
Molecular Orbital (MO) Theory
Overview
Source: Tamara M. Powers, Department of Chemistry, Texas A&M University
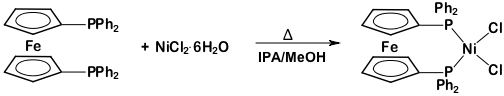
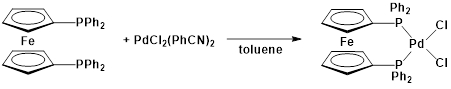
This protocol serves as a guide in the synthesis of two metal complexes featuring the ligand 1,1'-bis(diphenylphosphino)ferrocene (dppf): M(dppf)Cl2, where M = Ni or Pd. While both of these transition metal complexes are 4-coordinate, they exhibit different geometries at the metal center. Using molecular orbital (MO) theory in conjunction with 1H NMR and Evans method, we will determine the geometry of these two compounds.
Procedure
NOTE: For safety precautions, the Schlenk line safety should be reviewed prior to conducting the experiments. Glassware should be inspected for star cracks before using. Care should be taken to ensure that O2 is not condensed in the Schlenk line trap if using liquid N2. At liquid N2 temperature, O2 condenses and is explosive in the presence of organic solvents. If it is suspected that O2 has been condensed or a blue liquid is observed in the cold trap, leave the trap
Results
Pd(dppf)Cl2:
1H NMR (chloroform-d, 400 MHz, δ, ppm): 4.22 (alpha-H), 4.42 (beta-H), 7.89, 7.44, 7.54 (aromatic)3.
Ni(dppf)Cl2:
1H NMR (chloroform-d, 300 MHz, δ, ppm): 20.85, 10.04, 4.23, 3.98, 1.52, -3.31, -7.10.
Evans Method, looking at the 19F shift of trifluorotoluene:
Log in or to access full content. Learn more about your institution’s access to JoVE content here
Application and Summary
This video demonstrated how MO theory can be used as a model of bonding in transition metal complexes. We synthesized two complexes with the general formula M(dppf)Cl2. When M = Ni, the 4-coordinate complex exhibits a tetrahedral geometry. Replacing the Ni atom with a larger transition metal (Pd), the molecule takes on square planar geometry.
Previously, we learned about the important role ferrocene plays in the field of organometallic chemistry. Substituted ferrocenes, including dp
References
- Corain, B., Longato, B., Favero, G. Heteropolymetallic Complexes of 1,1’-Bis(diphenylphosphino)ferrocene (dppf). III*. Comparative Physicochemical Properties of (dppf)MCl2 (M = Co, Ni, Pd, Pt, Zn, Cd, Hg). Inorg Chim Acta. 157, 259-266 (1989).
- Cullen, W. R., Einstein, F. W. B., Jones, T., Kim, T.-J. Structures of three hydrogenation catalysts [(P-P)Rh(NBD)]ClO4 and some comparative rate studies where (P-P) = (η5-R1R2PC5H4)(η5-R3R4PC5H4)Fe (R1 = R2 = R3 = R4 = Ph, R1 = R2 = Ph, R3 = R4 = CMe3, R1 = R3 = Ph, R2 = R4 = CMe3). Organometallics. 4(2), 346-351 (1983).
- Colacot, T. J., C.-Olivares, R., H.-Ortega, S. Synthesis, X-ray, spectroscopic and a preliminary Suzuki coupling screening studies of a complete series of dppfMX2 (M = Pt, Pd, X = Cl, Br, I). J Organomet Chem. 637-639, 691-697 (2001).
- Rudie, A. W., Lichtenberg, D. W., Katcher, M. L., Davison, A. Comparative Study of 1,1’-bis(diphenylphosphino)cobaltocinium hexafluorophosphate and 1,1’-bis(dipenylphosphino)ferrocene as Bidentate Ligands. Inorg Chem. 17(10), 2859-2863, 1978.
Tags
Skip to...
Videos from this collection:

Now Playing
Molecular Orbital (MO) Theory
Inorganic Chemistry
35.3K Views

Synthesis Of A Ti(III) Metallocene Using Schlenk Line Technique
Inorganic Chemistry
31.6K Views

Glovebox and Impurity Sensors
Inorganic Chemistry
18.6K Views

Purification of Ferrocene by Sublimation
Inorganic Chemistry
54.6K Views

The Evans Method
Inorganic Chemistry
68.5K Views

Single Crystal and Powder X-ray Diffraction
Inorganic Chemistry
104.5K Views

Electron Paramagnetic Resonance (EPR) Spectroscopy
Inorganic Chemistry
25.4K Views

Mössbauer Spectroscopy
Inorganic Chemistry
22.0K Views

Lewis Acid-Base Interaction in Ph3P-BH3
Inorganic Chemistry
38.9K Views

Structure Of Ferrocene
Inorganic Chemistry
79.5K Views

Application of Group Theory to IR Spectroscopy
Inorganic Chemistry
45.2K Views

Quadruply Metal-Metal Bonded Paddlewheels
Inorganic Chemistry
15.3K Views

Dye-sensitized Solar Cells
Inorganic Chemistry
15.8K Views

Synthesis of an Oxygen-Carrying Cobalt(II) Complex
Inorganic Chemistry
51.7K Views

Photochemical Initiation Of Radical Polymerization Reactions
Inorganic Chemistry
16.7K Views
Copyright © 2025 MyJoVE Corporation. All rights reserved