
תורת מסלולית מולקולרית (MO)
Overview
מקור: תמרה מ. פאוורס, המחלקה לכימיה, אוניברסיטת טקסס A&M
פרוטוקול זה משמש כמדריך בסינתזה של שני מתחמי מתכת הכוללים את ליגנד 1,1'-bis (דיפנילפוספינו) פרוקן (dppf): M(dppf)Cl2, שבו M = Ni או Pd. בעוד ששני מתחמי מתכת המעבר האלה הם 4-קואורדינטות, הם מציגים גיאומטריות שונות במרכז המתכת. באמצעות תורת המסלול המולקולרי (MO) בשילוב עם שיטת 1H NMR ואוונס, אנו נקבע את הגיאומטריה של שתי תרכובות אלה.
Principles
ישנם מגוון מודלים שבהם משתמשים כימאים כדי לתאר מליטה במולקולות. חשוב לזכור כי מודלים הם ייצוגים של מערכות ולכן יש להם עוצמות אבל גם מגבלות חשובות. לדוגמה, מבני נקודות לואיס, השיטה הפשוטה ביותר לתיאור האופן שבו אטומים חולקים אלקטרונים, אינם לוקחים בחשבון את הגיאומטריה של האטומים במולקולה. תיאוריית הדחייה של זוג אלקטרונים של Valence Shell (VSEPR) אכן מתארת את הגיאומטריה של האטומים, אך היא אינה מספקת הסבר לתצפית כי מינים איזואלקטרוניים עם אותו מספר אלקטרונים ערכיים יכולים להציג גיאומטריות שונות. במיוחד עבור מתחמי מתכת מעבר, שני דגמים אלה נופלים בתיאור מליטה של מתכות. תורת שדה הגביש היא מודל מליטה ספציפי למעבר מתחמי מתכת. מודל זה בוחן את ההשפעות של שדה חשמלי של ליגנד על המסלולים האטומיים d או f של מרכז מתכת. האינטראקציה גורמת לניתוב של מסלולי המסלול האטומיים האלה.
בסרטון זה נתמקד בתיאוריית MO, שהיא מודל רב עוצמה שניתן להשתמש בו כדי לתאר מליטה לא רק במולקולות קבוצתיות עיקריות, אלא גם מתאימה למידול הקשר במתחמי מתכת מעבר. כאן, אנו להדגים כיצד ליצור דיאגרמת MO של תרכובות המכילות מתכת.
תורת MO:
תיאוריית MO מתארת מליטה כימית כשילוב ליניארי של מסלולי המסלול האטומיים (LCAO) של כל אטום בתרכובת נתונה. ה- MOs הנובעים מ- LCAOs מתארים הן את הגיאומטריה והן את האנרגיה של האלקטרונים המשותפים למספר אטומים במולקולה (כלומר,את הכיוון והעוצמה של הקשרים שנוצרו על ידי אטומים נתון).
כדי לסקור את היסודות של תיאוריית MO, שקולתחילה את המולקולה הדיאטומית F 2 (דיאגרמת MO מלאה באיור 1). אטום פלואור יש 4 מסלוליים אטומיים valence: 2s, 2px, 2py, ו 2pz. המסלול של 2 s נמוך יותר באנרגיה מאשר מסלולי האטום2 p, שלכולם יש את אותה אנרגיה. שילוב ליניארי של מסלוליים אטומיים יתרחש בין מסלוליים אטומיים של אנרגיה דומה לבין סימטריה תואמת. במקרה זה, מסלול 2s על אטום F אחד יקיים אינטראקציה עם 2s מסלולית על אטום F האחר. התוספת של שני מסלוליות אלה גורמת להיווצרות של Σ מליטה MO (איור 1). מליטה היא אינטראקציה מייצבת, ולכן, σ MO וכתוצאה מכך הוא נמוך יותר באנרגיה ביחס לאנרגיה של2 מסלולי האטום. הפחתה של מסלולי 2s גורמת לאינטראקציה אנטי-מליטה (מערערת), המוגדרת כ-σ*, שהיא גבוהה יותר באנרגיה ביחס למסלולים האטומייםשל 2 s (איור 1).

איור 1. דיאגרמת MO של F2.
כמו כן, מסלולי האטום 2p ישתלבו כדי ליצור אינטראקציות מליטה ואנטי מליטה. בדומה למסלולים האטומיים של 2 pz, המסלולים האטומייםשל 2pz (השוכבים לאורך קשר ה-F-F) יוצרים אינטראקציות σ ו-σ** . אם ניקח בחשבון את מסלולי המסלול האטומיים 2px ו- 2py, אנו רואים שהם יוצרים סוגים שונים של אינטראקציות מליטה ואנטי מליטה, הנקראות π ו-π*, בהתאמה ( איור1). קל להבחין בין קשרים σ π מכיוון σ מסלולי מליטה הם סימטריים באופן גלילי לגבי הציר הפנימי, בעוד π למסלולים יש מישור נדאל לאורך הציר הפנימי. החפיפה המרחבית בין מסלוליים אטומיים היוצרים קשרים σ גדולה מההי חפיפה המרחבית בין מסלוליים אטומיים היוצרים קשרים π. לכן, π ו-π* MOs המתקבלים הם פחות מיוצבים ומערערים, בהתאמה, בהשוואה ל- MOs σ ו- σ * שנוצרו על ידי מסלולי האטום 2pz. לאחר מכן נוכל למלא את MOs באלקטרונים הערכיים של שני אטומי ה-F.
כעת שקול מולקולה מורכבת יותר כגון [Co(NH3)6]Cl3 (איור 2). אם נשתמש באותו תהליך כמו לעיל (בהתחשב בחפיפה האטומית בין 2 אטומים בכל פעם), יצירת דיאגרמת MO של מולקולה זו תהיה מאתגרת ביותר. במקום זאת, אנו יכולים להשתמש בתורת הקבוצות כדי ליצור תחילה שילוב ליניארי מותאם סימטריה (SALC) של הליבנדים. לאחר מכן אנו יכולים להשתמש בסימטריה כדי לקבוע את אינטראקציות מליטה / אנטי מליטה שנוצרות בין מסלולי האטום על המתכת לבין SALCs וכתוצאה מכך.

| Oh | E | 8C3 | 6C2 | 6C4 | 3C2' | אני | 6S4 | 8S6 | 3σh | 3σd | ||
| גרם אחד | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | x2+y2+z2 | |
| 2 גרם | 1 | 1 | -1 | -1 | 1 | 1 | -1 | 1 | 1 | -1 | ||
| Eg | 2 | -1 | 0 | 0 | 2 | 2 | 0 | -1 | 2 | 0 | (2z2-x2-y2, x2-y2) | |
| T1g | 3 | 0 | -1 | 1 | -1 | 3 | 1 | 0 | -1 | -1 | (Rx, Ry, Rz) | |
| T2g | 3 | 0 | 1 | -1 | -1 | 3 | -1 | 0 | -1 | 1 | (xz, yz, xy) | |
| A 1u | 1 | 1 | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 | ||
| 2u | 1 | 1 | -1 | -1 | 1 | -1 | 1 | -1 | -1 | 1 | ||
| Eu | 2 | -1 | 0 | 0 | 2 | -2 | 0 | 1 | -2 | 0 | ||
| T1u | 3 | 0 | -1 | 1 | -1 | -3 | -1 | 0 | 1 | 1 | (x, Y, z) | |
| T2u | 3 | 0 | 1 | -1 | -1 | -3 | 1 | 0 | 1 | -1 | ||
| אדום Γ | 6 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 4 | 4 |
Γאדום =A1g + Eg + T1u
איור 2. שילוב ליניארי של מסלולי ליגנד אטומיים של [Co(NH3)6]Cl3.
כדי ליצור את SALCs עבור [Co(NH3)6]3+, אנו עוקבים אחר הליך דומה המתואר בסרטון "תורת הקבוצות" בסדרת הכימיה הלא אורגנית:
1. לקבוע את קבוצת הנקודות של המולקולה.
2. ליצור ייצוג מופחת של מסלולי האטום של ליגנד.
3. לצמצם את הייצוג המצומצם לייצוגים בלתי ניתנים לערעור.
[Co(NH3)6] 3+ נמצא בקבוצת הנקודות Oh. מכיוון שאנו מודאגים רק מההיקשרות במרכז המתכת, אנו יכולים פשוט לשקול את מסלולי המסלול האטומייםשל 2 על כל ליגנד NH3. אם נבצע את שלבים 1-3 עבור N 2s,מסלולית אנו מוצאים כי הייצוג מופחת הואΓ אדום =A 1g + Eg + T1u (איור 2). בעוד שסט A1g מייצג 1 SALC, ערכות Eg ו- T1u מייצגות למעשה 2 ו- 3 SALCs, בהתאמה, המעניקות סך של 6 SALCs (אותו מספר של ליגנדים בקטינה [Co(NH3)6]3+). 2 SALCs בסט Eg יש את אותה סימטריה וכתוצאה מכך MOs מנוון כאשר הם אינטראקציה עם המסלולים האטומיים של Co (אותו הדבר ניתן לומר על 3 SALCs בסט T1u). באמצעות טבלת התווים באיור 2, אנו יכולים לקבוע כיצד המסלולים האטומיים של Co משתנים בקבוצת Oh point. לדוגמה, מסלולי dz2 ו- dx2–y2 יוצרים ערכת Eg. מכיוון שיש לנו 2 סלקים ליגנד עם סימטריהE g, SALCs אלה ייצרו אינטראקציות מליטה / אנטי מליטה עם dz2 ו- dx2 –y2 Co מסלוליים אטומיים. בהמשך באותה צורה עבור כל מסלולי האטום הערכיות של Co, אנו יוצרים תרשים MO עבור קומפלקס מתכת המעבר, המוצג באיור 3. שים לב כי d-orbitals הנותרים (dxz, dyzו- dxy) לשנות כסט (T2g) אבל אין סימטריה מתאימה תואם SALC. מסלולים אטומיים אלה הופכים אפוא ל- MOs "לא מליטה". במילים אחרות, הם אינם משתתפים בקשר עם הליגנדים במתחם מתכת המעבר הזה.

איור 3. דיאגרמת MO עבור [Co(NH3)6]Cl3.
באיור 3 מודגשים מסלולי d-orbitals שאינם מליטה ואת מסלולי σ* עם תו d-orbital. כאשר קבוצה זו של MOs נחשבת בנפרד מדיאגרמת MO כולה היא נקראת דיאגרמת הפיצול d-orbital של קומפלקס מתכת מעבר. מכיוון שדיאגרמת הפיצול d-orbital מכילה את HOMO ואת LUMO, שהם בדרך כלל המסלולים החשובים ביותר להבנת הכימיה והספקטרוסקופיה של מתחמי התיאום, כימאים יתייחסו לעתים קרובות לדיאגרמת הפיצול d-orbitalבמקום לדיאגרמת MO כולה. בנוחות, ניתן למלא את דיאגרמת הפיצול d-orbitalבמספר de- במרכז המתכת, מכיוון שהאלקטרונים מבוססי הליגנד תמיד ממלאים את MOs מבוססי σ בדיאגרמת MO.
בהתחשב בדיאגרמות הפיצול d-orbital עבור M(dppf)Cl2:
שקול קומפלקס מתכת פשוט 4-קואורדינטות MX4. MX4 יכול להתקיים בשתי גיאומטריות: טטרהדרל או מתכנן מרובע. דיאגרמות הפיצול של d-orbitalעבור קבוצות הנקודות Td (tetrahedral) ו- D4h (מטען מרובע) מוצגות באיור 4. בעוד מתחמי המתכת הכלליים M(dppf)Cl 2 אין 4 ליגנדים מקבילים, ולכן אינם בקבוצות הנקודה Td או D4h, אנו עדיין יכולים להשתמש אלה d-מסלולי פיצולדיאגרמות כמודל לתאר את d-מסלולית MOs עבור שתי גיאומטריות אפשריות.
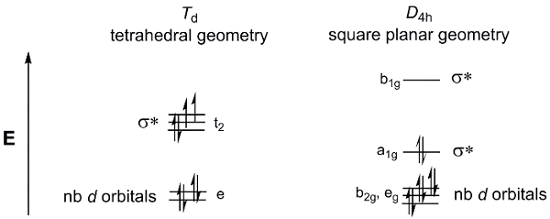
איור 4. דיאגרמות הפיצול של d-orbitalעבור קבוצות הנקודות Td (tetrahedral) ו- D4h (מתכנן מרובע).
עכשיו, שקול את ספירתהאלקטרונים d עבור M(dppf)Cl2. גם ני וגם פ.ד. נמצאים בקבוצה 10 של הטבלה המחזורית. לכן, לשניהם יהיה את אותו מצב חמצון (2+) וספירת אלקטרונים d(d8). אם נמלא את שתי דיאגרמות הפיצול של d-orbital למעלה ב-8 אלקטרונים, נראה כי הגיאומטריה המכוונת הריבועית גורמת לתסביך דימגנטי, בעוד שדיאגרמת שיטת הפעולה הטטרהדרלית עולה בקנה אחד עם מין פרמגנטי. ישנם מספר גורמים אשר נכנסים לקבוע איזו גיאומטריה מועדפת אנרגטית. בגיאומטריה המכוונת הריבועית, יש פחות אלקטרונים במסלולים אנטי מליטה, אשר יצביעו על כך הגיאומטריה המכוונת מרובעת היא יותר חיובית אלקטרונית. עם זאת, עלינו גם לשקול את האנרגיה הנדרשת לצמד אלקטרונים. אנרגיית זיווג האלקטרונים במולקולות מנסרות מרובעות גבוהה מזו שבמולקולות טטרהדרליות, שיש להן פחות מסלוליות מלאות לחלוטין. לבסוף, עלינו לשקול את כמות σ* d-orbitals הם מערערים. אטומי מתכת גדולים יותר יש חפיפה מרחבית גדולה יותר עם ליגנדים, וכתוצאה מכך אנרגיה גבוהה יותר σ * d-orbitals.
לבסוף, אנחנו גם צריכים לשקול את תרומת האנרגיה של דחיות steric. גיאומטריה Tetrahedral הוא מועדף יותר באופן סטרי (עם זוויות של 109.5 °) לעומת גיאומטריה מנומר מרובעת (90 °). לכן, ישנם מספר גורמים מנוגדים המשפיעים על איזו גיאומטריה מועדפת יותר, בהתחשב בזהותו של M ב- M(dppf)Cl2.
נוכל להבחין בין שתי הגיאומטריות האלה באמצעות NMR. אם המולקולה היא מנומר מרובע, נצפה באבחון 1H NMR של מין דימגנטי. אם המולקולה היא טטרהדרל, נצפה באותות פרמגנטיים ב-1H NMR. לבסוף, נשתמש בשיטת אוונס (ראה הסרטון "שיטת אוונס" לקבלת פרטים נוספים) כדי לקבוע את הפתרון של הרגע המגנטי של המינים הפרמגנטיים.
Procedure
הערה: כאמצעי זהירות, יש לבחון את בטיחות קו שלנק לפני ביצוע הניסויים. כלי זכוכית יש לבדוק עבור סדקים כוכב לפני השימוש. יש להקפיד על כך ש- O2 אינו מרוכז במלכודת הקו שלנק אם משתמשים בנוזל N2. בטמפרטורת N2 נוזלית, O2 מעבים והוא נפץ בנוכחות ממיסים אורגניים. אם יש חשד כי O2 כבר מרוכז או נוזל כחול נצפה במלכודת הקרה, להשאיר את המלכודת קרה תחת ואקום דינמי. אין להסיר את מלכודת N2 הנוזלית או לכבות את משאבת הוואקום. עם הזמן הנוזל O2 יתאדה לתוך המשאבה; זה רק בטוח להסיר את מלכודת N2 נוזלי פעם אחת כל O2 התאדה. לקבלת מידע נוסף, עיין בסרטון "סינתזה של מטלוקן טי(III) באמצעות טכניקת קו שלנק". 1
1. הגדרת קו שלנק לסינתזה של Ni(dppf)Cl2 ו- Pd(dppf)Cl2
הערה: לקבלת הליך מפורט יותר, אנא עיין בסרטון "העברת קווי שלנק של ממס" בסדרת יסודות הכימיה האורגנית).
- סגור את שסתום שחרור הלחץ.
- הפעל את גז N2 ואת משאבת ואקום.
- כאשר הוואקום של קו Schlenk מגיע ללחץ המינימלי שלו, להכין את המלכודת הקרה עם נוזל N2 או קרח / אצטון יבש.
- להרכיב את המלכודת הקרה.
2. סינתזה של Ni(dppf)Cl2 (איור 5) בתנאים אנאירוביים/אינרטיים
הערה בעוד הסינתזה של Ni(dppf)Cl2 יכול להתבצע בתנאים אירוביים, תשואות גבוהות יותר מתקבלות כאשר מתנהל בתנאים אנאירוביים.
- יש להוסיף 550 מ"ג dppf (1 mmol) ו-40 מ"ל של איזופרופנול לבקבוקון בעל שלושה צווארים.
הערה dppf ניתן לרכוש סיגמא אולדריץ 'או מסונתז באמצעות שיטות שנמצאו בספרות. 2 - התאימו את הצוואר המרכזי של הבקבוקון בעל שלושת הצווארים עם מחזק ומתאם ואקום. להתאים את שני הצווארים הנותרים עם פקק זכוכית אחד מחיצת גומי אחד.
- Degas הפתרון על ידי מבעבע גז N2 דרך הממס במשך 15 דקות. השתמש במתאם הוואקום בחלק העליון של המחזק כ"פתח אוורור ".
- חבר את מתאם הוואקום בחלק העליון של המחזק ל- N2 באמצעות קו Schlenk.
- התחל לחמם את הבקבוקון בעל שלושת הצווארים באמבט מים להגדיר 90 °C (50 °F).
- בבקבוקון התחתון העגול עם צוואר יחיד, יש להוסיף 237 מ"ג NiCl2·6H2O (1 mmol) ל-4 מ"ל של תערובת של איזופרופנול (כיתה ריאגנט) ומתנול (כיתה ריאגנט). Sonicate התערובת המתקבלת עד שכל מלח Ni נמס (כ 1 דקות).
הערה: אם Sonicator אינו זמין, לחמם בעדינות את התערובת באמבט מים. - Degas את תמיסת Ni על ידי מבעבע גז N2 דרך התערובת במשך 5 דקות.
- הוסיפו את תמיסה NiCl2·6H 2O לבקבוק התחתון העגול בעל שלושת הצווארים באמצעות העברת צינורית.
- אפשר את התגובה reflux עבור 2 שעות ב 90 °C (50 °F).
- אפשר לתגובה להתקרר באמבט קרח. לבודד את המשקעים הירוקים וכתוצאה מכך על ידי סינון ואקום דרך משפך fritted.
- לשטוף את המוצר עם 10 מ"ל של איזופרופנול קר, ואחריו 10 מ"ל של hexanes.
- אפשרו למוצר להתייבש באוויר לפני הכנת דגימת ה-NMR.
- קח 1H NMR של המוצר בכלורופורם-d.
- אם 1H NMR מעיד על מין פרמגנטי, להכין NMR עבור שיטת אוונס, בעקבות ההוראות בשלב 4.
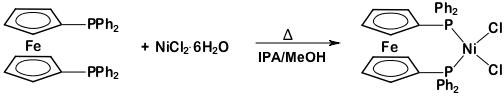
איור 5. סינתזה של Ni(dppf)Cl2.
3. סינתזה של Pd(dppf)Cl2 (איור 6)1
הערה: השתמש בטכניקות קו שלנק סטנדרטיות לסינתזה של Pd(dppf)Cl 2 (ראה את"סינתזה של Ti(III) Metallocene באמצעות טכניקת קו Schlenk" וידאו).
הערה בעוד הסינתזה של Pd(dppf)Cl2 יכול להתבצע בתנאים אירוביים, תשואות גבוהות יותר מתקבלות כאשר מתנהל בתנאים אנאירוביים.
- יש להוסיף 550 מ"ג (1 מ"מ) dppf ו-383 מ"ג (1 mmol) ביס (בנזונטריל)פלדיום(II) לכלוריד לבקבוק שלנק ולהכין את אגף שלנק להעברת הממס.
- הוסף 20 מ"ל של טולואן מנוטרל לבקבוק שלנק באמצעות העברת קנולה.
- אפשר לתגובה לערבב לפחות 12 שעות בטמפרטורת החדר.
- לבודד את המשקעים הכתומים וכתוצאה מכך על ידי סינון ואקום דרך משפך fritted.
- לשטוף את המוצר עם טולואן (10 מ"ל), ואחריו hexanes (10 מ"ל).
- אפשרו למוצר להתייבש באוויר לפני הכנת דגימת ה-NMR.
- קח 1H NMR של המוצר בכלורופורם-d.
- אם 1H NMR מעיד על מין פרמגנטי, להכין NMR עבור שיטת אוונס בעקבות ההוראות המתוארות בשלב 4.
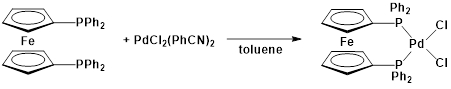
איור 6. סינתזה של Pd(dppf)Cl2.
4. הכנת מדגם שיטת אוונס
הערה: לקבלת הליך מפורט יותר, עיין בסרטון "שיטת אוונס".
- בלווייתן נוצץ, להכין פתרון 50:1 (נפח:נפח) של כלורופורם- d:trifluorotoluene. Pipette 2 מ"ל של ממס deuterated, וזה להוסיף 40 μL של trifluorotoluene. תכובע את ההון.
הערה: בדוגמה זו, אנו נשתמש 19F NMR כדי לצפות את השינוי של אות F ב trifluorotoluene בנוכחות המינים paramagnetic. - עם פתרון זה, להכין את ההוספה נימי.
- שוקלים 10-15 מ"ג של המדגם הפרמגנטי לתוך ממתקנה נוצצת חדשה ושימו לב למסה.
- Pipette ~ 600 μL של תערובת ממס מוכן לתוך הוויאל המכיל את המינים paramagnetic. שים לב למסה. ודא כי מוצק מתמוסס לחלוטין.
- בצינור NMR סטנדרטי, בזהירות טיפה את ההוספה נימי בזווית, כדי להבטיח שזה לא לשבור.
- פיפטה הפתרון המכיל את המינים העל-מגנטיים לתוך צינור NMR.
- לרכוש ולשמור ספקטרום NMR סטנדרטי 19F.
- שימו לב לטמפרטורת הגשוש.
- שים לב לתדר הרדיו.
Results
Pd(dppf)Cl2:
1 H NMR (כלורופורם-d,400 מגה-הרץ, δ, ppm): 4.22 (אלפא-H), 4.42 (בטא-H), 7.89, 7.44, 7.54 (ארומטי)3.
Ni(dppf)Cl2:
1 H NMR (כלורופורם-d,300 מגה-הרץ, δ, ppm): 20.85, 10.04, 4.23, 3.98, 1.52, -3.31, -7.10.
שיטת אוונס, מסתכל על משמרת 19F של trifluoroluene:
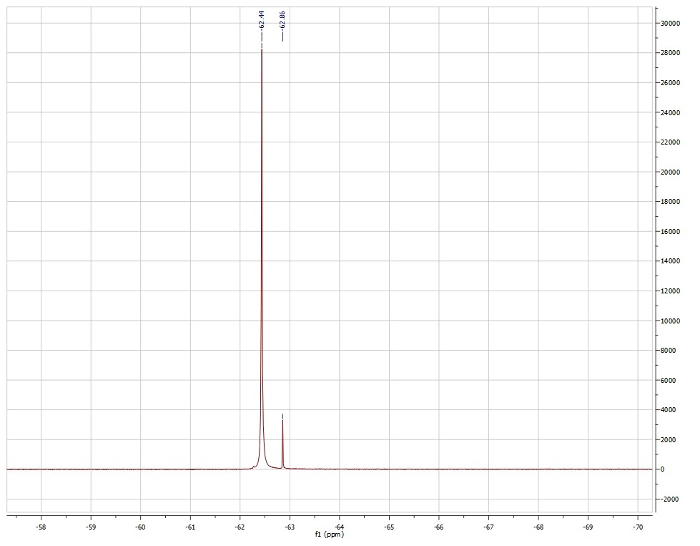
נצפתה μeff = 3.15 μb
מסה של המדגם: 9.5 מ"ג
מסת הפתרון (כלורופורם-d + טריפלואורוטולואן): 0.8365 גרם
טמפרטורת הגשוש: 296.3 K
שדה NMR (MHz): 470.06
דיווח μeff = 3.39 μb. 4
עבור S = 1 (חזוי המבוסס על גיאומטריה tetrahedral, איור 4), תיאורטי μeff = 2.83 μb.
עבור S = 3/2, μ תיאורטיeff = 3.46 μb.
בהתבסס על נתוני 1H NMR, אנו רואים כי Pd(dppf)Cl2 הוא דימגנטי ולכן מציג גיאומטריה מכוונת מרובעת. 1H NMR של Ni(dppf)Cl2 הוא paramagnetic ולכן הוא tetrahedral במרכז Ni. השיטה של אוון מאשרת כי Ni(dppf)Cl2 הוא paramagnetic, המציג רגע מגנטי פתרון של 3.15 μb, אשר קרוב לערך הספרות שדווח עבור תרכובת זו. מאז Ni הוא קטן, sterics עולה על כל ייצוב אלקטרוני הקשורים גיאומטריה מישור מרובע, מה שהופך Ni(dppf)Cl2 tetrahedral. מצד שני, PD הוא גדול, ולכן, יש אנרגיה גבוהה יותר σ * d-orbitals. במקרה זה, הייצוב האלקטרוני עולה בהרבה על הדחייה הסיסטרית, וכתוצאה מכך גיאומטריה מישורית מרובעת ב- PD(dppf)Cl2.
Application and Summary
וידאו זה הדגים כיצד תיאוריית MO יכולה לשמש כמודל של מליטה במתחמי מתכת מעבר. סינתזנו שני מתחמים עם הנוסחה הכללית M(dppf)Cl2. כאשר M = Ni, קומפלקס 4 הקואורדינטות מציג גיאומטריה טטרהדרלית. החלפת האטום Ni עם מתכת מעבר גדולה יותר (Pd), המולקולה לוקחת על גיאומטריה מנדרית מרובעת.
בעבר למדנו על התפקיד החשוב שפרוקן ממלא בתחום הכימיה האורגנומטלית. פרוקניות מוחלפות, כולל dppf, משמשות כלאטלינג ליגנדים עבור1 st,2 nd, ומתכות מעבר שורה3. המתחמים המתקבלים משמשים קטליזה הומוגנית(כלומר,[1,1'-bis (דיפנילפוספינו)פרוקן]פלדיום (II) דיכלוריד, Pd(dppf)Cl 2 , הואזרזלתגובות יוצרות קשר C-C ו- C-הטרואטום).
הבנת הקשר במתחמי מתכת המעבר חשובה להסבר המבנה והתגובה שלהם. אחת החוזקות של תיאוריית MO היא שהיא מספקת מודל טוב שניתן להשתמש בו כדי להסביר את התגובה של מתחמי מתכת מעבר. במקרים רבים, מרכז המתכת הוא המיקום של כל תגובתיות המוצגת על ידי המולקולה. לכן, חשוב שתהיה תמונה של צפיפות האלקטרונים במרכז המתכת, המסוכמת בתרשים הפיצול המקיף d-orbitalהנגזר מתורת MO (איור 3). שים לב כי לא רק MOs בדיאגרמת פיצול d-מסלולית התערוכה בעיקר d- אות מסלולית (מסלולי σ * הם הקרובים ביותר באנרגיה ל- d- מסלוליים אטומיים של המתכת ולכן רוב צפיפות האלקטרונים ב- MOs אלה מתמקדת באטום המתכת), אלא גם תרשים הפיצול מכיל את HOMO ו- LUMO של המולקולה. לכן, כל כימיה המתרחשת תשפיע ישירות על דיאגרמת הפיצול שלהמולקולה.
References
- Corain, B., Longato, B., Favero, G. Heteropolymetallic Complexes of 1,1’-Bis(diphenylphosphino)ferrocene (dppf). III*. Comparative Physicochemical Properties of (dppf)MCl2 (M = Co, Ni, Pd, Pt, Zn, Cd, Hg). Inorg Chim Acta. 157, 259-266 (1989).
- Cullen, W. R., Einstein, F. W. B., Jones, T., Kim, T.-J. Structures of three hydrogenation catalysts [(P-P)Rh(NBD)]ClO4 and some comparative rate studies where (P-P) = (η5-R1R2PC5H4)(η5-R3R4PC5H4)Fe (R1 = R2 = R3 = R4 = Ph, R1 = R2 = Ph, R3 = R4 = CMe3, R1 = R3 = Ph, R2 = R4 = CMe3). Organometallics. 4(2), 346-351 (1983).
- Colacot, T. J., C.-Olivares, R., H.-Ortega, S. Synthesis, X-ray, spectroscopic and a preliminary Suzuki coupling screening studies of a complete series of dppfMX2 (M = Pt, Pd, X = Cl, Br, I). J Organomet Chem. 637-639, 691-697 (2001).
- Rudie, A. W., Lichtenberg, D. W., Katcher, M. L., Davison, A. Comparative Study of 1,1’-bis(diphenylphosphino)cobaltocinium hexafluorophosphate and 1,1’-bis(dipenylphosphino)ferrocene as Bidentate Ligands. Inorg Chem. 17(10), 2859-2863, 1978.
Tags
Skip to...
Videos from this collection:

Now Playing
תורת מסלולית מולקולרית (MO)
Inorganic Chemistry
35.5K Views

סינתזה של מטלוקן Ti(III) בטכניקת קו שלנק
Inorganic Chemistry
31.6K Views

חיישני תא כפפות וטילמאה
Inorganic Chemistry
18.6K Views

טיהור פרוקן על ידי תת-הכרתיות
Inorganic Chemistry
54.7K Views

שיטת אוונס
Inorganic Chemistry
68.7K Views

עקיפה של קריסטל ואבקה
Inorganic Chemistry
105.1K Views

ספקטרוסקופיית תהודה פרמגנטית אלקטרונית (EPR)
Inorganic Chemistry
25.6K Views

Mössbauer Spectroscopy
Inorganic Chemistry
22.0K Views

אינטראקציה בסיס חומצה לואיס ב Ph3P-BH3
Inorganic Chemistry
39.0K Views

מבנה פרוקן
Inorganic Chemistry
79.7K Views

יישום תורת הקבוצות לספקטרוסקופיית IR
Inorganic Chemistry
45.8K Views

גלגלי משוטים מרופדים ממתכת מתכתית
Inorganic Chemistry
15.3K Views

תאים סולאריים רגישים לצבע
Inorganic Chemistry
16.0K Views

סינתזה של קומפלקס קובלט נושא חמצן(II)
Inorganic Chemistry
51.7K Views

ייזום פוטוכימי של תגובות פילמור רדיקליות
Inorganic Chemistry
17.1K Views
Copyright © 2025 MyJoVE Corporation. All rights reserved