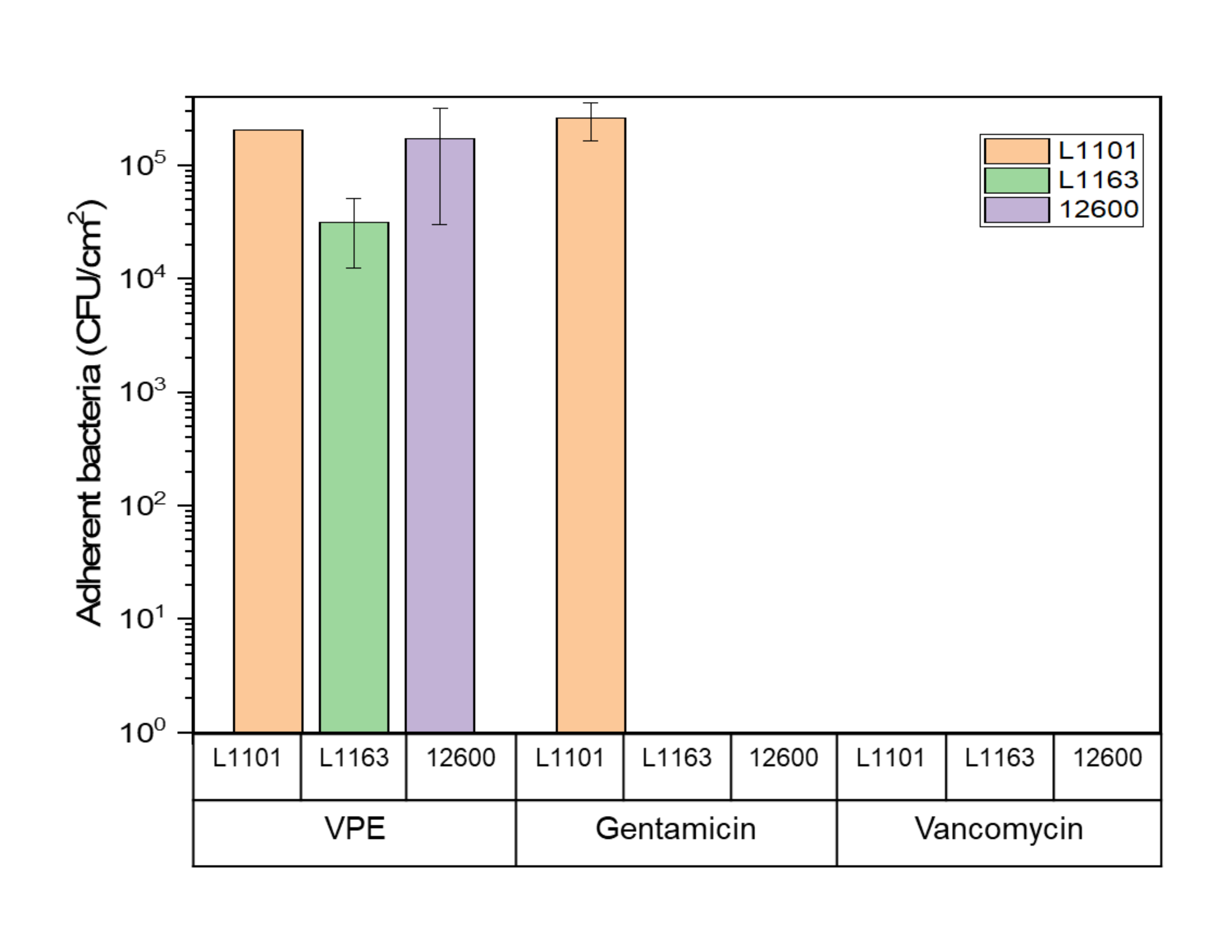
The surfaces of both gentamicin-eluting and vancomycin-eluting UHMWPE showed no viable adherent bacteria when exposed to susceptible and intermediate-resistant strains after day 7 or complete eradication, whichever came first. Some viable bacteria (1 x 105 CFU/mL) were present on gentamicin-eluting UHMWPE exposed to gentamicin-resistant L1101. Similarly, approximately 1 x 105 CFU/mL of viable adherent bacteria were recovered from the control virgin PE (Figure 5).

Figure 2: Time-dependent average antibiotic release from 7% w/w antibiotic-loaded UHMWPE strip. The average antibiotic release between time points from one 7% w/w gentamicin and vancomycin-loaded UHMWPE strip (3 mm3 x 5 mm3 x 10 mm3 ~ 2 cm2 surface area). The MIC against control strain ATCC 12600 is shown as a dotted line for gentamicin and a solid line for vancomycin. The error bars represent the standard deviation of the mean from six replicates (n = 6). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Real-time luminescent assay standard curve for all the S. aureus strains. Log (luminescence) was plotted against log (CFU/mL) to generate standard curves for control, (A) ATCC 12600, and clinical strains, (B) L1101 and (C) L1163. The equations describing the line of best fit and corresponding R2 values are indicated on the plots. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Bacterial viability determined using a luminescent assay for 7% w/w gentamicin-eluting and 7% w/w vancomycin-eluting UHMWPE. The time-dependent antibacterial activity of gentamicin and vancomycin eluted from UHMWPE against control strains, (A) ATCC 12600, and clinical strains, (B) L1163 and (C) L1101, are shown. Virgin 1020 PE served as a control for the experiment. The yellow line in the plots indicates the limit of detection for the respective S. aureus strain. Data are shown as mean ± standard deviation (n = 3). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Adherent bacteria viability determined using the luminescent assay Glo assay for 7% w/w gentamicin-eluting and 7% w/w vancomycin-eluting UHMWPE against all S. aureus strains. The bar chart indicates adherent bacteria (CFU) recovered per centimeter squared (cm2) of 7% gentamicin-loaded and 7% vancomycin-loaded UHMWPE at the end of the study period for all the strains tested. The bars show data as mean ± standard deviation (n = 3). Please click here to view a larger version of this figure.
{kind=link}
| Time points | Vancomycin | Gentamicin |
| Peak concentration (µg/mL) | Peak concentration (µg/mL) | |
| 0 - 6 h | 336 ± 72 | 263 ± 24 |
| 6 h -1 day | 57 ± 18 | 16 ± 2 |
| 1 day - 2 day | 60 ± 18 | 7 ± 1 |
| 2 day - 3 day | 23 ± 6 | 5 ± 0.4 |
| 3 day - 7 day | 49 ± 20 | 15 ± 1 |
Table 2: Peak drug concentration (μg/mL) at different time points. Data are shown as mean ± standard deviation (n = 6).
Supplementary Figure 2: Luminescence signal decay over a period of 10 min from the addition of assay reagent to the sample. A ±5% difference in the signal is shown as a dotted line Please click here to download this File.
Discussion
The localized sustained delivery of antibiotics is a necessary tool in the management of periprosthetic joint infections. Systemic antibiotics are the primary strategy in eradicating bacterial infection, and the local elution is used as a complementary tool to prevent the growth and colonization of any bacteria remaining after the implant removal and debridement of the tissue. The goal for the effective area under the curve (concentration over a period) for antibiotics with local administration is not well understood. The elution of antibiotics from such devices can be longitudinally quantified in vitro; however, to determine the translational value of these concentration profiles, a robust in vitro method to assess antibacterial activity is needed. In this paper, one such real-time method is described to determine the antibacterial activity of drug-eluting UHMWPE to be used as a sustained delivery device in joint replacements.
The real-time monitoring of bacterial viability is a crucial parameter of interest, and conventional microbiological methods lack the framework to accommodate this specific aspect of the study. The microbial viability assay used in this study was developed for the quantification of viable bacteria by measuring the luminescence corresponding to adenosine triphosphate (ATP). To directly investigate the time-dependent activity of the antibiotics eluted from the implant materials, three different strains with distinct antibiotic susceptibility profiles were incubated with them. The rationale for using laboratory and clinical strains with varying resistances to gentamicin and vancomycin was to understand the range of activity for a given implant formulation. Further, the antibacterial activity and efficacy against these distinct populations are dependent on the timing of administration. The method focuses on the feasibility of prophylaxis against these strains based on >70% of periprosthetic joint infections being caused by the contamination of the wound at the time of surgery24.
As a starting inoculum to develop this method, 1 x 105 CFU/mL was used. Different contaminating concentrations have been used for animal models although not much is known about the clinically relevant infection load for PJI. Animal infection models for PJI have been routinely established using 1 x 105 CFU, and a similar range is widely used in standardized methods (CLSI) to determine antibacterial activity25,26,27. Using 1 x 105 CFU/mL as an initial contaminating concentration allowed us to evaluate both the growth and eradication parameters at the same time.
Conventionally, MIC values are determined for a constant antibiotic concentration for a specific number of bacteria, and they fail to demonstrate the rate of antibacterial action. Due to this aspect, MIC values do not provide a quantitative differentiation to describe the antimicrobial activity profile28. The data from the current method emphasize the advantage of evaluating the strain-dependent killing kinetics of antibiotics rather than using the MIC to make dosing decisions. Using this method, it was possible to differentiate both the extent and the rate at which the implant materials affected the different strains. Gentamicin eluted from the implant material strips was effective in eradicating L1163 in 1 day and eradicating ATCC 12600 in 2-3 days, but it was ineffective in eradicating L1101 (Figure 4). In addition to the expected lack of activity of gentamicin elution against L1101 (MIC >32 µg/mL) due to its inherent gentamicin resistance (Figure 4C), the persistence of subpopulations was observed when exposed to vancomycin, for which L1101 exhibits intermediate resistance. In contrast, L1163 was definitively eradicated in the presence of vancomycin-eluting UHMWPE despite exhibiting similar intermediate resistance to vancomycin as L1101 (an MIC of 8 µg/mL has been observed for both strains).
The observations that the rate of activity of gentamicin against 12600 and L1163, which are gentamicin-susceptible with similar MIC values (an MIC of 1µg/mL has been observed for both strains), was different, as well as that the extent of activity of vancomycin against intermediate-resistant L1101 and L1163 was different (Figure 4A,B), supported the hypothesis that this real-time method in the presence of the eluting material could differentiate longitudinal differences in the activity.
In addition to the translational value of the results in interpreting how effective a given eluted concentration can be against these bacteria, there are several experimental methodological advantages. (1) The bacterial concentration is determined instantaneously at a given time, contrary to conventional methods in which the bacteria are incubated in broth or on agar for 18-24 h to determine viability. This period of growth can provide additional time for the bacteria to recover from antibiotic stress, introducing an additional possible source of error/variability. (2) The media is continually replaced while retaining the bacteria, which more closely resembles in vivo conditions than static conditions. (3) This assay inherently includes the drug release kinetics from the implant, which allows for better performance prediction. (4) The method has been developed using commonly available consumables without the need of any specific or expensive machinery.
Robust in vitro testing methods to evaluate drug-delivery applications are necessary before proceeding to in vivo animal studies and clinical trials. This assay can be modified and adapted to accommodate various approaches and drug delivery platforms such as particles, gels, films, and other drug-eluting materials to determine the efficiency of bacterial eradication in a simulated in vitro setup. Modifications can be performed for the sample setup by changing to a suitable in vitro medium environment, which has been shown to influence the activity of several antibiotics29,30.
The method also facilitates viable adherent bacteria determination, which is promising, as conventional methods to determine minimum biofilm eradication concentration is time-consuming and delivers inconsistent results. However, the method is to be rigorously tested on biofilms to develop a reliable and robust methodology to determine its sensitivity. The ATP-based luminescent method could be sensitive enough to detect viable forms of bacteria in biofilms including persisters, which may or may not be detected on an agar plate as visible colonies. Taken together, this versatile platform has the potential to incorporate relevant parameters to record real-time observations on the anti-bacterial and anti-biofilm activity of drugs of interest.
The efficacy of this method is governed by the following aspects:
Pre-determined elution characteristics and sample size
The elution profile of the antibiotic-eluting material can be identified in a separate experiment prior to this antibacterial activity measurement such that amount of material required to actively conduct the experiment within a stipulated time can be determined.
Container and volume determination
It is important to devise a setup in which the media volume of the experiment can accommodate the entire surface area of the same material and to ensure sufficient volume for the unobstructed release of the drug from the drug-loaded surface. The setup used was based on previous experimentation, ensuring "perfect sink" conditions for these hydrophilic drugs such that their diffusion is not hindered by solubility limitations.
Growth media characteristics
Growth media selection should be investigated to ensure the stability and the optimal performance of the selected drug(s)29. Cation-adjusted Mueller Hinton broth (CAMHB), which is widely used in the broth dilution method, was used to determine the MIC of known antibiotics. The medium enables optimal drug activity without the interference of toxic secondary metabolite accumulation31. The assay reagent has been tested and reported stable in different types of media, including those with serum components23,28. Although the relative luminescence unit values may vary across different media, the components of the media have been demonstrated to not interfere with the assay32,33. The experimental volume was further optimized to 1.5 mL, which is close to the synovial fluid volume present in an adult knee joint space34.
Temperature stability for the assay
The handling and addition of the luminescent reagent to the assay are to be performed in a consistent manner across experiments. Temperature changes alter the sensitivity of the assay, so it is important to incubate the reagent at room temperature for 2 h before adding it to the bacteria23.
Reagent incubation time
The luminescence from the reagent decays with time. The luminescent signal has a half-life of over 30 min, which is largely dependent on the medium and the type of bacterium used in the experiment23. Additionally, any differences in incubation time (i.e., the time between adding the reagent to the bacteria and reading the luminescence) will result in inconsistent readings for the same concentration of bacteria. A 5% difference was observed in the luminescent signal when taken within 1 min following the 5 min incubation time according to the manufacturer's instructions (Supplementary Figure 2). Taking this data into account, the luminescence readings were recorded within 1 min throughout the study to ensure the signal loss was not more than 5%. Further, it is important to limit the number of samples per plate to reduce the error introduced due to luminescence decay from the first well to the last well.
Maintenance of the bacterial population throughout the study
The method attempted to model the drug clearance and synovial volume turnover by continually separating the spent media from the bacterial population at each time point using high-speed centrifugation at 10,000 x g for 10 min35. This critical step ensures the sedimentation of all the viable and non-viable bacterial cells. Further to this, the sedimented bacteria are uniformly reconstituted in fresh MHB and transferred to the syringe setup, facilitating the complete carryover of the affected microbial population back to the experimental setup. The reproducibility and reliability of this method heavily rely upon simulating the sustained exposure of antibiotics to the microbial population derived from the initial inoculum.
A key limitation of this method is that it is a semi-static assay that does not accurately simulate drug half-lives and continuous synovial turnover. However, continual medium replacement partially compensates for this limitation. The sensitivity of the microbial viability assay was strain-dependent, ranging from 1 x 102-1 x 104 CFU/mL, which limits the detection capability. Furthermore, a standard curve needs to be plotted for each organism as the strain type contributes to the sensitivity and performance of the luminescent reagent. Both the bacteria growth dynamics and the activity of the used drug compound may be affected by the medium components, which should be further investigated.
Disclosures
The senior author (E.O.) receives royalties originating from the licensing of intellectual property to the following companies: Corin, Iconacy, Renovis, Arthrex, ConforMIS, Meril Healthcare, Exactech. There is no conflict of interest with any of the studies presented here.
Acknowledgements
This work was funded partially by National Institutes of Health Grant No. AR077023 (R01) and by the Harris Orthopaedic Laboratory. The authors thank Dr. Kerry Laplante and her team at the University of Rhode Island for providing the clinical strains L1101 and L1163.
Materials
| Name | Company | Catalog Number | Comments |
| 96 well plates - polystyrene, High Bind, white flat bottom wells, non-sterile, white, 100/cs | Corning, NY, USA | CLS3922-100EA | |
| 2-mercaptoethanol | Sigma Aldrich, Germany | ||
| ATCC 12600 | American Type culture Collection, VA, USA | ||
| BacTiter-Glo Microbial Cell Viability Assay | Promega Corporation, USA | G8231 | |
| BD Bacto Tryptic Soy Broth (Soybean-Casein Digest Medium) | Becton-Dickinson, USA | BD 211825 | purchased from Fisher Scientific, USA |
| BD Luer-Lok Syringe sterile, single use, 3 mL | BD, USA | 309657 | |
| BD Needle 5/8 in. single use, sterile, 25 G | BD, USA | 305122 | |
| BD BBL Dehydrated Culture Media: Mueller Hinton II Broth (Cation-Adjusted) | Becton-Dickinson, USA | B12322 | purchased from Fisher Scientific, USA |
| BD Difco Dehydrated Culture Media: Tryptic Soy Agar (Soybean-Casein Digest Agar) | Becton-Dickinson, USA | DF0369-17-6 | purchased from Fisher Scientific, USA |
| Boric Acid | Fisher Chemical, NJ, USA | ||
| Branson 1800 ultrasonic bath | Emerson, MO, USA | ||
| Corning Falcon Bacteriological Petri Dishes with Lid | Fisher Scientific, USA | 08-757-100D | |
| Gentamicin Sulfate | Fujian Fukang Pharmaceutical Co., Fuzhou, China | ||
| Greiner UV-Star 96 well plates | Sigma Aldrich, Germany | M3812-40EA | |
| Hydraulic press | Carver, Inc. Wabash, IN, USA | ||
| L1101 | Clinical strain from Dr Kerry Laplante, University of Rhode Island | ||
| L1163 | Clinical strain from Dr Kerry Laplante, University of Rhode Island | ||
| LSE benchtop shaking incubator | Corning, NY, USA | ||
| Methanol, Optima for HPLC, Fisher Chemical | Fisher Scientific, NJ, USA | A454-1 | |
| Napco CO2 6000 | Thermo Scientific, MA, USA | ||
| PBS, Phosphate Buffered Saline, 1X Solution, pH 7.4, Fisher BioReagents | Fisher Scientific, USA | BP24384 | |
| Phthaldiadehyde ≥97% (HPLC) | Sigma Aldrich, Germany | P1378-5g | |
| Plate reader (Synergy H1 | Biotek, VT, USA | ||
| press | Carver, Inc. Wabash, IN, USA | ||
| shaker Innova 2100 | New Brunswick Scientific, NJ, USA | ||
| ShopBot D2418 | ShopBot Tools, Inc., NC, USA | ||
| Sodium Hydroxide | Sigma Aldrich, Germany | ||
| Thermo Scientific Reagent Grade Deionized Water | Fisher Scientific, USA | 23-751628 | |
| UHMWPE | GUR1020, Celanese, TX, USA | ||
| Vancomycin Hydrochloride | Fagron, The Netherlands | 804148 | |
| WAB Turbula | WAB Turbula, Switzerland |
References
- Hunter, D. J., March, L., Chew, M. Osteoarthritis in 2020 and beyond: A Lancet Commission. Lancet. 396 (10264), 1711-1712 (2020).
- Sloan, M., Premkumar, A., Sheth, N. P. Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. The Journal of Bone and Joint Surgery. American Volume. 100 (17), 1455-1460 (2018).
- Gao, J., Xing, D., Dong, S., Lin, J. The primary total knee arthroplasty: A global analysis. Journal of Orthopaedic Surgery and Research. 15 (1), 190 (2020).
- . UpToDate. Prosthetic joint infection: Treatment Available from: https://www.uptodate.com/contents/prosthetic-joint-infection-treatment (2022)
- Dapunt, U., Radzuweit-Mihaljevic, S., Lehner, B., Haensch, G. M., Ewerbeck, V. Bacterial infection and implant loosening in hip and knee arthroplasty: Evaluation of 209 cases. Materials. 9 (11), 871 (2016).
- Kamath, A. F., et al. Quantifying the burden of revision total joint arthroplasty for periprosthetic infection. The Journal of Arthroplasty. 30 (9), 1492-1497 (2015).
- Tande, A. J., Patel, R. Prosthetic joint infection. Clinical Microbiology Reviews. 27 (2), 302-345 (2014).
- Davidson, D. J., Spratt, D., Liddle, A. D. Implant materials and prosthetic joint infection: The battle with the biofilm. EFORT Open Reviews. 4 (11), 633-639 (2019).
- de Vor, L., Rooijakkers, S. H. M., van Strijp, J. A. G. Staphylococci evade the innate immune response by disarming neutrophils and forming biofilms. FEBS Letters. 594 (16), 2556-2569 (2020).
- Dapunt, U., Giese, T., Stegmaier, S., Moghaddam, A., Hänsch, G. M. The osteoblast as an inflammatory cell: Production of cytokines in response to bacteria and components of bacterial biofilms. BMC Musculoskeletal Disorders. 17, 243 (2016).
- González, J. F., Hahn, M. M., Gunn, J. S. Chronic biofilm-based infections: Skewing of the immune response. Pathogens and Disease. 76 (3), 023 (2018).
- Dapunt, U., Giese, T., Prior, B., Gaida, M. M., Hänsch, G. M. Infectious versus non-infectious loosening of implants: activation of T lymphocytes differentiates between the two entities. International Orthopaedics. 38 (6), 1291-1296 (2014).
- Tai, D. B. G., Patel, R., Abdel, M. P., Berbari, E. F., Tande, A. J. Microbiology of hip and knee periprosthetic joint infections: A database study. Clinical Microbiology and Infection. 28 (2), 255-259 (2022).
- Foster, T. J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiology Reviews. 41 (3), 430-449 (2017).
- Kranjec, C., et al. Staphylococcal biofilms: challenges and novel therapeutic perspectives. Antibiotics. 10 (2), 131 (2021).
- Kahl, B. C., Becker, K., Löffler, B. Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clinical Microbiology Reviews. 29 (2), 401-427 (2016).
- Li, C., Renz, N., Trampuz, A. Management of periprosthetic joint infection. Hip & Pelvis. 30 (3), 138-146 (2018).
- Davis, J. S., et al. Predictors of treatment success after periprosthetic joint infection: 24-month follow up from a multicenter prospective observational cohort study of 653 patients. Open Forum Infectious Diseases. 9 (3), (2022).
- Suhardi, V. J., et al. A fully functional drug-eluting joint implant. Nature Biomedical Engineering. 1, 0080 (2017).
- Gil, D., Grindy, S., Muratoglu, O., Bedair, H., Oral, E. Antimicrobial effect of anesthetic-eluting ultra-high molecular weight polyethylene for post-arthroplasty antibacterial prophylaxis. Journal of Orthopaedic Research. 37 (4), 981-990 (2019).
- Robu, A., et al. Additives imparting antimicrobial properties to acrylic bone cements. Materials. 14 (22), 7031 (2021).
- Balouiri, M., Sadiki, M., Ibnsouda, S. K. Methods for in vitro evaluating antimicrobial activity: A review. Journal of Pharmaceutical Analysis. 6 (2), 71-79 (2016).
- BacTiter-Glo microbial cell viability assay instructions for use of products G8230, G8231, G8232 and G8233. <101/bactiter-glo-microbial-cell-viability-assay-protocol. Promega Corporation Available from: https://www.promega.com/resources/protocols/technical-bulletins/101/bactiter-glo-microbial-cell-viability-assay-protocol/ (2022)
- Izakovicova, P., Borens, O., Trampuz, A. Periprosthetic joint infection: Current concepts and outlook. EFORT Open Reviews. 4 (7), 482-494 (2019).
- López-Torres, I. I., Sanz-Ruíz, P., Navarro-García, F., León-Román, V. E., Vaquero-Martín, J. Experimental reproduction of periprosthetic joint infection: Developing a representative animal model. The Knee. 27 (3), 1106-1112 (2020).
- Carli, A. v., et al. Quantification of peri-implant bacterial load and in vivo biofilm formation in an innovative, clinically representative mouse model of periprosthetic joint infection. The Journal of Bone and Joint Surgery. American Volume. 99 (6), 25 (2017).
- Humphries, R. M., et al. CLSI Methods Development and Standardization Working Group best practices for evaluation of antimicrobial susceptibility tests. Journal of Clinical Microbiology. 56 (4), 01934 (2018).
- Mueller, M., de la Peña, A., Derendorf, H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: Kill curves versus MIC. Antimicrobial Agents and Chemotherapy. 48 (2), 369-377 (2004).
- Wijesinghe, G., et al. Influence of laboratory culture media on in vitro growth, adhesion, and biofilm formation of Pseudomonas aeruginosa and Staphylococcus aureus. Medical Principles and Practice. 28 (1), 28-35 (2019).
- Steixner, S. J. M., et al. Influence of nutrient media compared to human synovial fluid on the antibiotic susceptibility and biofilm gene expression of coagulase-negative Staphylococci In vitro. Antibiotics. 10 (7), 790 (2021).
- Sigma Aldrich. Mueller Hinton Broth (M-H Broth). Sigma Aldrich. , 70192 (2018).
- Thiriard, A., Raze, D., Locht, C. Development and standardization of a high-throughput Bordetella pertussis growth-inhibition assay. Frontiers in Microbiology. 11, 777 (2020).
- Clow, F., O'Hanlon, C. J., Christodoulides, M., Radcliff, F. J. Feasibility of using a luminescence-based method to determine serum bactericidal activity against Neisseria gonorrhoeae. Vaccines. 7 (4), 191 (2019).
- Kraus, V. B., Stabler, T. v., Kong, S. Y., Varju, G., McDaniel, G. Measurement of synovial fluid volume using urea. Osteoarthritis and Cartilage. 15 (10), 1217-1220 (2007).
- Gonçalves, F. D. A., de Carvalho, C. C. C. R. Phenotypic modifications in Staphylococcus aureus cells exposed to high concentrations of vancomycin and teicoplanin. Frontiers in Microbiology. 7, 13 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved