A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
خلية مفردة قابلة لإعادة الاستخدام للتحليلات الجينومية التكرارية
In This Article
Summary
يصف هذا البروتوكول طريقة الخلية الواحدة للتحليلات فوق الجينية التكرارية باستخدام خلية مفردة قابلة لإعادة الاستخدام. تسمح الخلية المفردة القابلة لإعادة الاستخدام بتحليل العلامات اللاجينية المتعددة في نفس الخلية المفردة والتحقق الإحصائي من النتائج.
Abstract
تم تصميم تحليلات epigenome أحادية الخلية الحالية للاستخدام مرة واحدة. يتم التخلص من الخلية بعد استخدام واحد ، مما يمنع تحليل العلامات اللاجينية المتعددة في خلية واحدة ويتطلب بيانات من خلايا أخرى لتمييز الإشارة عن ضوضاء الخلفية التجريبية في خلية واحدة. تصف هذه الورقة طريقة لإعادة استخدام نفس الخلية المفردة للتحليلات فوق الجينية التكرارية.
في هذه الطريقة التجريبية ، يتم تثبيت البروتينات الخلوية أولا على بوليمر بولي أكريلاميد بدلا من ربطها بالبروتين والحمض النووي ، مما يخفف من التحيز الهيكلي. تسمح هذه الخطوة الحاسمة بإجراء تجارب متكررة مع نفس الخلية المفردة. بعد ذلك ، يتم تلدين التمهيدي العشوائي مع تسلسل سقالة لربط القرب إلى الحمض النووي الجينومي ، ويتم إضافة التسلسل الجينومي إلى التمهيدي بالامتداد باستخدام بوليميراز الحمض النووي. بعد ذلك ، يرتبط الجسم المضاد ضد علامة جينية وتحكم IgG ، كل منها يحمل تحقيقات مختلفة للحمض النووي ، بالأهداف المعنية في نفس الخلية الواحدة.
يتم تحفيز ربط القرب بين التمهيدي العشوائي والجسم المضاد عن طريق إضافة الحمض النووي الموصل مع تسلسلات تكميلية إلى تسلسل سقالة التمهيدي العشوائي ومسبار الحمض النووي للجسم المضاد. يدمج هذا النهج معلومات الأجسام المضادة وتسلسل الجينوم القريب في منتج DNA واحد لربط القرب. من خلال تمكين التجارب المتكررة مع نفس الخلية المفردة ، تسمح هذه الطريقة بزيادة كثافة البيانات من خلية نادرة والتحليل الإحصائي باستخدام بيانات IgG والأجسام المضادة فقط من نفس الخلية. يمكن تخزين الخلايا المفردة القابلة لإعادة الاستخدام المحضرة بهذه الطريقة لبضعة أشهر على الأقل وإعادة استخدامها لاحقا لتوسيع التوصيف اللاجيني وزيادة كثافة البيانات. توفر هذه الطريقة المرونة للباحثين ومشاريعهم.
Introduction
تدخل تقنية الخلية الواحدة عصر تعدد الخلايا المفردة ، والذي يدمج تقنيات omics الفردية أحادية الخلية1. في الآونة الأخيرة ، تم دمج النسخ أحادي الخلية مع طرق للكشف عن إمكانية الوصول إلى الكروماتين (scNMT-seq2 و SHARE-seq3) أو تعديلات هيستون (Paired-seq4 و Paired-Tag5). في الآونة الأخيرة ، تم دمج النسخ أحادي الخلية والبروتينات مع إمكانية الوصول إلى الكروماتين (DOGMA-seq6). تستخدم هذه الطرق العلامات المستندة إلى transposase للكشف عن إمكانية الوصول إلى الكروماتين أو تعديلات الهستون.
تشق الأساليب القائمة على Transposase الحمض النووي الجينومي وتضيف رمزا شريطيا للحمض النووي في نهاية جزء الحمض النووي الجينومي. يمكن لكل جزء جينومي مشقوق قبول ما يصل إلى رمزين شريطيين للحمض النووي (= علامة جينية واحدة لكل موقع انقسام) ، ويتم فقد الحمض النووي الجينومي في موقع الانقسام. لذلك ، فإن النهج القائمة على الانقسام لها مفاضلة بين عدد العلامات اللاجينية التي تم اختبارها وكثافة الإشارة. هذا يعيق تحليل العلامات اللاجينية المتعددة في نفس الخلية الواحدة. تم تطوير طريقة فوق جينومية أحادية الخلية لا تشق الحمض النووي الجينومي للتغلب على هذه المشكلة 7,8.
بالإضافة إلى القضية المشتقة من الانقسام المذكورة أعلاه ، فإن النهج القائمة على الترانسبوزاز لها قيود أخرى. في تحليل epigenome أحادي الخلية ، من الأهمية بمكان معرفة موقع الهستونات والبروتينات المرتبطة بالحمض النووي على الجينوم. في الأساليب الحالية ، يتم تحقيق ذلك باستخدام خلايا مفردة غير ثابتة والاحتفاظ بتفاعلات البروتين والحمض النووي والبروتين والبروتين. ومع ذلك ، فإن هذا يولد تحيزا قويا لمناطق الكروماتين التي يمكن الوصول إليها ، حتى في تحليل تعديلات الهستون 9. يمكن الحفاظ على موقع الهستونات والبروتينات المرتبطة بالجينوم على الجينوم دون ربط البروتين - الحمض النووي والبروتين - البروتين ، باستخدام سقالة بولي أكريلاميد 7,8. يقلل هذا النهج من التحيز الهيكلي الملاحظ في الأساليب الحالية التي تعتمد على تفاعلات البروتين والحمض النووي والبروتين والبروتين.
يمكن للنهج القائمة على Transposase الحصول على إشارات مرة واحدة فقط من خلية واحدة. لذلك ، من الصعب تحديد epigenome الكامل لخلية واحدة بسبب انخفاض الإشارات. تم تطوير خلايا مفردة قابلة لإعادة الاستخدام للتغلب على القيود الحالية من خلال السماح بالتحليل الجيني التكراري في نفس الخلية المفردة.
Protocol
ملاحظة: يظهر تمثيل تخطيطي للطريقة في الشكل 1.

الشكل 1: تمثيل تخطيطي لسير عمل البروتوكول. يتم شرح الخطوات 7.2-13 من خلال التمثيلات التخطيطية. يشير كل صف إلى خطوة في البروتوكول. البروتين الخلوي الملون باللون الأخضر هو نيوكليوسوم بشري يتم إنشاؤه بناء على بنية بلورية (PDB: 6M4G). يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
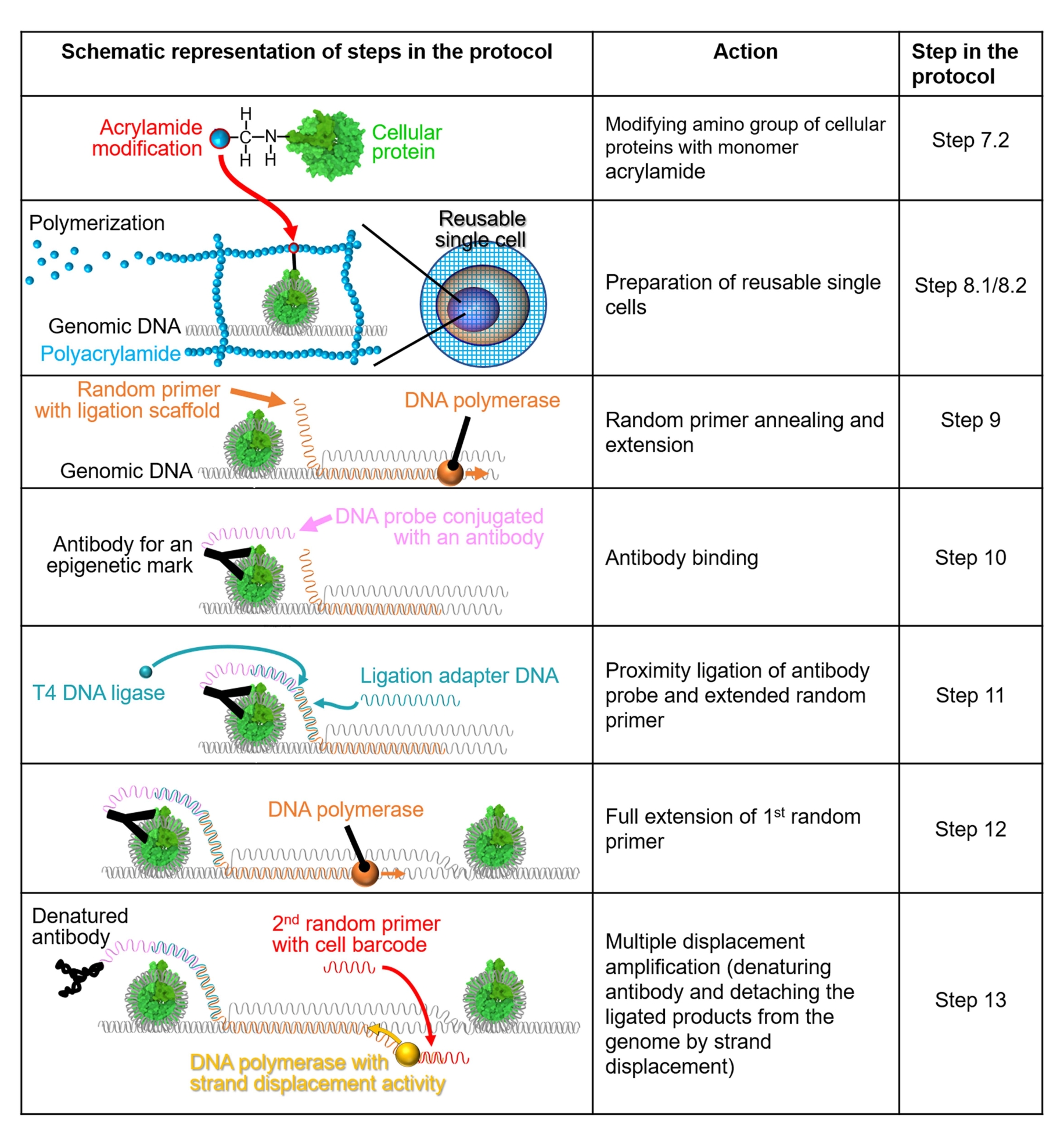{kind=link}
1. توازن الأعمدة المحلاة
ملاحظة: تتم موازنة أعمدة الدوران المحلاة كما هو موضح في الخطوات التالية. تستخدم أعمدة التحلية المتوازنة في الخطوات 2.1 و3.4 و4.6.
- قم بإزالة الإغلاق السفلي لعمود تحلية (قطع 7 كيلو دالتون ، حجم حبة راتنج 0.5 مل ، انظر جدول المواد) وقم بفك الغطاء الموجود أعلى عمود التحلية.
- ضع العمود في أنبوب منخفض الارتباط بالبروتين سعة 1.5 مل (أنبوب تجميع ، انظر جدول المواد) وأجهزة طرد مركزي عند 1500 × جم لمدة دقيقة واحدة في درجة حرارة الغرفة لإزالة محلول التخزين في العمود.
ملاحظة: استخدم جهاز طرد مركزي دوار متأرجح لتسطيح الجزء العلوي من سرير الخرز. - قم بإزالة التدفق في أنبوب 1.5 مل وأضف 300 ميكرولتر من 150 mM NaCl / 100 mM Phosphate Buffer ، الرقم الهيدروجيني 8.0 (انظر الجدول 1) ، أعلى طبقة الراتنج.
- أجهزة الطرد المركزي عند 1500 × جم لمدة 1 دقيقة في درجة حرارة الغرفة وإزالة التدفق في أنبوب التجميع.
- كرر الخطوات من 1.3 إلى 1.4 ثلاث مرات إضافية.
- تخلص من المخزن المؤقت من أنبوب التجميع وضع العمود في أنبوب تجميع جديد.
- استخدم عمود التحلية المتوازن في الخطوات 2.1 و3.4 و4.4.
2. التبادل العازلة للأجسام المضادة
ملاحظة: قم بإزالة الجلسرين والأرجينين وأزيد الصوديوم من anti-H3K27ac 10 و anti-H3K27me3 10 و anti-Med111 و anti-Pol II10 (انظر تكوين المخزن المؤقت الموضح في الجدول 1). يتم تنفيذ جميع الإجراءات التالية تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 1 ساعة
- ضع محلول الجسم المضاد (100 ميكرولتر ، انظر الجدول 2) على عمود تحلية متوازن تم إعداده بعد الخطوة 1.
- جهاز طرد مركزي عند 1500 × جم لمدة دقيقتين عند 4 درجات مئوية ونقل التدفق من أنبوب التجميع إلى أنبوب منخفض الارتباط سعة 1.5 مل.
- قم بقياس تركيز IgG باستخدام الامتصاص عند 280 نانومتر12 (استخدم مقياس الطيف الضوئي microvolume).
- انقل محلول الأجسام المضادة إلى كاسيت الترشيح الفائق (قطع الوزن الجزيئي 100 كيلو دالتون ، 0.5 مل ، انظر جدول المواد) وأجهزة الطرد المركزي عند 12000 × جم لمدة 5 دقائق عند 4 درجات مئوية.
- قم بقياس تركيز IgG باستخدام الامتصاص عند 280 نانومتر.
- كرر الخطوات 2.4-2.5 حتى يصل تركيز IgG إلى 1 مجم / مل.
3. تفعيل الأجسام المضادة
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 2.5 ساعة
- قم بإذابة 1 ملغ من succinimidyl 6-hydrazinonicotinate acetone hydrazone (S-HyNic، انظر جدول المواد) مع 100 ميكرولتر من N، N-dimethylformamide اللامائي (DMF، انظر جدول المواد).
ملاحظة: DMF هو مذيب عضوي قابل للاشتعال وسم كبد قوي يمتص من خلال الجلد. ارتد قفازات ونظارات واقية ومعطف مختبر. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - أضف 0.6 ميكرولتر من S-HyNic/DMF إلى 100 ميكرولتر من محلول الأجسام المضادة (1 ملغم/مل في 150 ملليمتر كلوريد الصوديوم/100 ملي مول فوسفات الصوديوم، pH8.0. انظر الجدول 2 لمعرفة الأجسام المضادة المستخدمة والتحكم في IgG.
- احتضان في درجة حرارة الغرفة لمدة 2 ساعة (حماية من الضوء).
- ضع 100 ميكرولتر من الجسم المضاد المتفاعل مع S-HyNic على الجزء العلوي من عمود التحلية المتوازن (انظر الخطوة 1) وأجهزة الطرد المركزي عند 1500 × جم لمدة دقيقتين عند 4 درجات مئوية لجمع العينة.
- تخلص من العمود المحلى بعد الاستخدام.
ملاحظة: يختلف استقرار مجموعات HyNic على البروتينات والجزيئات الحيوية الأخرى. يوصى باقتران الجزيئات الحيوية المعدلة HyNic على الفور.
4. تفعيل مسبار الحمض النووي
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 2.5 ساعة
- قم بإذابة حبيبات مسبار الحمض النووي المعدل بالأمين للأجسام المضادة أو التحكم في IgG (مسبار الأجسام المضادة ، الجدول 3) مع 20 ميكرولتر من 150 mM NaCl / 100 mM Sodium Phosphate buffer ، درجة الحموضة 8.0.
ملاحظة: ابحث عن حبيبة / فيلم رفيع وشفاف في الجزء السفلي من الأنبوب. ضع المخزن المؤقت مباشرة على الحبيبات. إذا كانت الحبيبات غير مرئية ، فقد تكون الحبيبات قد انفصلت والتقت بالجدار أو الغطاء. في هذه الحالة ، قم بطرد الأنبوب وابحث عن حبيبات في أسفل الأنبوب. - قم بإذابة 1 ملغ من succinimidyl 4-formylbenzoate (S-4FB ، انظر جدول المواد) مع 50 ميكرولتر من DMF اللامائي ، وأضف 10 ميكرولتر من DMF إلى مسبار الأجسام المضادة المذاب.
- أضف 4 ميكرولتر من S-4FB / DMF المحضرة في الخطوة 4.2 ، واخلطها واحتضانها في درجة حرارة الغرفة لمدة ساعتين (الحماية من الضوء).
- ضع 34 ميكرولتر من مسبار الأجسام المضادة المتفاعل مع S-4FB على الجزء العلوي من عمود التحلية المتوازن (انظر الخطوة 1).
- ضع 15 ميكرولتر من 150 mM NaCl / 100 mM Sodium phosphate buffer ، pH 8.0 ، على الجزء العلوي من طبقة الهلام بعد امتصاص العينة بالكامل ، وأجهزة الطرد المركزي عند 1500 × جم لمدة دقيقتين عند 4 درجات مئوية لجمع مسبار الأجسام المضادة المعدل S-4FB.
- استخدم التدفق من أجل الاقتران اللاحق ؛ قياس الامتصاص عند 260 نانومتر ؛ وحساب معدل استرداد مسبار الأجسام المضادة.
5. اقتران الأجسام المضادة المعدلة S-HyNic ومسبار الأجسام المضادة المعدل S-4FB
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 2 ساعة
- امزج الجسم المضاد المعدل S-HyNic ومسبار الأجسام المضادة المعدل S-4FB ، وقم بامتصاص المحلول لأعلى ولأسفل للخلط.
- احتضان لمدة 2 ساعة في درجة حرارة الغرفة (حماية من الضوء).
- لإخماد التفاعل ، أضف 478.8 ميكرولتر من محلول التبريد والتخزين (انظر الجدول 1).
- انقل محلول الأجسام المضادة المترافق بمسبار الأجسام المضادة إلى كاسيت الترشيح الفائق (قطع الوزن الجزيئي 100 كيلو دالتون).
- جهاز طرد مركزي لمدة 5 دقائق عند 12000 × جم ، 4 درجات مئوية.
- تحقق من حجم المحلول داخل الكاسيت عن طريق الماصة.
- كرر الخطوات 5.5-5.6 حتى يصل الحجم إلى 100 ميكرولتر ويخزن في -20 درجة مئوية.
ملاحظة: يتم قياس تركيز IgG بواسطة شطيرة ELISA13،14،15 باستخدام معيار IgG.
6. إعداد الخرز المغناطيسي الأساسية
ملاحظة: في هذه الطريقة ، يتم تضمين خلية واحدة في حبة أكريلاميد ثنائية الطبقات (انظر الشكل 2). النواة هي حبة بولي أكريلاميد المغناطيسية. الطبقة الخارجية هي بولي أكريلاميد وحده. يتم إنشاء الخرزات المغناطيسية الأساسية في هذا القسم. هذا القسم ليس ضروريا للتجربة. الوقت: 3 ساعات

الشكل 2: بنية حبة بولي أكريلاميد ثنائية الطبقات للرؤية وسهولة التعامل في تجارب REpi-seq. أ: الجسيمات النانوية المغناطيسية من الخطوة 6.6 بعد الطرد المركزي. يتم تعديل الجسيمات النانوية المغناطيسية باستخدام مادة الأكريلاميد الأحادية ودمجها في حبة بولي أكريلاميد المغناطيسية الموضحة في B. (ب) تمثيل تخطيطي لخلية مفردة قابلة لإعادة الاستخدام مع حبة مغناطيسية بولي أكريلاميد. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
- امزج 50 ميكرولتر من محلول بيكربونات الصوديوم 1 متر ، ودرجة الحموضة 8.5 ، و 450 ميكرولتر من محلول الأكريلاميد 40٪.
ملاحظة: مادة الأكريلاميد هي سم عصبي. ارتد قفازات وواقي للعين ومعطف مختبر. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - 1 جم من 30 نانومتر من مسحوق أكسيد الحديد الذي يعمل بالإستر NHS (انظر جدول المواد) في محلول الأكريلاميد / بيكربونات الصوديوم واحتضانه عند 4 درجات مئوية طوال الليل.
ملاحظة: نظرا لأن حبات الأكريلاميد شفافة ، فقد يكون من الصعب رؤية موضع الخرزات والتلاعب به. يؤدي تضمين حبة أساسية مصنوعة من أكسيد الحديد إلى تحسين الرؤية وتسهيل التلاعب لأنه يمكن التحكم في موضع الخرزات باستخدام المغناطيس. ومع ذلك ، إذا كان المستخدمون على دراية بتجارب REpi-seq ، فيمكن تخطي استخدام حبات بولي أكريلاميد المغناطيسية الأساسية. - انقل معلق nanobead إلى أنبوبين (1.5 مل) ، وأجهزة طرد مركزي عند 21300 × جم لمدة ساعة واحدة (استخدم دوارا بزاوية) عند 4 درجات مئوية ، وقم بإزالة المادة الطافية.
- الملاط السفلي ب 1 مل من 40٪ أكريلاميد / بيس أكريلاميد (19: 1 ، انظر جدول المواد).
ملاحظة: بيس أكريلاميد هو سم عصبي. ارتد قفازات ومعطف مختبر. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - أجهزة طرد مركزي عند 21300 × جم لمدة 1 ساعة (استخدم دوارا زاويا) عند 4 درجات مئوية.
- جهاز طرد مركزي عند 5000 × جم لمدة 30 دقيقة (استخدم دوار متأرجح بدون فرامل) عند 4 درجات مئوية.
- قم بإزالة المادة الطافية باستخدام ماصة P1000 ذات الشفط البطيء السرعة.
- اضبط مستوى الصوت على 400 ميكرولتر من 40٪ أكريلاميد / بيس أكريلاميد (19: 1 ، انظر جدول المواد).
- أضف 25 ميكرولتر من محلول كبريتات الأمونيوم 10٪ (انظر الجدول 1).
ملاحظة: بيرسلفات الأمونيوم هو عامل مؤكسد قوي. قد يؤدي الغبار المحمول جوا الذي يحتوي على بيرسلفات الأمونيوم إلى تهيج العين والأنف والحلق والرئة والجلد عند ملامسته. ارتد قفازات ونظارات واقية ومعطف مختبر. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - لتوليد حبات بولي أكريلاميد مغناطيسية أساسية ، قم بنقل 0.5 ميكرولتر من معلق أكسيد الحديد المعدل بالأكريلاميد إلى أنبوب PCR.
- أضف 50 ميكرولتر من 4٪ N، N ، N'، N'-TETRAMETHYLETHYLENEDIAMINE / زيت معدني (TEMED، انظر الجدول 1) واحتضان طوال الليل في درجة حرارة الغرفة.
ملاحظة: TEMED مذيب قابل للاشتعال. العمل تحت غطاء محرك السيارة. لا تستنشق. الابتعاد عن اللهب المكشوف والأسطح الساخنة ومصادر الاشتعال. ارتد قفازات ونظارات واقية ومعطف مختبر. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية.
7. تعديل المجموعة الأمينية من البروتينات الخلوية مع مونومر أكريلاميد
ملاحظة: تم تصميم REpi-seq لتحليل الجينوم للفأر والخلايا البشرية على مستوى الخلية الواحدة. يجب تحسين كل خطوة عند استخدام هذه الطريقة على خلايا الأنواع الأخرى غير الفأر أو الإنسان.
- حصاد الخلايا
ملاحظة: الوقت: 30 دقيقة- قم بقياس تركيز الخلية وصلاحيتها باستخدام عداد الخلية (انظر جدول المواد).
ملاحظة: تؤثر صلاحية الخلايا في هذه الخطوة على عدد الخلايا التي تعتبر خلايا حية في تحليل البيانات. - اضبط تركيز الخلية على 1 × 105 خلايا / مل مع وسط زراعة (*) يحتوي على 10٪ مصل بقري جنيني.
ملاحظة: * وسط الاستزراع هو وسط استزراع مثالي للخلايا محل الاهتمام. - نقل 1 مل من معلق الخلية إلى أنبوب 1.5 مل.
- قم بطرد مركزي تعليق الخلية عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية وإزالة المادة الطافية.
- أضف 1 مل من محلول ملحي مخزن بالفوسفات (PBS) ، واخلط الخلايا عن طريق السحب اللطيف ، وطرد مركزي تعليق الخلية عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية.
- إزالة الطاف.
ملاحظة: يجب تنفيذ جميع الخطوات المذكورة أعلاه في غطاء تنظيف التدفق الصفحي لمنع التلوث.
- قم بقياس تركيز الخلية وصلاحيتها باستخدام عداد الخلية (انظر جدول المواد).
- تعديل المجموعة الأمينية من البروتينات الخلوية مع مادة الأكريلاميد الأحادية
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 1.5 ساعة- أضف 1 مل من محلول تعديل المجموعة الأمينية (انظر الجدول 1) إلى حبيبات الخلية وقم بتعليق حبيبات الخلية عن طريق السحب اللطيف.
- احتضان الأنبوب على الجليد لمدة 1 ساعة والطرد المركزي تعليق الخلية عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية.
- قم بإزالة المادة الطافية وأعد تعليق الخلايا التي تحتوي على 100 مل من 4٪ أكريلاميد / 1 مللي متر EDTA / PBS تحتوي على صبغة مقحمة للحمض النووي (انظر الجدول 1 ، خلية واحدة / ميكرولتر).
8. إعداد خلايا مفردة قابلة لإعادة الاستخدام
- إعداد خلايا مفردة قابلة لإعادة الاستخدام (نسخة يدوية)
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 9 ساعات / 96 خلية- امزج 1 مل من معلق الخلية (خلية واحدة / ميكرولتر) و 199 مل من 1 مللي متر EDTA / PBS تحتوي على صبغة إقحامية للحمض النووي (انظر الجدول 1).
- نقل 200 ميكرولتر من تعليق الخلية إلى كل بئر من ألواح 96 بئرا مسطحة القاع (إجمالي 10 ألواح ، انظر جدول المواد).
- ضع الغطاء على لوحة 96 بئرا وامسح 10 لوحات باستخدام مجهر المسح (انظر جدول المواد) لتحديد الآبار التي تحتوي على خلية واحدة.
- نقل محتويات البئر التي تحتوي على خلية واحدة إلى أنبوب PCR.
- قم بإمالة اللوحة (باتجاه المشغل) ، وانتظر بضع دقائق حتى تغرق الخلية المفردة إلى الحافة السفلية للبئر.
- ضع طرف الماصة في الزاوية السفلية وانقل الخلية المفردة والمخزن المؤقت (نضح 210 ميكرولتر / بئر = 200 ميكرولتر من المخزن المؤقت + 10 ميكرولتر من الهواء) إلى أنبوب PCR.
ملاحظة: تأكد من استخدام طرف P200 منخفض الاستبقاء. - افحص البئر باستخدام مجهر مضان لتأكيد النقل.
- إذا كانت خلية واحدة لا تزال في البئر ، أضف 200 ميكرولتر / بئر من 1 mM EDTA / PBS تحتوي على صبغة إقحامية للحمض النووي.
- ثم كرر النقل.
- جهاز طرد مركزي أنبوب PCR عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية باستخدام دوار متأرجح بدون فرامل.
ملاحظة: يتسبب الكبح في تدفق دوامة الماء في الأنبوب ، مما يعطل ترسيب الخلية المفردة. عند الطرد المركزي باستخدام دوار متأرجح بدون فرامل ، ستغرق الخلية المفردة دائما في قاع الأنبوب. ومع ذلك ، إذا تم استخدام دوار بزاوية ، فإن الخلية المفردة تعلق على الجدار الجانبي لأنبوب PCR ويمكن أن تضيع. - قم بإزالة 195 ميكرولتر من المادة الطافية بسرعة سحب بطيئة جدا.
- أضف 195 ميكرولتر / أنبوب من محلول الأكريلاميد / ثنائي الأكريلاميد / APS (انظر الجدول 1).
- جهاز طرد مركزي أنبوب PCR عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية باستخدام دوار متأرجح بدون فرامل.
- قم بإزالة 195 ميكرولتر من المادة الطافية بسرعة سحب بطيئة جدا.
- انقل الجزء السفلي 3 ميكرولتر إلى أنبوب تفاعل البوليميراز المتسلسل الذي يحتوي على 4٪ زيت معدني / زيت معدني وحبة بولي أكريلاميد مغناطيسية (تم إنشاؤها في الخطوة 6).
ملاحظة: تأكد من استخدام طرف P10 منخفض الاستبقاء. الاستغناء عن الخلية بالقرب من حبة بولي أكريلاميد المغناطيسية. في الزيت المعدني ، يلتصق السائل الذي يحتوي على خلية مفردة بسطح حبة بولي أكريلاميد المغناطيسية عن طريق التوتر السطحي. - احتضان بين عشية وضحاها في درجة حرارة الغرفة.
ملاحظة: تولد هذه العملية حبة جل أكريلاميد ثنائية الطبقة. النواة هي حبة هلام مغناطيسي. الطبقة الخارجية عبارة عن هلام بولي أكريلاميد يحتوي على خلية واحدة. يتم تضمين الخلية المفردة في الطبقة الخارجية من الجل. نقطة توقف آمنة: بعد غسل الخلايا المفردة القابلة لإعادة الاستخدام باستخدام 50٪ جلسرين / 1 مللي متر EDTA / 0.05٪ توين 20 / 0.5٪ BSA / TBS عازلة ، يمكن تخزين الخلايا المفردة القابلة لإعادة الاستخدام عند -20 درجة مئوية لمدة تصل إلى 6 أشهر.
- إعداد خلايا مفردة قابلة لإعادة الاستخدام (نسخة شبه آلية)
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 3 ساعات / 96 خلية- امزج 1 مل من معلق الخلية (خلية واحدة / ميكرولتر) و 199 مل من 1 مللي متر EDTA / PBS تحتوي على صبغة إقحامية للحمض النووي (انظر الجدول 1).
- انقل 200 ميكرولتر من تعليق الخلية إلى كل بئر من لوحة نانوويل 4 نانو لتر (انظر الشكل 3 وجدول المواد) وضع لوحة نانوويل 4 نانو على روبوت آلي أحادي الخلية (انظر جدول المواد).
- ضع لوحة PCR ذات 96 بئرا كلوحة وجهة على روبوت التقاط الخلية الواحدة الآلي. تأكد من أن كل بئر يحتوي على 200 ميكرولتر / بئر محلول أكريلاميد / ثنائي أكريلاميد / APS (انظر الجدول 1).
- انقل خلية واحدة من بئر نانوي سعة 4 نانو لتر إلى بئر من لوحة تفاعل البوليميراز المتسلسل ذات 96 بئرا (انظر الفيديو التكميلي S1).
- ضع غطاء فوق لوحة PCR ذات 96 بئرا وأجهزة الطرد المركزي للوحة 96 بئرا عند 240 × جم لمدة 5 دقائق عند 4 درجات مئوية باستخدام دوار متأرجح بدون فرامل.
- ضع لوحة PCR المكونة من 96 بئرا على سطح روبوت أوتوماتيكي لمعالجة السوائل (انظر جدول المواد ، الشكل 4 ، والرمز التكميلي 1).
- اسحب الكود التكميلي 1 وأفلته في نافذة البرنامج (انظر جدول المواد) لروبوت معالجة السوائل.
- قم بتشغيل البرنامج على روبوت معالجة السوائل التلقائي (انظر الفيديو التكميلي S2 والفيديو التكميلي S3).
- قم بإزالة 195 ميكرولتر من المادة الطافية بسرعة سحب بطيئة جدا.
- أضف 50 ميكرولتر من 4٪ زيت معدني / زيت معدني.
ملاحظة: يمكن تنفيذ الخطوات من 8.2.8.1 إلى 8.2.8.2 عن طريق السحب اليدوي.
- احتضان بين عشية وضحاها في درجة حرارة الغرفة (الشكل 5). نقطة توقف آمنة: بعد غسل الخلايا المفردة القابلة لإعادة الاستخدام باستخدام 50٪ جلسرين / 1 مللي متر EDTA / 0.05٪ توين 20 / 0.5٪ BSA / TBS عازلة ، قم بتخزين الخلايا المفردة القابلة لإعادة الاستخدام عند -20 درجة مئوية لمدة تصل إلى 6 أشهر.

الشكل 3: الانتقاء التلقائي أحادي الخلية ونقله إلى لوحة PCR ذات 96 بئرا في الخطوة 8.2 . (أ) نظرة عامة على نظام انتقاء الخلية الواحدة. يوجد روبوت واحد لالتقاط الخلايا في غطاء نظيف للتدفق الصفحي لتجنب التلوث. (ب) صفيحة من 24 بئرا بها 4 nL نانوآبار داخل البئر. ج: توزيع الخلايا في بئر من الصفيحة التي تحتوي على 24 بئرا. النقاط الخضراء هي خلايا يتم تحديدها كخلية واحدة في كل بئر نانوي 4 nL. النقاط الأرجوانية هي خلايا يتم تحديدها على أنها مضاعفة أو مضاعفات للخلايا. (د) صورة برايتفيلد للبئر في الصفيحة المكونة من 24 بئرا. المربع الأخضر هو بئر نانوي 4 nL يحتوي على خلية واحدة. المربع الأرجواني هو بئر نانوي 4 nL يحتوي على خلايا متعددة. (ه) حقل مكبرة لنحو 4 آبار نانوية نانوية. النقاط الساطعة هي خلايا مفردة في 4 نانو آبار نانوية. يحدد نظام انتقاء الخلية المفردة الآبار النانوية التي تحتوي على خلية واحدة من خلال الحصول على صور برايت فيلد ومضان للخلايا ذات تلطيخ DAPI. يتم نقل الخلايا المفردة المحددة من بئر نانو 4 nL إلى بئر من لوحة PCR ذات 96 بئرا. قضبان المقياس = 2 مم (C ، D) ، 100 ميكرومتر (E). اختصار = DAPI = 4',6-دياميدينو-2-فينيليندول. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}

الشكل 4: توليد خلايا مفردة قابلة لإعادة الاستخدام باستخدام روبوت مناولة السوائل. أ: سطح روبوت مناولة السوائل. يحتوي السطح على 11 فتحة لرفوف طرف الماصة (طرف P300: فتحات 1-3 ، طرف P20: فتحات 5-6) ، 2 مل لوحة بئر عميق 96 بئر (الفتحة 4) ، لوحان PCR 96 بئرا يحتويان على خلية واحدة لكل بئر (الفتحتان 7 و 10) ، ولوحان مسطحان القاع 96 بئرا للنفايات السائلة (الفتحتان 8 و 11). (ب) السطح بعد وضع أدوات المختبر. (ج) التمثيل التخطيطي للسحب الآلي في الخطوة 8.8.1. يزيل البرنامج المادة الطافية دون استنشاق خلية واحدة من أسفل لوحة PCR المكونة من 96 بئرا. (د) خلايا مفردة قابلة لإعادة الاستخدام تم إنشاؤها باستخدام الكود التكميلي 1. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
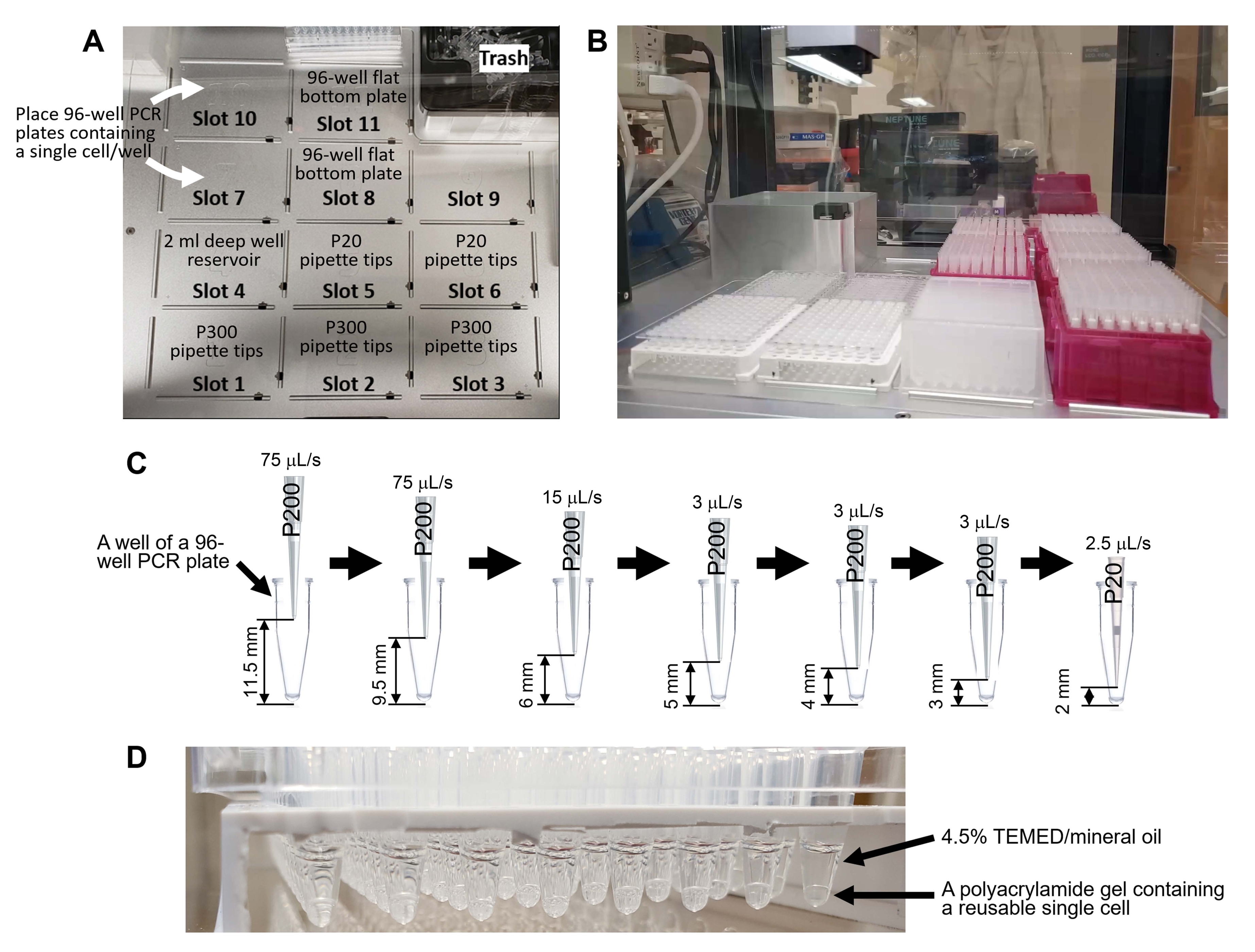{kind=link}
9. التلدين التمهيدي العشوائي والتمديد
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. تشير العلامات النجمية (*) في الخطوات التالية إلى أنه يمكن استخدام فاصل مغناطيسي للتحكم في موضع حبات بولي أكريلاميد التي تحتوي على خلية مفردة قابلة لإعادة الاستخدام في الأنبوب. ومع ذلك ، فإن استخدام الفاصل المغناطيسي ليس ضروريا. عن طريق تحريك طرف الماصة ببطء على طول جدار الأنبوب ، يتم دفع الخرزات لأعلى للغسيل أو التبادل العازل. الوقت: 9 ساعات
- قم بإزالة الزيت المعدني عن طريق سحب الماصة (*) واغسل (*) 5 مرات باستخدام 200 ميكرولتر من 1x TP Mg (-) buffer (تمييع 10x TP Mg (-) buffer إلى 1x buffer بماء عالي النقاء ، انظر الجدول 1).
- قم بإزالة المادة الطافية (*) وأضف 15 ميكرولتر من المخزن المؤقت للتلدين (انظر الجدول 1).
- احتضان لمدة 1 ساعة على الجليد.
ملاحظة: الغرض من هذه الحضانة هو اختراق البلازما والأغشية النووية وتوصيل التمهيدي العشوائي إلى نواة الخلية. - ضع أنابيب PCR على جهاز تدوير حراري وقم بتسخينه على حرارة 94 درجة مئوية لمدة 3 دقائق.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 20 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. - نقل الأنابيب إلى كتلة معدنية مبردة بالثلج واحتضانها لمدة 2 دقيقة.
- أضف 4 ميكرولتر من مزيج MgSO4 / NaCl / dNTP (انظر الجدول 1) واخلطه بخلاط دوامة بسرعة متوسطة.
- أضف 1 ميكرولتر من بوليميراز Bst ، جزء كبير (انظر جدول المواد) واخلطه باستخدام خلاط دوامة بسرعة متوسطة.
- احتضان لمدة 4 ساعات على شاكر (600 دورة في الدقيقة عند 4 درجات مئوية).
ملاحظة: الغرض من هذه الحضانة هو توصيل بوليميراز Bst إلى نواة الخلية. - ضع أنبوب PCR على جهاز تدوير حراري وقم بتشغيل أحد البرامج التالية.
- قم بتشغيل برنامج 4 ساعات: 10 درجات مئوية لمدة 30 دقيقة ، و 20 درجة مئوية لمدة 30 دقيقة ، و 25 درجة مئوية لمدة 180 دقيقة.
- بدلا من ذلك ، قم بتشغيل برنامج ليلي: 4 درجات مئوية لمدة 4 ساعات ، 10 درجات مئوية لمدة 2 ساعة ، 20 درجة مئوية لمدة 2 ساعة ، 25 درجة مئوية لمدة 4 ساعات ، واستمر عند 4 درجات مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 25 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي 105 °C. نقطة توقف آمنة: أوقف التجربة هنا لمدة تصل إلى 1 يوم عن طريق تخزين الخلايا المفردة القابلة لإعادة الاستخدام عند 4 °C.
10. ربط الأجسام المضادة
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 1.5 ساعة
- أضف 1.625 ميكرولتر من محلول NaCl / EDTA / BSA (انظر الجدول 1) واخلطه عن طريق الدوامة بسرعة منخفضة.
ملاحظة: تهدف هذه الخطوة إلى 1) تسهيل استخلاب Mg2+ بواسطة EDTA ، 2) تثبيت ارتباط التمهيديالعشوائي الممتد 1 بواسطة 300 mM NaCl ، و 3) منع الارتباط غير النوعي للأجسام المضادة باستخدام ألبومين مصل الأبقار (BSA) في التفاعل اللاحق. - احتضان لمدة 1 ساعة على الجليد وإضافة 0.1 ميكروغرام / مل لكل من الجسم المضاد والتحكم IgG مترافق مع مسبار الأجسام المضادة (أعدت في الخطوة 5).
- احتضان بين عشية وضحاها على الجليد مع هز لطيف على شاكر.
11. ربط القرب لمسبار الأجسام المضادة والتمهيدي العشوائي الممتد عن قرب
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. تشير العلامات النجمية (*) في الخطوات التالية إلى المكان الذي يمكن فيه استخدام فاصل مغناطيسي للتحكم في موضع حبات بولي أكريلاميد التي تحتوي على خلية مفردة قابلة لإعادة الاستخدام في الأنبوب. ومع ذلك ، فإن استخدام الفاصل المغناطيسي ليس ضروريا. من خلال تحريك طرف الماصة ببطء على طول جدار الأنبوب ، يمكن دفع الخرزات لأعلى للغسيل أو التبادل العازل. الوقت: 6 ساعات
- اغسل (*) مرتين باستخدام 200 ميكرولتر من 1x TPM-T buffer (حضانة 20 دقيقة في كل غسلة) على الثلج. تمييع 10x TPM-T العازلة (Tris-HCl / كلوريد البوتاسيوم / كبريتات المغنيسيوم / Triton X-100) إلى 1x بالماء عالي النقاء).
- قم بإزالة (*) المادة الطافية واغسلها (*) مرة واحدة باستخدام 1x T4 DNA ligase buffer (انظر جدول المواد).
- قم بإزالة (*) المادة الطافية وأضف 19 ميكرولتر من محلول محول الربط (انظر الجدول 1).
- احتضان لمدة 1 ساعة عند 25 درجة مئوية.
- أضف 1 ميكرولتر T4 DNA ligase (انظر جدول المواد) وامزج الأنبوب على خلاط دوامة بسرعة متوسطة.
- ضع الأنبوب على جهاز تدوير حراري وقم بتشغيل البرنامج التالي: برنامج ربط القرب ، 4 ساعات عند 16 درجة مئوية و 30 دقيقة عند 25 درجة مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 25 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي درجة حرارة الغرفة. - اغسل (*) مرتين لفترة وجيزة باستخدام 200 ميكرولتر من 1x Bst Mg(-) EDTA (+) buffer (انظر الجدول 1؛ قم بإعداد مخزن مؤقت واحد × من مخزن مؤقت 10x) وقم بتخزين الخلية عند 4 درجات مئوية، طوال الليل. نقطة توقف آمنة: أوقف التجربة هنا لمدة تصل إلى 1 يوم عن طريق تخزين الخلايا المفردة القابلة لإعادة الاستخدام عند 4 °C.
12. التمديد الكامل للبرايمر العشوائي الأول
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 4.5 ساعة
- اغسل مرتين باستخدام 200 ميكرولتر 1x Bst Mg (-) EDTA (-) buffer (انظر الجدول 1 ، تحضير مخزن مؤقت 1x من مخزن مؤقت 10x) ، وإزالة المادة الطافية ، وإضافة 20 ميكرولتر من خليط Bst / dNTPs / MgSO4 (انظر الجدول 1).
- تخلط مع خلاط دوامة بسرعة متوسطة واحتضانها لمدة 4 ساعات على شاكر مداري (عند 6 درجات مئوية و 500 دورة في الدقيقة).
- ضع الأنبوب على جهاز تدوير حراري وقم بتشغيل البرنامج التالي: برنامج التمديد الكامل: 10 درجات مئوية لمدة 1 ساعة ، 20 درجة مئوية لمدة 1 ساعة ، 30 درجة مئوية لمدة 1 ساعة ، 40 درجة مئوية لمدة 1 ساعة ، 50 درجة مئوية لمدة 1 ساعة ، 65 درجة مئوية لمدة 1 ساعة ، 94 درجة مئوية لمدة 10 دقائق ، واستمر عند 4 درجات مئوية.
ملاحظة: نقطة التوقف الآمنة: يمكن إيقاف التجربة هنا لمدة تصل إلى 1 يوم عن طريق تخزين الخلايا المفردة القابلة لإعادة الاستخدام عند 4 °C.
13. تضخيم الإزاحة المتعددة
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 2.5 ساعة (الخطوات 13.1-13.2) + 15 دقيقة (الخطوات 13.3-13.4) + يوم واحد (الخطوات 13.5-13.10)
- أضف 0.4 ميكرولتر من 100 ميكرومتر 2ثانية التمهيدي العشوائي (انظر الجدول 3) واخلطه مع خلاط دوامة بسرعة متوسطة.
- احتضان لمدة 2 ساعة عند 6 درجات مئوية و 500 دورة في الدقيقة على شاكر مداري ، ووضع الأنبوب على دورة حرارية وتسخينه عند 94 درجة مئوية لمدة 3 دقائق.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 25.4 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. - ضع الأنابيب في كتلة معدنية مبردة بالثلج وأضف 1 ميكرولتر / أنبوب من بوليميراز الحمض النووي Bst.
- دوامة بسرعة بطيئة ، ضع الأنابيب على دورة حرارية ، وقم بتشغيل البرنامج التالي:
4 درجة مئوية لمدة 4 ساعات ، 10 درجة مئوية لمدة 30 دقيقة ، 20 درجة مئوية لمدة 30 دقيقة ، 30 درجة مئوية لمدة 30 دقيقة ، 40 درجة مئوية لمدة 30 دقيقة ، 50 درجة مئوية لمدة 30 دقيقة ، 65 درجة مئوية لمدة 60 دقيقة ، 94 درجة مئوية لمدة 3 دقائق ، وعقد عند 4 درجات مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 26.4 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي 105 °C. نقطة توقف آمنة: أوقف التجربة هنا لمدة تصل إلى 1 يوم عن طريق تخزين الخلايا المفردة القابلة لإعادة الاستخدام عند 4 °C. - جمع طاف (حوالي 20 ميكرولتر) ، ونقله إلى أنبوب PCR.
- قم بتخزين المادة الطافية المجمعة في درجة حرارة -80 درجة مئوية.
- أضف 20 ميكرولتر / أنبوب من 0.05٪ Tween 20 / 0.1x TE buffer إلى أنبوب PCR يحتوي على خلية مفردة قابلة لإعادة الاستخدام (انظر الجدول 1) واحتضان الخلية المفردة القابلة لإعادة الاستخدام عند 4 درجات مئوية ، طوال الليل. نقطة توقف آمنة: أوقف التجربة هنا لبضعة أيام عن طريق تمديد وقت الحضانة.
- اجمع المادة الطافية وادمج المادة الطافية مع العينة التي تم جمعها في الخطوة 13.5.
- كرر الخطوات من 13.7 إلى 13.8 مرة أخرى. انقع الخلية المفردة القابلة لإعادة الاستخدام في 50٪ جلسرين / 5 مللي متر EDTA / 0.5٪ BSA / 0.05٪ Tween20 / TBS عازلة واحتضانها لمدة 30 دقيقة على شاكر مداري (4 درجات مئوية ، 600 دورة في الدقيقة). قم بتخزين الخلية المفردة القابلة لإعادة الاستخدام في -20 درجة مئوية حتى التجربة التالية.
- أضف 40 ميكرولتر من مزيج Exo- master (انظر الجدول 1) وضع الأنبوب على جهاز تدوير حراري وقم بتشغيل البرنامج التالي: ط) 95 درجة مئوية لمدة 5 دقائق ، ب) 95 درجة مئوية لمدة 30 ثانية ، ج) 60 درجة مئوية 30 ثانية ، د) 72 درجة مئوية لمدة 30 ثانية ، ت) كرر الخطوات من الثاني إلى الرابع 19 مرة ، سادسا) 72 درجة مئوية لمدة 5 دقائق ، vii) عقد عند 4 درجات مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول حوالي 45 ميكرولتر ، بما في ذلك حجم حبة بولي أكريلاميد. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. نقطة توقف آمنة: يمكن إيقاف التجربة هنا لبضعة أيام على الأقل عن طريق تخزين العينة عند -80 درجة مئوية.
14. تنقية الفينول كلوروفورم وهطول الأمطار البولي ايثيلين جلايكول
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 1.5 ساعة
- انقل المنتج إلى أنبوب منخفض الارتباط للحمض النووي سعة 0.5 مل وأضف 100 ميكرولتر من الفينول: الكلوروفورم: كحول الأيزو أميل.
ملاحظة: الفينول: الكلوروفورم: كحول الأيزو أميل يسبب تهيجا وربما حروقا عن طريق الاتصال. ارتد قفازات ونظارات واقية ومعطف مختبر. استخدمه فقط مع التهوية الكافية ، أو ارتد جهاز تنفس مناسب. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - رج لمدة 30 ثانية باليد وأجهزة الطرد المركزي عند 12000 × جم لمدة 10 دقائق عند 4 درجات مئوية.
- اجمع الطور المائي (80 ميكرولتر) في أنبوب منخفض الارتباط للحمض النووي سعة 0.5 مل ، وأضف 40.84 ميكرولتر / أنبوب من خليط الأكريلاميد الخطي / MgCl2 (انظر الجدول 1).
- أضف 47.06 ميكرولتر / أنبوب من 50٪ (وزن / حجم) PEG8000 (خال من RNase) واخلطه عن طريق الماصة.
- احتضان لمدة 20 دقيقة في درجة حرارة الغرفة وأجهزة الطرد المركزي في 240 × غرام لمدة 10 دقائق في درجة حرارة الغرفة.
- قم بإزالة المادة الطافية وإضافة 400 ميكرولتر / أنبوب من الإيثانول بنسبة 80٪ (EtOH).
- اغسل باستخدام 80٪ EtOH ، وقم بإزالة المادة الطافية باستخدام شفاط ، وجفف الحبيبات بالهواء.
- الحبيبات ب 20 ميكرولتر من 1 مللي مول EDTA / 10 مللي متر Tris-HCl ، ودرجة الحموضة 7.4 عازلة ، وقم بتخزين المحلول عند -80 درجة مئوية.
ملاحظة: قم بقياس تركيز الحمض النووي باستخدام صبغة إقحامية مزدوجة الشريط خاصة بالحمض النووي (انظر جدول المواد). نقطة توقف آمنة: يمكن إيقاف التجربة بأمان هنا لمدة أسبوع على الأقل.
15. النسخ في المختبر
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase و RNase. الوقت: 5 ساعات
- قم بإذابة الحمض النووي للمنتجات المشتقة من خلية واحدة (من الخطوة 14) وقم بإعداد مكتبة مختلطة من المنتجات المشتقة من خلية واحدة عن طريق خلط 2 ميكرولتر / خلية من منتجات الحمض النووي (الحجم الإجمالي 20 ميكرولتر).
- أضف 26 ميكرولتر من المزيج الرئيسي للنسخ في المختبر (انظر جدول المواد وبروتوكول الشركة المصنعة) واخلطه عن طريق الماصة.
- ضع أنبوب PCR على جهاز تدوير حراري واحتضانه لمدة 4 ساعات عند 37 درجة مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول 46 ميكرولتر. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. - أضف 5 ميكرولتر من 10x DNase I buffer (انظر جدول المواد) وأضف 4 ميكرولتر من DNase I (خال من RNase ، 4 U ، انظر جدول المواد).
- امزج واحتضن لمدة 15 دقيقة عند 37 درجة مئوية ، وانقل العينة إلى أنبوب سعة 0.5 مل.
- أضف 300 ميكرولتر / أنبوب من ثيوسيانات الغوانيدينيوم - الفينول - الكلوروفورم (انظر جدول المواد) واخلطه عن طريق الدوامة اللطيفة.
ملاحظة: يمكن أن يؤدي ثيوسيانات الغوانيدينيوم الفينول كلوروفورم إلى حروق كيميائية خطيرة عن طريق الاتصال. ارتد قفازات ونظارات واقية ومعطف مختبر. استخدم فقط مع التهوية الكافية أو ارتداء جهاز تنفس مناسب. اتبع إرشادات السلامة المؤسسية. تخلص من أدوات المختبر المستخدمة وفقا للإرشادات المؤسسية. - احتضان لمدة 5 دقائق في R.T. على شاكر ثم قم بتخزين العينات في -80 درجة مئوية لمدة تصل إلى 3 أيام.
ملاحظة: نقطة توقف آمنة: يمكن إيقاف التجربة بأمان هنا لمدة تصل إلى 3 أيام.
16. تنقية الحمض النووي الريبي
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase و RNase. الوقت: 2 ساعة
- أضف 80 ميكرولتر / أنبوب من الكلوروفورم إلى العينة من الخطوة 15.7 ورج الأنبوب بقوة باليد لمدة 15 ثانية.
- احتضان العينة لمدة 2-15 دقيقة في درجة حرارة الغرفة والطرد المركزي عند 12000 × جم لمدة 15 دقيقة عند 4 درجات مئوية.
- جمع ونقل المرحلة المائية من العينة (~ 200 ميكرولتر) إلى أنبوب جديد 1.5 مل.
- أضف 600 ميكرولتر / أنبوب من ثيوسيانات الغوانيدينيوم - الفينول - الكلوروفورم وانقل العينة إلى أنبوب سعة 1.5 مل.
- أضف 180 ميكرولتر / أنبوب من الكلوروفورم ورج الأنبوب بقوة باليد لمدة 15 ثانية.
- أجهزة الطرد المركزي العينة عند 12000 × جم لمدة 10 دقائق عند 4 درجات مئوية.
- جمع ونقل المرحلة المائية للعينة (~ 450 ميكرولتر) إلى أنبوب 1.5 مل.
- أضف 60 ميكرولتر / أنبوب من مادة الأكريلاميد الخطية (5 ميكروغرام / ميكرولتر) وأضف 400 ميكرولتر / أنبوب من الأيزوبروبانول بنسبة 100٪ إلى المرحلة المائية.
- احتضان الأنبوب في درجة حرارة الغرفة لمدة 10 دقائق وطرده مركزيا عند 12000 × جم لمدة 10 دقائق عند 4 درجات مئوية.
- قم بإزالة المادة الطافية بعناية.
- اغسل حبيبات الحمض النووي الريبي 3 مرات ب 200 ميكرولتر من الإيثانول بنسبة 75٪ (ماء خال من EtOH / RNase) ، وأزل المادة الطافية ، وجفف حبيبات الحمض النووي الريبي بالهواء.
ملاحظة: لا تدع الحمض النووي الريبي يجف تماما لأن الحبيبات يمكن أن تفقد قابليتها للذوبان. - أعد تعليق حبيبات الحمض النووي الريبي في 20 ميكرولتر من الماء الخالي من RNase الذي يحتوي على مثبط RNAse (1 ميكرولتر / 20 ميكرولتر) وقم بإذابة الحبيبات عن طريق الماصة.
- قياس الامتصاص عند 260 نانومتر.
17. النسخ العكسي
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 1 ساعة
- أضف 7 ميكرولتر / أنبوب من التمهيدي للنسخ العكسي + مزيج dNTP (انظر الجدول 1 والجدول 3) وضع الأنابيب على جهاز تدوير حراري.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول 27 ميكرولتر. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. - يسخن على حرارة 65 درجة مئوية لمدة 5 دقائق ويوضع الأنابيب على الثلج لمدة 1 دقيقة على الأقل.
- أضف 14 ميكرولتر / أنبوب من مزيج النسخ العكسي الرئيسي (انظر الجدول 1) واخلطه عن طريق الماصة.
- ضع الأنابيب على جهاز تدوير حراري ، واحتضنها على حرارة 55 درجة مئوية لمدة 10 دقائق ثم عند 80 درجة مئوية لمدة 10 دقائق.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول 60 ميكرولتر. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية.
18. توليف الخيط الثاني
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 2.5 ساعة
- أضف 40 ميكرولتر / أنبوب من الماء عالي النقاء وقسم محلول 100 ميكرولتر إلى أنبوبين PCR سعة 0.2 مل (50 ميكرولتر / أنبوب).
- أضف 60 ميكرولتر / أنبوب من مزيج تخليق الخيط الثاني (انظر الجدول 1) وضع الأنابيب على جهاز تدوير حراري وقم بتشغيل البرنامج التالي: ط) 95 درجة مئوية لمدة 5 دقائق ، ب) 95 درجة مئوية لمدة 30 ثانية ، ج) 60 درجة مئوية لمدة 30 ثانية ، د) 72 درجة مئوية لمدة 30 ثانية ، ت) كرر الخطوات من الثاني إلى الرابع 20 مرة ، و سادسا) استمر عند 4 درجات مئوية.
ملاحظة: حجم الأنبوب 0.2 مل. حجم المحلول 110 ميكرولتر. درجة حرارة غطاء التدوير الحراري هي 105 درجة مئوية. - أضف 4.4 ميكرولتر / أنبوب من 0.5 متر EDTA (20 مللي متر نهائي) وخزنه في -80 درجة مئوية طوال الليل.
ملاحظة: نقطة التوقف الآمنة: يمكن إيقاف التجربة بأمان هنا لمدة تصل إلى بضعة أيام. - تنقية الحمض النووي عن طريق تنقية الفينول كلوروفورم وترسيب البولي إيثيلين جلايكول (الموصوف في الخطوة 14).
ملاحظة: نقطة التوقف الآمنة: يمكن إيقاف التجربة بأمان هنا لمدة أسبوع على الأقل.
19. تقييد هضم الإنزيم واختيار الحجم
ملاحظة: يتم تنفيذ الإجراء التالي تحت غطاء نظيف لتجنب تلوث DNase. الوقت: 3 ساعات (الخطوات 19.1-19.7)
- قم بقياس تركيز الحمض النووي للحمض النووي المنقى (من الخطوة 18.4) عن طريق قياس الامتصاص عند 260 نانومتر.
- انقل 6 ميكروغرام من الحمض النووي إلى أنبوب PCR ، وأضف 30 ميكرولتر من 10x Digestion buffer ، واضبط الحجم على 294 ميكرولتر بماء فائق النقاء.
- أضف 6 ميكرولتر من إنزيم تقييد BciVI واحتضانه عند 37 درجة مئوية لمدة 1 ساعة.
- أداء هطول الأمطار EtOH مع بولي أكريلاميد الخطي.
- أضف 60 ميكرولتر / أنبوب من 3 M أسيتات الصوديوم (درجة الحموضة 5.2) ثم أضف 40 ميكرولتر / أنبوب من مادة الأكريلاميد الخطية (5 مجم / مل ، انظر جدول المواد).
- أضف 400 ميكرولتر / أنبوب من EtOH واحتضان طوال الليل عند -20 درجة مئوية.
ملاحظة: نقطة توقف آمنة: يمكن إيقاف التجربة بأمان هنا ليوم واحد. - جهاز طرد مركزي 12000 × جم لمدة 10 دقائق عند 4 درجات مئوية وإزالة المادة الطافية.
- اغسل الحبيبات مرتين بنسبة 80٪ EtOH وجفف الحبيبات.
- قم بإذابة الحبيبات ب 20 ميكرولتر من المخزن المؤقت 1xTE وأضف 4 ميكرولتر / أنبوب من المخزن المؤقت لتحميل الجل 6x.
- قم بتحميل العينة في محلول أغاروز 5٪ / 0.5x TAE مؤقت وقم بإجراء رحلان كهربائي (50 فولت لمدة 40 دقيقة).
- قم بقص وجمع الجل الذي يزيد عن 50 نقطة أساس (انظر الشكل 6) واستخرج الحمض النووي باستخدام مجموعة استخراج الجل.
ملاحظة: نقطة التوقف الآمنة: يمكن إيقاف التجربة بأمان هنا لمدة أسبوع على الأقل. - قم ببناء مكتبة التسلسل باستخدام مجموعة إعداد مكتبة الحمض النووي (انظر جدول المواد)16,17.
النتائج
تم إنشاء خلايا مفردة K562 باستخدام البروتوكول الموضح في الخطوة 8 (انظر الشكل 5). تم تضمين خلايا مفردة في الطبقة الخارجية من حبة بولي أكريلاميد. تم تلطيخ الحمض النووي للخلية وتصوره باستخدام صبغة مقحمة لتلطيخ الحمض النووي.
Discussion
توضح هذه المقالة البروتوكول خطوة بخطوة للتحليل متعدد الخلايا أحادية الخلية الذي تم الإبلاغ عنه مؤخرا باستخدام خلايا مفردة قابلة لإعادة الاستخدام7. في الفقرات اللاحقة ، نناقش النقاط الحرجة ، مع التركيز على القيود المحتملة في البروتوكول.
إحدى النقاط الحرجة في ?...
Disclosures
الدكتور Ohnuki و Tosato هما مخترعان مشاركان في براءة اختراع بعنوان "طرق تحضير خلية مفردة قابلة لإعادة الاستخدام وطرق لتحليل epigenome و transcriptome وجينوم خلية واحدة" (EP3619307 و US20200102604). تم تقديم طلب البراءة جزئيا بناء على النتائج الأولية المتعلقة بالتكنولوجيا الموصوفة في المخطوطة الحالية. تم إجراء الاختراع أو الاختراعات الموصوفة والمطالب بها في طلب براءة الاختراع هذا بينما كان المخترعون موظفين بدوام كامل في حكومة الولايات المتحدة. لذلك ، بموجب 45 قانون اللوائح الفيدرالية الجزء 7 ، تم أو ينبغي تعيين جميع الحقوق والملكية والمصلحة في طلب براءة الاختراع هذا بموجب القانون إلى حكومة الولايات المتحدة. تنقل حكومة الولايات المتحدة جزءا من الإتاوات التي تتلقاها إلى مخترعيها الموظفين بموجب 15 قانون الولايات المتحدة § 3710c.
Acknowledgements
نشكر الدكتور ديفيد سانشيز مارتن وكريستوفر ب. باك على تعليقاتهما خلال مرحلة وضع المفاهيم للمشروع. كما نشكر مركز الجينوم ، ومركز أبحاث السرطان ، والمعهد الوطني للسرطان ، والمعاهد الوطنية للصحة للمساعدة في التجارب الأولية ، وموارد المعلوماتية الحيوية التعاونية ، و CCR ، و NCI ، و NIH للحصول على المشورة في التحليل الحسابي. نشكر السيدة آنا وورد على المساعدة في تحسين بوليميراز الحمض النووي المستخدم في الطريقة. استخدم هذا العمل الموارد الحسابية لمجموعة NIHHPC Biowulf (http://hpc.nih.gov). يتم دعم هذا المشروع من قبل البرنامج الداخلي لمركز أبحاث السرطان ، والمعهد الوطني للسرطان ، والمعاهد الوطنية للصحة ، وجائزة الابتكار لمدير المعهد الوطني للسرطان (# 397172) ، والأموال الفيدرالية من المعهد الوطني للسرطان بموجب العقد رقم HHSN261200800001E. نشكر الدكاترة توم ميستيلي وكارول ثيل ودوغلاس آر لوي وجميع أعضاء مختبر الأورام الخلوية على تعليقاتهم المثمرة.
Materials
| Name | Company | Catalog Number | Comments |
| 10x CutSmart buffer | New England BioLabs | B6004 | 10x Digestion buffer |
| 200 proof ethanol | Warner-Graham Company | 200 proof | Ethanol |
| 5-Hydroxymethylcytosine (5-hmC) Monoclonal Antibody [HMC/4D9] | Epigentek | A-1018-100 | Anti-5hmC |
| Acridine Orange/Propidium Iodide Stain | Logos Biosystems | F23001 | Cell counter |
| Acrylamide solution, 40% in H2O, for molecular biology | MilliporeSigma | 01697-500ML | 40% acrylamide solution |
| All-in-One Fluorescence Microscope BZ-X710 | Keyence | BZ-X710 | Scanning microscope |
| Amicon Ultra-0.5 Centrifugal Filter Unit | MilliporeSigma | UFC510024 | Ultrafiltration cassette |
| Ammonium persulfate for molecular biology | MilliporeSigma | A3678-100G | Ammonium persulfate powder |
| Anhydrous DMF | Vector laboratories | S-4001-005 | Anhydrous N,N-dimethylformamide (DMF) |
| Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) antibody [4H8] | Abcam | ab5408 | Anti-Pol II |
| Anti-TRAP220/MED1 (phospho T1457) antibody | Abcam | ab60950 | Anti-Med1 |
| BciVI | New England BioLabs | R0596L | BciVI |
| Bovine Serum Albumin solution, 20 mg/mL in H2O, low bioburden, protease-free, for molecular biology | MilliporeSigma | B8667-5ML | 20% BSA (Table 7) |
| Bst DNA Polymerase, Large Fragment | New England BioLabs | M0275L | Bst DNA polymerase |
| BT10 Series 10 µl Barrier Tip | NEPTUNE | BT10 | P10 low-retention tip |
| CellCelector | Automated Lab Solutions | N/A | Automated single cell picking robot |
| CellCelector 4 nl nanowell plates for single cell cloning, Plate S200-100 100K, 24 well,ULA | Automated Lab Solutions | CC0079 | 4 nL nanowell plate |
| Chloroform | MilliporeSigma | Chloroform | |
| Corning Costar 96-Well, Cell Culture-Treated, Flat-Bottom Microplate | Corning | 3596 | Flat-bottom 96-well plates |
| Deep Vent (exo-) DNA Polymerase | New England BioLabs | M0259L | Exo- DNA polymerase |
| DNA LoBind Tubes, 0.5 mL | Eppendorf | 30108035 | 0.5 mL DNA low-binding tube |
| DNA Oligo, 1st random primer | Integrated DNA Technologies | N/A, see Table 3 | 1st random primer |
| DNA Oligo, 2nd random primer Cell#01 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#02 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#03 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#04 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#05 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#06 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#07 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#08 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#09 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#10 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#11 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#12 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd synthesis primer | Integrated DNA Technologies | N/A, see Table 3 | 2nd synthesis primer |
| DNA Oligo, Ligation Adaptor | Integrated DNA Technologies | N/A, see Table 3 | Ligation Adaptor |
| DNA Oligo, Reverse Transcription primer | Integrated DNA Technologies | N/A, see Table 3 | Reverse Transcription primer |
| DNase I (RNase-free) | New England BioLabs | M0303L | DNase I (RNase-free, 4 U). |
| DNase I Reaction Buffer | New England BioLabs | B0303S | 10x DNase I buffer (NEB) |
| dNTP Mix (10 mM each) | Thermo Fisher | R0192 | 10 mM dNTPs |
| Fetal Bovine Serum, USA origin, Heat-inactivated | MilliporeSigma | F4135-500ML | Fetal bovine serum |
| HiScribe T7 High Yield RNA Synthesis Kit | New England BioLabs | E2040S | In-vitro-transcription master mix |
| Histone H3K27ac antibody | Active motif | 39133 | Anti-H3K27ac |
| Histone H3K27me3 antibody | Active motif | 39155 | Anti-H3K27me3 |
| IgG from rabbit serum | Millipore Sigma | I5006-10MG | Control IgG |
| Iron oxide(II,III) magnetic nanopowder, 30 nm avg. part. size (TEM), NHS ester functionalized | MilliporeSigma | 747467-1G | NHS ester functionalized 30 nm iron oxide powder |
| K-562 | American Type Culture Collection (ATCC) | CCL-243 | cells |
| Linear Acrylamide (5 mg/mL) | Thermo Fisher | AM9520 | Linear Acrylamide |
| LUNA-FL Dual Fluorescence Cell Counter | Logos Biosystems | L20001 | Cell counter |
| LUNA Cell Counting Slides, 50 Slides | Logos Biosystems | L12001 | Cell counter |
| Mineral oil, BioReagent, for molecular biology, light oil | MilliporeSigma | M5904-500ML | Mineral oil |
| N,N,N′,N′-Tetramethylethylenediamine for molecular biology | MilliporeSigma | T7024-100ML | N,N,N′,N′-Tetramethylethylenediamine |
| NaCl (5 M), RNase-free | Thermo Fisher | AM9760G | 5M NaCl |
| NanoDrop Lite | Thermo Fisher | 2516 | Microvolume spectrophotometer |
| NEST 2 mL 96-Well Deep Well Plate, V Bottom | Opentrons | N/A | 2 mL deep well 96-well plate |
| Non-skirted 96-well PCR plate | Genesee Scientific | 27-405 | 96-well PCR plate |
| NuSive GTG Agarose | Lonza | 50081 | Agarose |
| OmniPur Acrylamide: Bis-acrylamide 19:1, 40% Solution | MilliporeSigma | 1300-500ML | 40%Acrylamide/Bis-acrylamide |
| OT-2 lab robot | Opentrons | OT2 | Automated liquid handling robot |
| Paraformaldehyde, EM Grade, Purified, 20% Aqueous Solution | Electron Microscopy Sciences | 15713 | 20% Pararmaldehyde |
| PBS (10x), pH 7.4 | Thermo Fisher | 70011044 | 10x PBS |
| PIPETMAN Classic P1000 | GILSON | F123602 | A P1000 pipette |
| Protein LoBind Tubes, 1.5 mL | Eppendorf | 925000090 | 1.5 mL Protein low-binding tube |
| QIAgen Gel Extraction kit | Qiagen | 28706 | A P1000 pipette |
| Quant-iT PicoGreen dsDNA Assay | Thermo Fisher | P11495 | dsDNA specific intercalator dye |
| Quick Ligation kit | New England BioLabs | M2200L | T4 DNA ligase (NEB) |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher | 10777019 | RNAse inhibitor |
| S-4FB Crosslinker (DMF-soluble) | Vector laboratories | S-1004-105 | Succinimidyl 4-formylbenzoate (S-4FB) |
| S-HyNic | Vector laboratories | S-1002-105 | Succinimidyl 6-hydrazinonicotinate acetone hydrazone (S-HyNic) |
| Sodium Acetate, 3 M, pH 5.2, Molecular Biology Grade | MilliporeSigma | 567422-100ML | 3M Sodium acetate (pH 5.2) |
| Sodium bicarbonate, 1M buffer soln., pH 8.5 | Alfa Aesar | J60408 | 1M sodium bicarbonate buffer, pH 8.5 |
| Sodium phosphate dibasic for molecular biology | MilliporeSigma | S3264-250G | Na2HPO4 |
| Sodium phosphate monobasic for molecular biology | MilliporeSigma | S3139-250G | NaH2PO4 |
| SuperScript IV reverse transcriptase | Thermo Fisher | 18090050 | Reverse transcriptase |
| SYBR Gold Nucleic Acid Gel Stain (10,000x Concentrate in DMSO) | Thermo Fisher | S11494 | An intercalator dye for DNA |
| T4 DNA Ligase Reaction Buffer | New England BioLabs | B0202S | 10x T4 DNA ligase reaction buffer |
| ThermoPol Reaction Buffer Pack | New England BioLabs | B9004S | 10x TPM-T buffer (Tris-HCl/Pottasium chloride/Magnesium sulfate/Triton X-100) |
| TRIzol LS reagent | Thermo Fisher | 10296-028 | Guanidinium thiocyanate-phenol-chloroform extraction |
| TruSeq Nano DNA library prep kit | Illumina | 20015965 | A DNA library preparation kit (see also the manufacturer's instruction) |
| Ultramer DNA Oligo, Anti-5hmC_Ab#005 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-H3K27ac_Ab#002 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-H3K27me3_Ab#003 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-Med1_Ab#004 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-Pol II_Ab#006 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Control IgG_Ab#001 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for control IgG |
| UltraPure 0.5 M EDTA, pH 8.0 | Thermo Fisher | 15575020 | 0.5M EDTA, pH 8.0 |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fisher | 10977023 | Ultrapure water |
| Zeba Splin Desalting Columns, 7K MWCO, 0.5 mL | Thermo Fisher | 89882 | Desalting column |
References
- Perkel, J. M. Single-cell analysis enters the multiomics age. Nature. 595 (7868), 614-616 (2021).
- Clark, S. J., et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature Communications. 9 (1), 781 (2018).
- Ma, S., et al. Chromatin potential identified by shared single-cell profiling of RNA and Chromatin. Cell. 183 (4), 1103-1116 (2020).
- Zhu, C., et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nature Structural & Molecular Biology. 26 (11), 1063-1070 (2019).
- Zhu, C., et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nature Methods. 18 (3), 283-292 (2021).
- Mimitou, E. P., et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nature Biotechnology. 39 (10), 1246-1258 (2021).
- Ohnuki, H., Venzon, D. J., Lobanov, A., Tosato, G. Iterative epigenomic analyses in the same single cell. Genome Research. 31 (10), 1819-1830 (2021).
- Tosato, G., Ohnuki, H. . Methods of preparing a re-usable single cell and methods fro analyzing the epigenome, transcriptome, and genome of a single cell. , (2021).
- Harada, A., et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nature Cell Biology. 21 (2), 287-296 (2019).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nature Structural & Molecular Biology. 18 (1), 91-93 (2011).
- Marina, R. J., et al. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. EMBO Journal. 35 (3), 335-355 (2016).
- Weast, R. C., Weast, R. C., Astle, M. J., Beyer, W. H., et al. . Handbook of chemistry and physics: a ready-reference book of chemical and physical data. 56th edn. , (1975).
- Aydin, S. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides. 72, 4-15 (2015).
- Gan, S. D., Patel, K. R. Enzyme immunoassay and enzyme-linked immunosorbent assay. Journal of Investigative Dermatology. 133 (9), 12 (2013).
- Porstmann, T., Kiessig, S. T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Chao, H. P., et al. Systematic evaluation of RNA-Seq preparation protocol performance. BMC Genomics. 20 (1), 571 (2019).
- Song, Y., et al. A comparative analysis of library prep approaches for sequencing low input translatome samples. BMC Genomics. 19 (1), 696 (2018).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Ku, W. L., et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nature Methods. 16 (4), 323-325 (2019).
- Carter, B., et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nature Communications. 10 (1), 3747 (2019).
- Maskell, D. P., et al. Structural basis for retroviral integration into nucleosomes. Nature. 523 (7560), 366-369 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved