Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Wiederverwendbare Einzelzelle für iterative epigenomische Analysen
In diesem Artikel
Zusammenfassung
Das vorliegende Protokoll beschreibt eine Einzelzellmethode für iterative epigenomische Analysen unter Verwendung einer wiederverwendbaren Einzelzelle. Die wiederverwendbare Einzelzelle ermöglicht die Analyse mehrerer epigenetischer Markierungen in derselben Einzelzelle und die statistische Validierung der Ergebnisse.
Zusammenfassung
Aktuelle Einzelzell-Epigenomanalysen sind für den einmaligen Gebrauch konzipiert. Die Zelle wird nach einmaliger Verwendung verworfen, wodurch die Analyse mehrerer epigenetischer Markierungen in einer einzelnen Zelle verhindert wird und Daten von anderen Zellen benötigt werden, um das Signal vom experimentellen Hintergrundrauschen in einer einzelnen Zelle zu unterscheiden. In diesem Artikel wird eine Methode beschrieben, mit der dieselbe Einzelzelle für iterative epigenomische Analysen wiederverwendet werden kann.
Bei dieser experimentellen Methode werden zelluläre Proteine zunächst an einem Polyacrylamid-Polymer verankert, anstatt sie mit Protein und DNA zu vernetzen, wodurch strukturelle Verzerrungen verringert werden. Dieser kritische Schritt ermöglicht wiederholte Experimente mit derselben Einzelzelle. Als nächstes wird ein zufälliger Primer mit einer Gerüstsequenz für die Proximity-Ligation an die genomische DNA geglüht, und die genomische Sequenz wird dem Primer durch Erweiterung unter Verwendung einer DNA-Polymerase hinzugefügt. Anschließend werden ein Antikörper gegen einen epigenetischen Marker und Kontroll-IgG, die jeweils mit unterschiedlichen DNA-Sonden markiert sind, an die jeweiligen Ziele in derselben Einzelzelle gebunden.
Die Proximity-Ligation wird zwischen dem Random-Primer und dem Antikörper induziert, indem eine Konnektor-DNA mit komplementären Sequenzen an die Gerüstsequenz des Random-Primers und der Antikörper-DNA-Sonde angehängt wird. Dieser Ansatz integriert Antikörperinformationen und benachbarte Genomsequenzen in einem einzigen DNA-Produkt der Proximity-Ligation. Durch die Ermöglichung wiederholter Experimente mit derselben Einzelzelle ermöglicht diese Methode eine Erhöhung der Datendichte einer seltenen Zelle und eine statistische Analyse, bei der nur IgG- und Antikörperdaten derselben Zelle verwendet werden. Die auf diese Weise hergestellten wiederverwendbaren Einzelzellen können mindestens einige Monate gelagert und später wiederverwendet werden, um die epigenetische Charakterisierung zu erweitern und die Datendichte zu erhöhen. Diese Methode bietet Forschern und ihren Projekten Flexibilität.
Einleitung
Die Single-Cell-Technologie tritt in die Ära der Single-Cell-Multiomics ein, bei der einzelne Single-Cell-Omics-Technologien integriertwerden 1. In jüngster Zeit wurde die Einzelzell-Transkriptomik mit Methoden zum Nachweis der Chromatinzugänglichkeit (scNMT-seq2 und SHARE-seq3) oder Histonmodifikationen (Paired-seq4 und Paired-Tag5) kombiniert. In jüngerer Zeit wurden Einzelzell-Transkriptomik und Proteomik in die Chromatin-Zugänglichkeit integriert (DOGMA-seq6). Diese Methoden verwenden Transposase-basiertes Tagging, um die Zugänglichkeit von Chromatin oder Histonmodifikationen zu erkennen.
Transposase-basierte Ansätze spalten genomische DNA und fügen am Ende des genomischen DNA-Fragments einen DNA-Barcode hinzu. Jedes gespaltene genomische Fragment kann nur bis zu zwei DNA-Barcodes (= eine epigenetische Markierung pro Spaltstelle) aufnehmen, und die genomische DNA an der Spaltstelle geht verloren. Daher haben spaltungsbasierte Ansätze einen Kompromiss zwischen der Anzahl der getesteten epigenetischen Markierungen und der Signaldichte. Dies erschwert die Analyse mehrerer epigenetischer Markierungen in derselben Zelle. Um dieses Problem zu lösen, wurde eine einzelzellige epigenomische Methode entwickelt, bei der die genomische DNA nicht gespalten wird 7,8.
Zusätzlich zu dem oben erwähnten Problem der Spaltung weisen Transposase-basierte Ansätze weitere Einschränkungen auf. Bei der Einzelzell-Epigenomanalyse ist es von entscheidender Bedeutung, die Position von Histonen und DNA-assoziierten Proteinen im Genom zu kennen. In aktuellen Ansätzen wird dies durch die Verwendung unfixierter Einzelzellen und die Beibehaltung von Protein-DNA- und Protein-Protein-Interaktionen erreicht. Dies führt jedoch zu einer starken Verzerrung zugänglicher Chromatinregionen, selbst bei der Analyse von Histonmodifikationen 9. Die Lage von Histonen und genomassoziierten Proteinen auf dem Genom kann ohne Vernetzung von Protein-DNA und Protein-Protein unter Verwendung eines Polyacrylamid-Gerüsts erhalten werden 7,8. Dieser Ansatz reduziert die strukturelle Verzerrung, die bei aktuellen Ansätzen beobachtet wird, die von Protein-DNA- und Protein-Protein-Interaktionen abhängen.
Transposase-basierte Ansätze können Signale nur einmal von einer einzelnen Zelle erfassen. Daher ist es aufgrund des Abfalls der Signale schwierig, das gesamte Epigenom einer einzelnen Zelle abzugrenzen. Wiederverwendbare Einzelzellen wurden entwickelt, um die derzeitigen Einschränkungen zu überwinden, indem sie eine iterative epigenomische Analyse in derselben Einzelzelle ermöglichen.
Protokoll
HINWEIS: Eine schematische Darstellung des Verfahrens ist in Abbildung 1 dargestellt.

Abbildung 1: Schematische Darstellung des Protokoll-Workflows. Die Schritte 7.2-13 werden anhand schematischer Darstellungen erläutert. Jede Zeile gibt einen Schritt im Protokoll an. Ein grün eingefärbtes zelluläres Protein ist ein menschliches Nukleosom, das auf der Grundlage einer Kristallstruktur (PDB: 6M4G) erzeugt wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
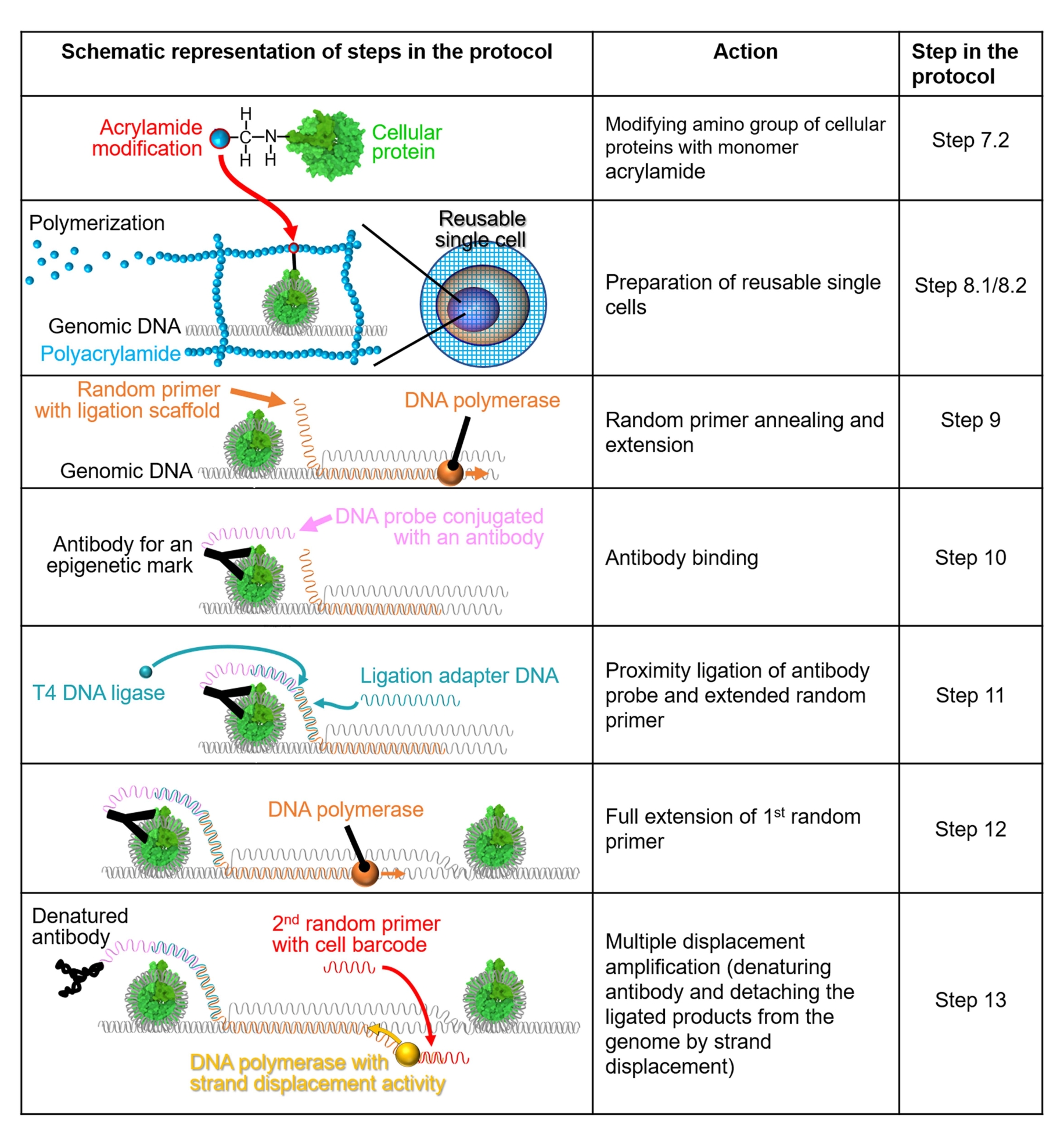{kind=link}
1. Gleichgewicht der Entsalzungskolonnen
HINWEIS: Entsalzungs-Spin-Säulen werden wie in den folgenden Schritten beschrieben ausgeglichen. Die äquilibrierten Entsalzungskolonnen werden in den Schritten 2.1, 3.4 und 4.6 verwendet.
- Entfernen Sie den unteren Verschluss einer Entsalzungskolonne (7 kDa Cut-off, 0,5 mL Harzraupenvolumen, siehe Materialtabelle) und lösen Sie die Kappe an der Oberseite der Entsalzungskolonne.
- Legen Sie die Säule in ein 1,5-ml-Proteinröhrchen mit niedriger Bindung (ein Sammelröhrchen, siehe Materialtabelle) und zentrifugieren Sie bei 1.500 × g für 1 Minute bei Raumtemperatur, um die Speicherlösung in der Säule zu entfernen.
Anmerkungen: Verwenden Sie eine Ausschwingrotorzentrifuge, um die Oberseite des Perlenbetts zu glätten. - Entfernen Sie den Durchfluss im 1,5-ml-Röhrchen und geben Sie 300 μl 150 mM NaCl/100 mM Phosphatpuffer mit einem pH-Wert von 8,0 (siehe Tabelle 1) auf das Harzbett.
- Bei 1.500 × g 1 min bei Raumtemperatur zentrifugieren und den Durchfluss im Auffangröhrchen entfernen.
- Wiederholen Sie die Schritte 1.3-1.4 noch dreimal.
- Entsorgen Sie den Puffer aus dem Auffangröhrchen und setzen Sie die Säule in ein neues Auffangröhrchen ein.
- Verwenden Sie die äquilibrierte Entsalzungskolonne in den Schritten 2.1, 3.4 und 4.4.
2. Pufferaustausch von Antikörpern
Anmerkungen: Entfernen Sie Glycerin, Arginin und Natriumazid aus Anti-H3K27ac 10, Anti-H3K27me3 10, Anti-Med111 und Anti-PolII 10 (siehe Pufferzusammensetzung in Tabelle 1). Alle folgenden Verfahren werden unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 1 h
- Tragen Sie die Antikörperlösung (100 μl, siehe Tabelle 2) auf eine äquilibrierte Entsalzungssäule auf, die nach Schritt 1 vorbereitet wurde.
- Zentrifugieren Sie bei 1.500 × g für 2 min bei 4 °C und übertragen Sie den Durchfluss aus dem Sammelröhrchen in ein 1,5-ml-Proteinröhrchen mit niedriger Bindung.
- Messen Sie die IgG-Konzentration anhand der Absorption bei 280 nm12 (verwenden Sie ein Mikrovolumen-Spektralphotometer).
- Die Antikörperlösung wird in eine Ultrafiltrationskassette (Molekulargewichtsgrenzwert 100 kDa, 0,5 ml, siehe Materialtabelle) überführt und bei 12.000 × g für 5 min bei 4 °C zentrifugiert.
- Messen Sie die IgG-Konzentration anhand der Absorption bei 280 nm.
- Wiederholen Sie die Schritte 2.4-2.5, bis die IgG-Konzentration 1 mg/ml erreicht.
3. Aktivierung von Antikörpern
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 2,5 h
- Lösen Sie 1 mg Succinimidyl-6-hydrazinonicotinat-Acetonhydrazon (S-HyNic, siehe Stofftabelle) mit 100 μl wasserfreiem N,N-Dimethylformamid (DMF, siehe Stofftabelle) auf.
HINWEIS: DMF ist ein brennbares organisches Lösungsmittel und ein starkes Lebergift, das über die Haut aufgenommen wird. Tragen Sie Handschuhe, eine Schutzbrille und einen Laborkittel. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - 0,6 μl S-HyNic/DMF zu 100 μl der Antikörperlösung (1 mg/ml in 150 mM NaCl/100 mM Natriumphosphat, pH8,0. Siehe Tabelle 2 für die verwendeten Antikörper und Kontroll-IgG.
- Bei Raumtemperatur 2 h inkubieren (lichtgeschützt).
- Tragen Sie 100 μl S-HyNic-reagierten Antikörper auf die Oberseite der äquilibrierten Entsalzungssäule auf (siehe Schritt 1) und zentrifugieren Sie bei 1.500 × g für 2 min bei 4 °C, um die Probe zu entnehmen.
- Entsorgen Sie die Entsalzungskolonne nach Gebrauch.
HINWEIS: Die Stabilität von HyNic-Gruppen auf Proteinen und anderen Biomolekülen variiert. Es wird empfohlen, HyNic-modifizierte Biomoleküle sofort zu konjugieren.
4. Aktivierung der DNA-Sonde
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 2,5 h
- Lösen Sie ein Pellet einer aminmodifizierten DNA-Sonde für Antikörper- oder Kontroll-IgG (Antikörpersonde, Tabelle 3) mit 20 μl 150 mM NaCl/100 mM Natriumphosphatpuffer, pH 8,0, auf.
Anmerkungen: Achten Sie auf ein dünnes, transparentes Pellet/eine dünne Folie am Boden des Röhrchens. Tragen Sie den Puffer direkt auf das Pellet auf. Wenn das Pellet nicht sichtbar ist, hat sich das Pellet möglicherweise gelöst und an der Wand oder dem Deckel haftet. Zentrifugieren Sie in diesem Fall das Röhrchen und suchen Sie nach einem Pellet am Boden des Röhrchens. - Lösen Sie 1 mg Succinimidyl-4-formylbenzoat (S-4FB, siehe Materialtabelle) mit 50 μl wasserfreiem DMF auf und geben Sie 10 μl DMF in die gelöste Antikörpersonde.
- Fügen Sie 4 μl S-4FB/DMF hinzu, mischen Sie es und inkubieren Sie es 2 h lang bei Raumtemperatur (vor Licht schützen).
- Tragen Sie 34 μl S-4FB-reagierte Antikörpersonde auf die Oberseite der äquilibrierten Entsalzungssäule auf (siehe Schritt 1).
- Tragen Sie 15 μl 150 mM NaCl/100 mM Natriumphosphatpuffer, pH 8,0, auf die Oberseite des Gelbetts auf, nachdem die Probe vollständig absorbiert wurde, und zentrifugieren Sie bei 1.500 × g für 2 min bei 4 °C, um die S-4FB-modifizierte Antikörpersonde zu sammeln.
- Verwenden Sie den Durchfluss für die nachfolgende Konjugation. Messung der Absorption bei 260 nm; und berechnen Sie die Wiederfindungsrate der Antikörpersonde.
5. Konjugation von S-HyNic-modifiziertem Antikörper und S-4FB-modifizierter Antikörpersonde
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 2 h
- Mischen Sie den S-HyNic-modifizierten Antikörper und die S-4FB-modifizierte Antikörpersonde und pipettieren Sie die Lösung zum Mischen auf und ab.
- 2 h bei Raumtemperatur (lichtgeschützt) inkubieren.
- Um die Reaktion zu unterdrücken, werden 478,8 μl Abschreck- und Lagerlösung hinzugefügt (siehe Tabelle 1).
- Die Antikörpersonden-konjugierte Antikörperlösung wird in eine Ultrafiltrationskassette (Molekulargewichtsgrenzwert 100 kDa) überführt.
- 5 min bei 12.000 × g, 4 °C zentrifugieren.
- Überprüfen Sie das Volumen der Lösung in der Kassette durch Pipettieren.
- Wiederholen Sie die Schritte 5.5-5.6, bis das Volumen 100 μL erreicht hat, und lagern Sie es bei -20 °C.
HINWEIS: Die IgG-Konzentration wird mit einem Sandwich-ELISA13,14,15 unter Verwendung eines IgG-Standards gemessen.
6. Vorbereitung der magnetischen Kernperlen
HINWEIS: Bei dieser Methode wird eine einzelne Zelle in eine zweischichtige Acrylamidperle eingebettet (siehe Abbildung 2). Der Kern ist eine magnetische Polyacrylamidperle. Die äußere Schicht besteht ausschließlich aus Polyacrylamid. In diesem Abschnitt werden die magnetischen Kernperlen erzeugt. Dieser Abschnitt ist für das Experiment nicht unbedingt erforderlich. Dauer: 3 h

Abbildung 2: Struktur eines zweilagigen Polyacrylamid-Beads für Sichtbarkeit und einfache Handhabung in REpi-seq-Experimenten . (A) Magnetische Nanopartikel aus Schritt 6.6 nach der Zentrifugation. Die magnetischen Nanopartikel werden mit monomerem Acrylamid modifiziert und in die in B gezeigte Polyacrylamid-Magnetperle integriert. (B) Schematische Darstellung einer wiederverwendbaren Einzelzelle mit einer Polyacrylamid-Magnetperle. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
- Mischen Sie 50 μl 1 M Natriumbicarbonatpuffer, pH 8,5, und 450 μl 40%ige Acrylamidlösung.
HINWEIS: Acrylamid ist ein Nervengift. Tragen Sie Handschuhe, einen Augenschutz und einen Laborkittel. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - 1 g NHS-ester-funktionalisiertes 30 nm Eisenoxidpulver (siehe Materialtabelle) im Acrylamid/Natriumhydrogencarbonat-Puffer suspendieren und über Nacht bei 4 °C inkubieren.
Anmerkungen: Da Acrylamidperlen transparent sind, kann es schwierig sein, die Position der Perlen zu sehen und zu manipulieren. Die Aufnahme einer Kernperle aus Eisenoxid verbessert die Sichtbarkeit und erleichtert die Manipulation, da die Position der Perlen mit einem Magneten gesteuert werden kann. Wenn die Benutzer jedoch mit den REpi-seq-Experimenten vertraut sind, kann auf die Verwendung der magnetischen Polyacrylamid-Kernkügelchen verzichtet werden. - Die Nanobead-Suspension wird in 2 Röhrchen (1,5 ml) überführt, bei 4 °C bei 21.300 × g zentrifugiert (abgewinkelter Rotor verwenden) und den Überstand entfernt.
- Suspendieren Sie die untere Aufschlämmung mit 1 ml 40%igem Acrylamid/Bisacrylamid (19:1, siehe Materialtabelle).
HINWEIS: Bisacrylamid ist ein Nervengift. Tragen Sie Handschuhe und einen Laborkittel. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - Zentrifugieren Sie bei 21.300 × g für 1 h (mit abgewinkeltem Rotor) bei 4 °C.
- Zentrifugieren Sie bei 5.000 × g für 30 min (verwenden Sie einen Schwungrotor ohne Bremse) bei 4 °C.
- Entfernen Sie den Überstand mit einer P1000-Pipette mit langsamer Aspiration.
- Stellen Sie das Volumen auf 400 μl 40 % Acrylamid/Bis-Acrylamid ein (19:1, siehe Materialtabelle).
- Fügen Sie 25 μl 10%ige Ammoniumpersulfatlösung hinzu (siehe Tabelle 1).
Anmerkungen: Ammoniumpersulfat ist ein starkes Oxidationsmittel. Staub in der Luft, der Ammoniumpersulfat enthält, kann bei Kontakt Augen, Nase, Rachen, Lunge und Haut reizen. Tragen Sie Handschuhe, eine Schutzbrille und einen Laborkittel. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - Um magnetische Polyacrylamid-Kernkügelchen zu erzeugen, werden 0,5 μl der acrylamidmodifizierten Eisenoxid-Suspension in ein PCR-Röhrchen übertragen.
- 50 μl 4% N,N,N',N'-Tetramethylethylendiamin/Mineralöl (TEMED, siehe Tabelle 1) zugeben und über Nacht bei Raumtemperatur inkubieren.
HINWEIS: TEMED ist ein brennbares Lösungsmittel. Arbeiten Sie unter einer Haube. Nicht einatmen. Von offenen Flammen, heißen Oberflächen und Zündquellen fernhalten. Tragen Sie Handschuhe, eine Schutzbrille und einen Laborkittel. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien.
7. Modifikation der Aminogruppe zellulärer Proteine mit Monomer Acrylamid
HINWEIS: REpi-seq wurde entwickelt, um das Epigenom von Maus- und menschlichen Zellen auf Einzelzellebene zu analysieren. Jeder Schritt muss optimiert werden, wenn diese Methode auf Zellen anderer Spezies als Maus oder Mensch angewendet wird.
- Ernte der Zellen
HINWEIS: Dauer: 30 min- Messen Sie die Zellkonzentration und Viabilität mit einem Zellzähler (siehe Materialtabelle).
HINWEIS: Die Viabilität der Zellen in diesem Schritt wirkt sich darauf aus, wie viele Zellen in der Datenanalyse lebende Zellen sind. - Stellen Sie die Zellkonzentration auf 1 × 105 Zellen/ml mit Nährmedium (*) ein, das 10 % fötales Kälberserum enthält.
HINWEIS: *Das Nährmedium ist ein optimales Nährmedium für die zu untersuchenden Zellen. - Übertragen Sie 1 ml der Zellsuspension in ein 1,5-ml-Röhrchen.
- Die Zellsuspension bei 240 × g für 5 min bei 4 °C zentrifugieren und den Überstand entfernen.
- Fügen Sie 1 ml phosphatgepufferte Kochsalzlösung (PBS) hinzu, mischen Sie die Zellen durch sanftes Pipettieren und zentrifugieren Sie die Zellsuspension bei 240 × g für 5 Minuten bei 4 °C.
- Entfernen Sie den Überstand.
Anmerkungen: Alle oben genannten Schritte sollten in einer Laminar-Flow-Clean-Haube durchgeführt werden, um eine Kontamination zu vermeiden.
- Messen Sie die Zellkonzentration und Viabilität mit einem Zellzähler (siehe Materialtabelle).
- Modifikation der Aminogruppe zellulärer Proteine mit monomerem Acrylamid
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 1,5 h- Geben Sie 1 ml Aminogruppenmodifikationslösung (siehe Tabelle 1) in das Zellpellet und suspendieren Sie das Zellpellet durch sanftes Pipettieren.
- Das Röhrchen wird 1 h auf Eis inkubiert und die Zellsuspension bei 240 × g für 5 min bei 4 °C zentrifugiert.
- Entfernen Sie den Überstand und resuspendieren Sie die Zellen mit 100 ml 4%igem Acrylamid/1 mM EDTA/PBS, das einen Interkalatorfarbstoff für DNA enthält (siehe Tabelle 1, 1 Zelle/μl).
8. Aufbereitung von wiederverwendbaren Einzelzellen
- Aufbereitung von wiederverwendbaren Einzelzellen (manuelle Version)
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 9 h/96 Zellen- Mischen Sie 1 ml der Zellsuspension (1 Zelle/μl) und 199 ml 1 mM EDTA/PBS, das einen Interkalatorfarbstoff für DNA enthält (siehe Tabelle 1).
- Übertragen Sie 200 μl der Zellsuspension in jede Vertiefung von 96-Well-Platten mit flachem Boden (insgesamt 10 Platten, siehe Materialtabelle).
- Setzen Sie die Abdeckung auf die 96-Well-Platte und scannen Sie die 10 Platten mit einem Rastermikroskop (siehe Materialtabelle), um Wells zu identifizieren, die eine einzelne Zelle enthalten.
- Übertragen Sie den Inhalt der Vertiefung, die eine einzelne Zelle enthält, in ein PCR-Röhrchen.
- Kippen Sie die Platte (in Richtung des Bedieners) und warten Sie einige Minuten, bis die einzelne Zelle an den unteren Rand des Bohrlochs gesunken ist.
- Setzen Sie die Pipettenspitze auf die untere Ecke und übertragen Sie die einzelne Zelle und den Puffer (Aspirat 210 μL/Well = 200 μL Puffer + 10 μL Luft) in ein PCR-Röhrchen.
Anmerkungen: Achten Sie darauf, eine P200-Spitze mit geringer Retention zu verwenden. - Überprüfen Sie die Vertiefung mit einem Fluoreszenzmikroskop, um die Übertragung zu bestätigen.
- Wenn sich noch eine einzelne Zelle in der Vertiefung befindet, fügen Sie 200 μL/Well 1 mM EDTA/PBS hinzu, die einen Interkalatorfarbstoff für die DNA enthält.
- Wiederholen Sie dann die Übertragung.
- Zentrifugieren Sie das PCR-Röhrchen bei 240 × g für 5 min bei 4 °C mit einem Schwenkrotor ohne Bremse.
Anmerkungen: Das Bremsen verursacht einen wirbelnden Wasserstrom in der Röhre, der die Ausfällung der einzelnen Zelle stört. Beim Zentrifugieren mit einem schwingenden Rotor ohne Bremse sinkt die einzelne Zelle immer auf den Boden des Rohres. Wird jedoch ein abgewinkelter Rotor verwendet, heftet sich die einzelne Zelle an die Seitenwand des PCR-Röhrchens und kann verloren gehen. - Entfernen Sie 195 μl des Überstands mit einer sehr langsamen Pipettiergeschwindigkeit.
- Fügen Sie 195 μl/Tube Acrylamid/Bis-acrylamid/APS-Lösung hinzu (siehe Tabelle 1).
- Zentrifugieren Sie das PCR-Röhrchen bei 240 × g für 5 min bei 4 °C mit einem Schwenkrotor ohne Bremse.
- Entfernen Sie 195 μl des Überstands mit sehr langsamer Pipettiergeschwindigkeit.
- Übertragen Sie die unteren 3 μl in das PCR-Röhrchen mit 4 % TEMED/Mineralöl und einem magnetischen Polyacrylamid-Bead (generiert in Schritt 6).
Anmerkungen: Achten Sie darauf, eine P10-Spitze mit geringer Retention zu verwenden. Dosieren Sie die Zelle in der Nähe der magnetischen Polyacrylamidperle. Im Mineralöl haftet die Flüssigkeit, die die Einzelzelle enthält, durch Oberflächenspannung an der Oberfläche der magnetischen Polyacrylamidperle. - Über Nacht bei Raumtemperatur inkubieren.
Anmerkungen: Bei diesem Verfahren wird eine zweischichtige Acrylamid-Gelperle erzeugt. Der Kern ist eine magnetische Gelperle. Die äußere Schicht besteht aus Polyacrylamid-Gel, das eine einzelne Zelle enthält. Die einzelne Zelle ist in die äußere Schicht des Gels eingebettet. Sicherer Haltepunkt: Nach dem Waschen der wiederverwendbaren Einzelzellen mit 50% Glycerin/1 mM EDTA/0,05% Tween 20/0,5% BSA/TBS Puffer können die wiederverwendbaren Einzelzellen bei -20 °C bis zu 6 Monate gelagert werden.

Abbildung 3: Automatisches Einzelzell-Picking und Transfer in eine 96-Well-PCR-Platte in Schritt 8.2 . (A) Überblick über ein Einzelzell-Picking-System. Ein Einzelzellen-Picking-Roboter befindet sich in einer Laminar-Flow-Reinigungshaube, um eine Kontamination zu vermeiden. (B) Eine 24-Well-Platte mit 4 nL-Nanowells im Inneren des Wells. (C) Zellverteilung in einer Vertiefung von der 24-Well-Platte. Grüne Punkte sind Zellen, die als eine einzelne Zelle in jedem 4-ml-Nanowell identifiziert werden. Magenta-Punkte sind Zellen, die als Dubletten oder Multipletts von Zellen identifiziert werden. (D) Hellfeldbild des Wells in der 24-Well-Platte. Ein grünes Quadrat ist ein 4-nL-Nanotopf, das eine einzelne Zelle enthält. Ein magentafarbenes Quadrat ist ein 4-nL-Nanotopf, das mehrere Zellen enthält. (E) Vergrößertes Feld von einigen 4 nL Nanowells. Helle Punkte sind einzelne Zellen in 4 nL Nanowells. Das Einzelzell-Picking-System identifiziert Nanowells, die eine einzelne Zelle enthalten, indem es Hellfeld- und Fluoreszenzbilder von Zellen mit DAPI-Färbung aufnimmt. Identifizierte Einzelzellen werden aus dem 4 nL Nanowell in ein Well einer 96-Well PCR Platte überführt. Maßstabsbalken = 2 mm (C, D), 100 μm (E). Abkürzung = DAPI = 4',6-Diamidino-2-phenylindol. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Erzeugung wiederverwendbarer Einzelzellen mit Hilfe eines Liquid-Handling-Roboters. (A) Ein Deck des Liquid-Handling-Roboters. Das Deck verfügt über 11 Steckplätze für Pipettenspitzengestelle (P300-Spitze: Schlitze 1-3, P20-Spitze: Schlitze 5-6), 2-ml-Deep-Well-96-Well-Platte (Schlitz 4), zwei 96-Well-PCR-Platten mit einer einzelnen Zelle pro Well (Schlitze 7 und 10) und zwei 96-Well-Platten mit flachem Boden für flüssige Abfälle (Schlitze 8 und 11). (B) Das Deck nach dem Platzieren der Laborgeräte. (C) Schematische Darstellung des robotergestützten Pipettierens in Schritt 8.8.1. Das Programm entfernt den Überstand, ohne eine einzige Zelle vom Boden der 96-Well-PCR-Platte abzusaugen. (D) Wiederverwendbare Einzelzellen, die unter Verwendung des Ergänzungscodes 1 generiert wurden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
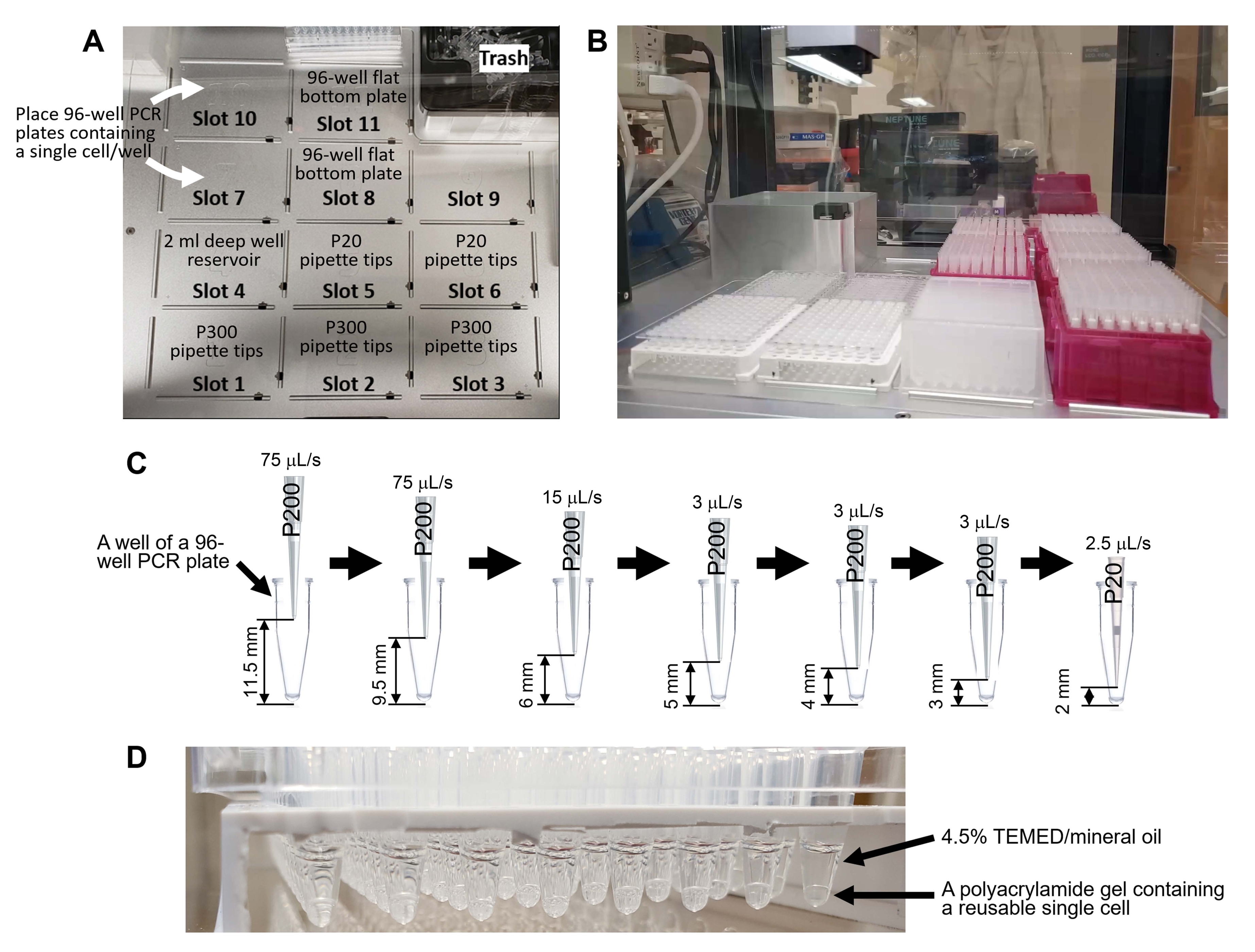{kind=link}
9. Zufälliges Glühen und Verlängern des Primers
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Sternchen (*) bei den folgenden Schritten zeigen an, dass ein Magnetabscheider verwendet werden kann, um die Position der Polyacrylamidperlen zu steuern, die eine wiederverwendbare Einzelzelle im Röhrchen enthalten. Der Einsatz des Magnetabscheiders ist jedoch nicht zwingend erforderlich. Durch langsames Herunterbewegen der Pipettenspitze entlang der Röhrchenwand werden die Kügelchen zum Waschen oder Pufferaustausch nach oben gedrückt. Dauer: 9 h
- Entfernen Sie das Mineralöl durch Pipettieren(*) und waschen (*) Sie 5 Mal mit 200 μL 1x TP Mg(-)-Puffer (verdünnen Sie 10x TP Mg(-)-Puffer auf 1x Puffer mit Reinstwasser, siehe Tabelle 1).
- Entfernen Sie den Überstand (*) und fügen Sie 15 μl Glühpuffer hinzu (siehe Tabelle 1).
- 1 h auf Eis inkubieren.
HINWEIS: Der Zweck dieser Inkubation besteht darin, das Plasma und die Kernmembranen zu permeabilisieren und den zufälligen Primer an den Zellkern abzugeben. - Legen Sie die PCR-Röhrchen auf einen Thermocycler und erhitzen Sie sie 3 Minuten lang auf 94 °C.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ungefähr 20 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. - Übertragen Sie die Röhrchen auf einen eisgekühlten Metallblock und inkubieren Sie sie 2 Minuten lang.
- Fügen Sie 4 μlMgSO4/NaCl/dNTP-Mischung hinzu (siehe Tabelle 1) und mischen Sie mit einem Wirbelmischer bei mittlerer Geschwindigkeit.
- Fügen Sie 1 μl Bst-Polymerase und ein großes Fragment hinzu (siehe Materialtabelle) und mischen Sie es mit einem Wirbelmischer bei mittlerer Geschwindigkeit.
- 4 h auf einem Schüttler (600 U/min bei 4 °C) inkubieren.
HINWEIS: Der Zweck dieser Inkubation besteht darin, die Bst-Polymerase in den Zellkern zu bringen. - Legen Sie das PCR-Röhrchen auf einen Thermocycler und führen Sie eines der folgenden Programme aus.
- Führen Sie ein 4-stündiges Programm durch: 10 °C für 30 Minuten, 20 °C für 30 Minuten und 25 °C für 180 Minuten.
- Alternativ können Sie ein Nachtprogramm durchführen: 4 °C für 4 h, 10 °C für 2 h, 20 °C für 2 h, 25 °C für 4 h und bei 4 °C halten.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ca. 25 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. Sicherer Haltepunkt: Stoppen Sie das Experiment hier für bis zu 1 Tag, indem Sie die wiederverwendbaren Einzelzellen bei 4 °C lagern.
10. Antikörperbindung
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 1,5 h
- Fügen Sie 1,625 μl NaCl/EDTA/BSA-Lösung hinzu (siehe Tabelle 1) und mischen Sie durch Vortexen bei niedriger Geschwindigkeit.
HINWEIS: Dieser Schritt zielt darauf ab, 1) die Chelatierung von Mg2+ durch EDTA zu erleichtern, 2) die Bindung des erweiterten 1. zufälligen Primers durch 300 mM NaCl zu stabilisieren und 3) die unspezifische Bindung von Antikörpern mit Rinderserumalbumin (BSA) in der nachfolgenden Reaktion zu blockieren. - 1 Stunde auf Eis inkubieren und jeweils 0,1 μg/ml Antikörper und Kontroll-IgG konjugiert mit Antikörpersonde (in Schritt 5 hergestellt) zugeben.
- Über Nacht auf Eis unter leichtem Schütteln auf einem Shaker inkubieren.
11. Proximity-Ligation von Antikörpersonde und proximal erweitertem Zufallsprimer
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Sternchen (*) in den folgenden Schritten zeigen an, wo ein Magnetabscheider verwendet werden kann, um die Position von Polyacrylamid-Kügelchen zu steuern, die eine wiederverwendbare Einzelzelle im Röhrchen enthalten. Der Einsatz des Magnetabscheiders ist jedoch nicht zwingend erforderlich. Durch langsames Herunterbewegen der Pipettenspitze entlang der Röhrchenwand können die Kügelchen zum Waschen oder Pufferaustausch nach oben gedrückt werden. Dauer: 6 h
- Waschen (*) zweimal mit 200 μL 1x TPM-T-Puffer (20 min Inkubation in jeder Wäsche) auf Eis. 10x TPM-T-Puffer (Tris-HCl/Kaliumchlorid/Magnesiumsulfat/Triton X-100) auf 1x mit Reinstwasser verdünnen).
- Entfernen (*) Sie den Überstand und waschen (*) einmal mit 1x T4-DNA-Ligase-Puffer (siehe Materialtabelle).
- Entfernen (*) Sie den Überstand und fügen Sie 19 μl Ligationsadapterlösung hinzu (siehe Tabelle 1).
- 1 h bei 25 °C inkubieren.
- Fügen Sie 1 μL T4-DNA-Ligase hinzu (siehe Materialtabelle) und mischen Sie das Röhrchen auf einem Wirbelmischer bei mittlerer Geschwindigkeit.
- Legen Sie das Röhrchen auf einen Thermocycler und führen Sie das folgende Programm durch: Proximity-Ligationsprogramm, 4 h bei 16 °C und 30 min bei 25 °C.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ca. 25 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels entspricht Raumtemperatur. - Waschen (*) zweimal kurz mit 200 μL 1x Bst Mg(-)EDTA(+)-Puffer (siehe Tabelle 1; bereiten Sie 1x Puffer aus 10x Stammpuffer vor) und lagern Sie die Zelle bei 4 °C über Nacht. Sicherer Haltepunkt: Stoppen Sie das Experiment hier für bis zu 1 Tag, indem Sie die wiederverwendbaren Einzelzellen bei 4 °C lagern.
12. Vollständige Erweiterung des 1. zufälligen Primers
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 4,5 h
- Zweimal mit 200 μL 1x Bst Mg(-) EDTA(-)-Puffer waschen (siehe Tabelle 1, 1x Puffer aus 10x Stammpuffer vorbereiten), Überstand entfernen und 20 μL Bst/dNTPs/MgSO4-Gemisch hinzufügen (siehe Tabelle 1).
- Mit einem Wirbelmischer bei mittlerer Drehzahl mischen und 4 h auf einem Orbitalschüttler (bei 6 °C und 500 U/min) inkubieren.
- Legen Sie das Röhrchen auf einen Thermocycler und führen Sie das folgende Programm aus: Vollauszugsprogramm: 10 °C für 1 h, 20 °C für 1 h, 30 °C für 1 h, 40 °C für 1 h, 50 °C für 1 h, 65 °C für 1 h, 94 °C für 10 min und bei 4 °C halten.
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier für bis zu 1 Tag gestoppt werden, indem die wiederverwendbaren Einzelzellen bei 4 °C gelagert werden.
13. Mehrfache Auslenkungsverstärkung
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 2,5 h (Schritte 13.1-13.2) + 15 min (Schritte 13.3-13.4) + 1 Tag (Schritte 13.5-13.10)
- Fügen Sie 0,4 μL 100 μM2 Random Primer hinzu (siehe Tabelle 3) und mischen Sie mit einem Wirbelmischer bei mittlerer Geschwindigkeit.
- 2 h bei 6 °C und 500 U/min auf einem Orbitalschüttler inkubieren, das Röhrchen auf einen Thermocycler legen und 3 min bei 94 °C erhitzen.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ungefähr 25,4 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. - Legen Sie die Röhrchen in einen eisgekühlten Metallblock und fügen Sie 1 μl/Röhrchen Bst-DNA-Polymerase hinzu.
- Bewegen Sie die Röhrchen bei langsamer Geschwindigkeit, legen Sie sie auf einen Thermocycler und führen Sie das folgende Programm aus:
4 °C für 4 h, 10 °C für 30 min, 20 °C für 30 min, 30 °C für 30 min, 40 °C für 30 min, 50 °C für 30 min, 65 °C für 60 min, 94 °C für 3 min und bei 4 °C halten.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ungefähr 26,4 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. Sicherer Haltepunkt: Stoppen Sie das Experiment hier für bis zu 1 Tag, indem Sie die wiederverwendbaren Einzelzellen bei 4 °C lagern. - Sammeln Sie den Überstand (ca. 20 μl) und übertragen Sie ihn in ein PCR-Röhrchen.
- Lagern Sie den gesammelten Überstand bei -80 °C.
- Geben Sie 20 μl/Röhrchen 0,05 % Tween 20/0,1x TE-Puffer in ein PCR-Röhrchen, das die wiederverwendbare Einzelzelle enthält (siehe Tabelle 1), und inkubieren Sie die wiederverwendbare Einzelzelle bei 4 °C über Nacht. Sicherer Haltepunkt: Stoppen Sie das Experiment hier für einige Tage, indem Sie die Inkubationszeit verlängern.
- Sammeln Sie den Überstand und kombinieren Sie ihn mit der in Schritt 13.5 entnommenen Probe.
- Wiederholen Sie die Schritte 13.7-13.8 noch einmal. Die wiederverwendbare Einzelzelle wird in 50 % Glycerin/5 mM EDTA/0,5 % BSA/0,05 % Tween20/TBS-Puffer eingeweicht und 30 min auf einem Orbitalschüttler (4 °C, 600 U/min) inkubiert. Lagern Sie die wiederverwendbare Einzelzelle bis zum nächsten Experiment bei -20 °C.
- Fügen Sie 40 μl Exo-Mastermix hinzu (siehe Tabelle 1), legen Sie das Röhrchen auf einen Thermocycler und führen Sie das folgende Programm aus: i) 95 °C für 5 min, ii) 95 °C für 30 s, iii) 60 °C 30 s, iv) 72 °C für 30 s, v) Wiederholen Sie die Schritte ii-iv 19 Mal, vi) 72 °C für 5 min, vii) bei 4 °C halten.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt ungefähr 45 μl, einschließlich des Volumens der Polyacrylamidperle. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. Sicherer Haltepunkt: Das Experiment kann hier für mindestens einige Tage gestoppt werden, indem die Probe bei -80 °C gelagert wird.
14. Phenol-Chloroform-Reinigung und Polyethylenglykol-Fällung
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 1,5 h
- Übertragen Sie das Produkt in ein 0,5-ml-DNA-Röhrchen mit niedriger Bindung und fügen Sie 100 μl Phenol:Chloroform:Isoamylalkohol hinzu.
Anmerkungen: Phenol:Chloroform:Isoamylalkohol verursacht Reizungen und kann bei Kontakt zu Verbrennungen führen. Tragen Sie Handschuhe, eine Schutzbrille und einen Laborkittel. Nur bei ausreichender Belüftung verwenden oder eine geeignete Atemschutzmaske tragen. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - 30 s von Hand schütteln und bei 12.000 × g 10 min bei 4 °C zentrifugieren.
- Sammeln Sie die wässrige Phase (80 μl) in einem 0,5-ml-DNA-Röhrchen mit geringer Bindung und fügen Sie 40,84 μl/Röhrchen lineares Acrylamid/MgCl2-Gemisch hinzu (siehe Tabelle 1).
- Fügen Sie 47,06 μl/Röhrchen 50 % (w/v) PEG8000 (RNase-frei) hinzu und mischen Sie durch Pipettieren.
- 20 min bei Raumtemperatur inkubieren und bei 240 × g 10 min bei Raumtemperatur zentrifugieren.
- Entfernen Sie den Überstand und fügen Sie 400 μl/Röhrchen mit 80%igem Ethanol (EtOH) hinzu.
- Mit 80 % EtOH waschen, den Überstand mit einem Absauger entfernen und das Pellet an der Luft trocknen lassen.
- Das Pellet wird mit 20 μl 1 mM EDTA/10 mM Tris-HCl, pH 7,4 Puffer suspendiert und die Lösung bei -80 °C gelagert.
Anmerkungen: Messen Sie die DNA-Konzentration mit einem doppelsträngigen DNA-spezifischen Interkalatorfarbstoff (siehe Materialtabelle). Sicherer Haltepunkt: Das Experiment kann hier sicher für mindestens eine Woche gestoppt werden.
15. In-vitro-Transkription
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase- und RNase-Kontamination zu vermeiden. Dauer: 5 h
- Tauen Sie die DNA von Einzelzellprodukten auf (ab Schritt 14) und stellen Sie eine gemischte Bibliothek von Einzelzellprodukten her, indem Sie 2 μL/Zelle der DNA-Produkte mischen (Gesamtvolumen 20 μL).
- Fügen Sie 26 μl In-vitro-Transkriptions-Mastermix hinzu (siehe Materialtabelle und Protokoll des Herstellers) und mischen Sie durch Pipettieren.
- Legen Sie das PCR-Röhrchen auf einen Thermocycler und inkubieren Sie es 4 h lang bei 37 °C.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt 46 μL. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. - Fügen Sie 5 μl 10x DNase I-Puffer hinzu (siehe Materialtabelle) und fügen Sie 4 μl DNase I (RNase-frei, 4 HE, siehe Materialtabelle) hinzu.
- Mischen und 15 Minuten bei 37 °C inkubieren und die Probe in ein 0,5-ml-Röhrchen überführen.
- Fügen Sie 300 μl/Röhrchen Guanidiniumthiocyanat-Phenol-Chloroform hinzu (siehe Materialtabelle) und mischen Sie es durch sanftes Vortexen.
HINWEIS: Guanidiniumthiocyanat-Phenol-Chloroform kann bei Kontakt zu schweren Verätzungen führen. Tragen Sie Handschuhe, eine Schutzbrille und einen Laborkittel. Nur bei ausreichender Belüftung verwenden oder eine geeignete Atemschutzmaske tragen. Befolgen Sie die institutionellen Sicherheitsrichtlinien. Entsorgen Sie die gebrauchten Laborgeräte gemäß den institutionellen Richtlinien. - Inkubieren Sie für 5 min bei R.T. auf einem Schüttler und lagern Sie die Proben dann bis zu 3 Tage bei -80 °C.
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier für bis zu 3 Tage gefahrlos gestoppt werden.
16. RNA-Aufreinigung
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase- und RNase-Kontamination zu vermeiden. Dauer: 2 h
- Geben Sie 80 μl/Röhrchen Chloroform in die Probe aus Schritt 15.7 und schütteln Sie das Röhrchen 15 s lang kräftig von Hand.
- 2-15 min bei Raumtemperatur inkubieren und die Probe bei 12.000 × g für 15 min bei 4 °C zentrifugieren.
- Sammeln Sie die wässrige Phase der Probe (~200 μl) und übertragen Sie sie in ein neues 1,5-ml-Röhrchen.
- Fügen Sie 600 μl/Röhrchen Guanidiniumthiocyanat-Phenol-Chloroform hinzu und überführen Sie die Probe in ein 1,5-ml-Röhrchen.
- Fügen Sie 180 μL/Röhrchen Chloroform hinzu und schütteln Sie das Röhrchen 15 s lang kräftig von Hand.
- Zentrifugieren Sie die Probe bei 12.000 × g für 10 min bei 4 °C.
- Sammeln Sie die wässrige Phase der Probe (~450 μL) und übertragen Sie sie in ein 1,5-ml-Röhrchen.
- Fügen Sie 60 μl/Tube lineares Acrylamid (5 μg/μl) hinzu und fügen Sie der wässrigen Phase 400 μl/Tube 100%iges Isopropanol hinzu.
- Das Röhrchen wird 10 min bei Raumtemperatur inkubiert und bei 12.000 × g für 10 min bei 4 °C zentrifugiert.
- Entfernen Sie den Überstand vorsichtig.
- Waschen Sie das RNA-Pellet 3 Mal mit 200 μl 75%igem Ethanol (EtOH/RNase-freies Wasser), entfernen Sie den Überstand und trocknen Sie das RNA-Pellet an der Luft.
Anmerkungen: Lassen Sie die RNA nicht vollständig trocknen, da das Pellet an Löslichkeit verlieren kann. - Resuspendieren Sie das RNA-Pellet in 20 μl RNase-freiem Wasser, das einen RNASE-Inhibitor (1 μL/20 μL) enthält, und lösen Sie das Pellet durch Pipettieren auf.
- Messen Sie die Absorption bei 260 nm.
17. Umgekehrte Transkription
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 1 h
- Fügen Sie 7 μl/Röhrchen Reverse Transkriptionsprimer + dNTP-Mischung hinzu (siehe Tabelle 1 und Tabelle 3) und legen Sie die Röhrchen auf einen Thermocycler.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt 27 μL. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. - 5 min bei 65 °C erhitzen und die Röhrchen für mindestens 1 min auf Eis legen.
- Fügen Sie 14 μl/Röhrchen Reverse-Transkriptase-Mastermix hinzu (siehe Tabelle 1) und mischen Sie durch Pipettieren.
- Legen Sie die Röhrchen auf einen Thermocycler und inkubieren Sie sie bei 55 °C für 10 min und dann bei 80 °C für 10 min.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt 60 μL. Die Temperatur des Thermocyclerdeckels beträgt 105 °C.
18. Synthese des zweiten Strangs
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 2,5 h
- Fügen Sie 40 μl/Röhrchen Reinstwasser hinzu und teilen Sie die 100 μl-Lösung in zwei 0,2-ml-PCR-Röhrchen (50 μl/röhrchen) auf.
- Fügen Sie 60 μl/Röhrchen der Zweitstrangsynthesemischung hinzu (siehe Tabelle 1), legen Sie die Röhrchen auf einen Thermocycler und führen Sie das folgende Programm aus: i) 95 °C für 5 min, ii) 95 °C für 30 s, iii) 60 °C für 30 s, iv) 72 °C für 30 s, v) Wiederholen Sie die Schritte ii-iv 20 Mal und vi) halten Sie sie bei 4 °C.
Anmerkungen: Die Röhrchengröße beträgt 0,2 ml. Das Volumen der Lösung beträgt 110 μL. Die Temperatur des Thermocyclerdeckels beträgt 105 °C. - 4,4 μl/Röhrchen 0,5 M EDTA (letzte 20 mM) zugeben und bei -80 °C über Nacht lagern.
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier bis zu einigen Tagen gefahrlos abgebrochen werden. - Reinigen Sie die DNA durch Phenol-Chloroform-Reinigung und Polyethylenglykolfällung (beschrieben in Schritt 14).
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier sicher für mindestens eine Woche gestoppt werden.
19. Restriktionsenzymverdauung und Größenauswahl
Anmerkungen: Das folgende Verfahren wird unter einer sauberen Haube durchgeführt, um eine DNase-Kontamination zu vermeiden. Dauer: 3 h (Schritte 19.1-19.7)
- Messen Sie die DNA-Konzentration der gereinigten DNA (ab Schritt 18.4), indem Sie die Absorption bei 260 nm messen.
- Übertragen Sie 6 μg DNA in ein PCR-Röhrchen, fügen Sie 30 μl 10x Aufschlusspuffer hinzu und stellen Sie das Volumen mit Reinstwasser auf 294 μl ein.
- 6 μl BciVI-Restriktionsenzym zugeben und bei 37 °C 1 h inkubieren.
- Führen Sie die EtOH-Fällung mit linearem Polyacrylamid durch.
- Fügen Sie 60 μl/Tube 3 M Natriumacetat (pH 5,2) und dann 40 μl/Tube lineares Acrylamid (5 mg/ml, siehe Materialtabelle) hinzu.
- Fügen Sie 400 μl/Röhrchen EtOH hinzu und inkubieren Sie es über Nacht bei -20 °C.
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier getrost für einen Tag abgebrochen werden. - 12.000 × g für 10 min bei 4 °C zentrifugieren und den Überstand entfernen.
- Waschen Sie das Pellet zweimal mit 80% EtOH und trocknen Sie das Pellet.
- Lösen Sie das Pellet mit 20 μL 1xTE-Puffer auf und fügen Sie 4 μL/Röhrchen frischen 6x Gelladepuffer hinzu.
- Laden Sie die Probe in einen 5%igen Agarosegel/0,5-fachen TAE-Puffer und führen Sie eine Elektrophorese durch (50 V für 40 min).
- Schneiden und sammeln Sie das Gel über 50 bp (siehe Abbildung 6) und extrahieren Sie die DNA mit einem Gelextraktionskit.
HINWEIS: Sicherer Haltepunkt: Das Experiment kann hier sicher für mindestens eine Woche gestoppt werden. - Konstruieren Sie die Sequenzierungsbibliothek mit einem DNA-Bibliotheks-Vorbereitungskit (siehe Materialtabelle)16,17.
Ergebnisse
K562-Einzelzellen wurden unter Verwendung des in Schritt 8 beschriebenen Protokolls erzeugt (siehe Abbildung 5). Einzelne Zellen wurden in die äußere Schicht des Polyacrylamid-Beads eingebettet. Die Zell-DNA wurde gefärbt und mit einem Interkalatorfarbstoff für die DNA-Färbung visualisiert.

Abbildung 5: Ge...
Diskussion
Dieser Artikel beschreibt das Schritt-für-Schritt-Protokoll für die kürzlich berichtete multiepigenomische Einzelzellanalyse mit wiederverwendbaren Einzelzellen7. In den folgenden Abschnitten diskutieren wir kritische Punkte und betonen mögliche Einschränkungen des Protokolls.
Einer der kritischen Punkte im gesamten Protokoll (aus den Schritten 7.2-13) ist die Vermeidung einer DNase-Kontamination. Eine einzelne Zelle hat nur zwei Kopien der genomischen DNA. Daher ...
Offenlegungen
Dr. Ohnuki und Dr. Tosato sind Miterfinder eines Patents mit dem Titel "Verfahren zur Herstellung einer wiederverwendbaren Einzelzelle und Verfahren zur Analyse des Epigenoms, Transkriptoms und Genoms einer einzelnen Zelle" (EP3619307 und US20200102604). Die Patentanmeldung wurde zum Teil auf der Grundlage vorläufiger Ergebnisse im Zusammenhang mit der im vorliegenden Manuskript beschriebenen Technologie eingereicht. Die Erfindung oder Erfindungen, die in dieser Patentanmeldung beschrieben und beansprucht werden, wurden gemacht, während die Erfinder Vollzeitangestellte der US-Regierung waren. Daher wurden gemäß 45 Code of Federal Regulations Part 7 alle Rechte, Titel und Interessen an dieser Patentanmeldung per Gesetz an die US-Regierung abgetreten oder sollten gesetzlich übertragen werden. Die US-Regierung überträgt einen Teil der Lizenzgebühren, die sie erhält, gemäß 15 U.S. Code § 3710c an ihre angestellten Erfinder.
Danksagungen
Wir danken Dr. David Sanchez-Martin und Dr. Christopher B. Buck für ihre Kommentare während der Konzeptphase des Projekts. Wir danken auch dem Genomics Core, dem Center for Cancer Research, dem National Cancer Institute, den National Institutes of Health für die Hilfe bei vorläufigen Experimenten und der Collaborative Bioinformatics Resource, CCR, NCI, NIH für die Beratung bei der computergestützten Analyse. Wir danken Frau Anna Word für ihre Hilfe bei der Optimierung der in der Methode verwendeten DNA-Polymerasen. Diese Arbeit nutzte die Rechenressourcen des NIHHPC-Biowulf-Clusters (http://hpc.nih.gov). Dieses Projekt wird durch das Intramurale Programm des Center for Cancer Research, des National Cancer Institute, der National Institutes of Health, des NCI Director's Innovation Award (#397172) und Bundesmittel des National Cancer Institute unter der Vertragsnummer HHSN261200800001E unterstützt. Wir danken Dr. Tom Misteli, Dr. Carol Thiele, Dr. Douglas R. Lowy und allen Mitgliedern des Laboratory of Cellular Oncology für ihre produktiven Kommentare.
Materialien
| Name | Company | Catalog Number | Comments |
| 10x CutSmart buffer | New England BioLabs | B6004 | 10x Digestion buffer |
| 200 proof ethanol | Warner-Graham Company | 200 proof | Ethanol |
| 5-Hydroxymethylcytosine (5-hmC) Monoclonal Antibody [HMC/4D9] | Epigentek | A-1018-100 | Anti-5hmC |
| Acridine Orange/Propidium Iodide Stain | Logos Biosystems | F23001 | Cell counter |
| Acrylamide solution, 40% in H2O, for molecular biology | MilliporeSigma | 01697-500ML | 40% acrylamide solution |
| All-in-One Fluorescence Microscope BZ-X710 | Keyence | BZ-X710 | Scanning microscope |
| Amicon Ultra-0.5 Centrifugal Filter Unit | MilliporeSigma | UFC510024 | Ultrafiltration cassette |
| Ammonium persulfate for molecular biology | MilliporeSigma | A3678-100G | Ammonium persulfate powder |
| Anhydrous DMF | Vector laboratories | S-4001-005 | Anhydrous N,N-dimethylformamide (DMF) |
| Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) antibody [4H8] | Abcam | ab5408 | Anti-Pol II |
| Anti-TRAP220/MED1 (phospho T1457) antibody | Abcam | ab60950 | Anti-Med1 |
| BciVI | New England BioLabs | R0596L | BciVI |
| Bovine Serum Albumin solution, 20 mg/mL in H2O, low bioburden, protease-free, for molecular biology | MilliporeSigma | B8667-5ML | 20% BSA (Table 7) |
| Bst DNA Polymerase, Large Fragment | New England BioLabs | M0275L | Bst DNA polymerase |
| BT10 Series 10 µl Barrier Tip | NEPTUNE | BT10 | P10 low-retention tip |
| CellCelector | Automated Lab Solutions | N/A | Automated single cell picking robot |
| CellCelector 4 nl nanowell plates for single cell cloning, Plate S200-100 100K, 24 well,ULA | Automated Lab Solutions | CC0079 | 4 nL nanowell plate |
| Chloroform | MilliporeSigma | Chloroform | |
| Corning Costar 96-Well, Cell Culture-Treated, Flat-Bottom Microplate | Corning | 3596 | Flat-bottom 96-well plates |
| Deep Vent (exo-) DNA Polymerase | New England BioLabs | M0259L | Exo- DNA polymerase |
| DNA LoBind Tubes, 0.5 mL | Eppendorf | 30108035 | 0.5 mL DNA low-binding tube |
| DNA Oligo, 1st random primer | Integrated DNA Technologies | N/A, see Table 3 | 1st random primer |
| DNA Oligo, 2nd random primer Cell#01 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#02 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#03 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#04 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#05 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#06 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#07 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#08 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#09 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#10 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#11 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd random primer Cell#12 | Integrated DNA Technologies | N/A, see Table 3 | 2nd random primer |
| DNA Oligo, 2nd synthesis primer | Integrated DNA Technologies | N/A, see Table 3 | 2nd synthesis primer |
| DNA Oligo, Ligation Adaptor | Integrated DNA Technologies | N/A, see Table 3 | Ligation Adaptor |
| DNA Oligo, Reverse Transcription primer | Integrated DNA Technologies | N/A, see Table 3 | Reverse Transcription primer |
| DNase I (RNase-free) | New England BioLabs | M0303L | DNase I (RNase-free, 4 U). |
| DNase I Reaction Buffer | New England BioLabs | B0303S | 10x DNase I buffer (NEB) |
| dNTP Mix (10 mM each) | Thermo Fisher | R0192 | 10 mM dNTPs |
| Fetal Bovine Serum, USA origin, Heat-inactivated | MilliporeSigma | F4135-500ML | Fetal bovine serum |
| HiScribe T7 High Yield RNA Synthesis Kit | New England BioLabs | E2040S | In-vitro-transcription master mix |
| Histone H3K27ac antibody | Active motif | 39133 | Anti-H3K27ac |
| Histone H3K27me3 antibody | Active motif | 39155 | Anti-H3K27me3 |
| IgG from rabbit serum | Millipore Sigma | I5006-10MG | Control IgG |
| Iron oxide(II,III) magnetic nanopowder, 30 nm avg. part. size (TEM), NHS ester functionalized | MilliporeSigma | 747467-1G | NHS ester functionalized 30 nm iron oxide powder |
| K-562 | American Type Culture Collection (ATCC) | CCL-243 | cells |
| Linear Acrylamide (5 mg/mL) | Thermo Fisher | AM9520 | Linear Acrylamide |
| LUNA-FL Dual Fluorescence Cell Counter | Logos Biosystems | L20001 | Cell counter |
| LUNA Cell Counting Slides, 50 Slides | Logos Biosystems | L12001 | Cell counter |
| Mineral oil, BioReagent, for molecular biology, light oil | MilliporeSigma | M5904-500ML | Mineral oil |
| N,N,N′,N′-Tetramethylethylenediamine for molecular biology | MilliporeSigma | T7024-100ML | N,N,N′,N′-Tetramethylethylenediamine |
| NaCl (5 M), RNase-free | Thermo Fisher | AM9760G | 5M NaCl |
| NanoDrop Lite | Thermo Fisher | 2516 | Microvolume spectrophotometer |
| NEST 2 mL 96-Well Deep Well Plate, V Bottom | Opentrons | N/A | 2 mL deep well 96-well plate |
| Non-skirted 96-well PCR plate | Genesee Scientific | 27-405 | 96-well PCR plate |
| NuSive GTG Agarose | Lonza | 50081 | Agarose |
| OmniPur Acrylamide: Bis-acrylamide 19:1, 40% Solution | MilliporeSigma | 1300-500ML | 40%Acrylamide/Bis-acrylamide |
| OT-2 lab robot | Opentrons | OT2 | Automated liquid handling robot |
| Paraformaldehyde, EM Grade, Purified, 20% Aqueous Solution | Electron Microscopy Sciences | 15713 | 20% Pararmaldehyde |
| PBS (10x), pH 7.4 | Thermo Fisher | 70011044 | 10x PBS |
| PIPETMAN Classic P1000 | GILSON | F123602 | A P1000 pipette |
| Protein LoBind Tubes, 1.5 mL | Eppendorf | 925000090 | 1.5 mL Protein low-binding tube |
| QIAgen Gel Extraction kit | Qiagen | 28706 | A P1000 pipette |
| Quant-iT PicoGreen dsDNA Assay | Thermo Fisher | P11495 | dsDNA specific intercalator dye |
| Quick Ligation kit | New England BioLabs | M2200L | T4 DNA ligase (NEB) |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher | 10777019 | RNAse inhibitor |
| S-4FB Crosslinker (DMF-soluble) | Vector laboratories | S-1004-105 | Succinimidyl 4-formylbenzoate (S-4FB) |
| S-HyNic | Vector laboratories | S-1002-105 | Succinimidyl 6-hydrazinonicotinate acetone hydrazone (S-HyNic) |
| Sodium Acetate, 3 M, pH 5.2, Molecular Biology Grade | MilliporeSigma | 567422-100ML | 3M Sodium acetate (pH 5.2) |
| Sodium bicarbonate, 1M buffer soln., pH 8.5 | Alfa Aesar | J60408 | 1M sodium bicarbonate buffer, pH 8.5 |
| Sodium phosphate dibasic for molecular biology | MilliporeSigma | S3264-250G | Na2HPO4 |
| Sodium phosphate monobasic for molecular biology | MilliporeSigma | S3139-250G | NaH2PO4 |
| SuperScript IV reverse transcriptase | Thermo Fisher | 18090050 | Reverse transcriptase |
| SYBR Gold Nucleic Acid Gel Stain (10,000x Concentrate in DMSO) | Thermo Fisher | S11494 | An intercalator dye for DNA |
| T4 DNA Ligase Reaction Buffer | New England BioLabs | B0202S | 10x T4 DNA ligase reaction buffer |
| ThermoPol Reaction Buffer Pack | New England BioLabs | B9004S | 10x TPM-T buffer (Tris-HCl/Pottasium chloride/Magnesium sulfate/Triton X-100) |
| TRIzol LS reagent | Thermo Fisher | 10296-028 | Guanidinium thiocyanate-phenol-chloroform extraction |
| TruSeq Nano DNA library prep kit | Illumina | 20015965 | A DNA library preparation kit (see also the manufacturer's instruction) |
| Ultramer DNA Oligo, Anti-5hmC_Ab#005 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-H3K27ac_Ab#002 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-H3K27me3_Ab#003 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-Med1_Ab#004 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Anti-Pol II_Ab#006 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for antibody |
| Ultramer DNA Oligo, Control IgG_Ab#001 | Integrated DNA Technologies | N/A, see Table 3 | An amine-modified DNA probe for control IgG |
| UltraPure 0.5 M EDTA, pH 8.0 | Thermo Fisher | 15575020 | 0.5M EDTA, pH 8.0 |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fisher | 10977023 | Ultrapure water |
| Zeba Splin Desalting Columns, 7K MWCO, 0.5 mL | Thermo Fisher | 89882 | Desalting column |
Referenzen
- Perkel, J. M. Single-cell analysis enters the multiomics age. Nature. 595 (7868), 614-616 (2021).
- Clark, S. J., et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature Communications. 9 (1), 781 (2018).
- Ma, S., et al. Chromatin potential identified by shared single-cell profiling of RNA and Chromatin. Cell. 183 (4), 1103-1116 (2020).
- Zhu, C., et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nature Structural & Molecular Biology. 26 (11), 1063-1070 (2019).
- Zhu, C., et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nature Methods. 18 (3), 283-292 (2021).
- Mimitou, E. P., et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nature Biotechnology. 39 (10), 1246-1258 (2021).
- Ohnuki, H., Venzon, D. J., Lobanov, A., Tosato, G. Iterative epigenomic analyses in the same single cell. Genome Research. 31 (10), 1819-1830 (2021).
- Tosato, G., Ohnuki, H. . Methods of preparing a re-usable single cell and methods fro analyzing the epigenome, transcriptome, and genome of a single cell. , (2021).
- Harada, A., et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nature Cell Biology. 21 (2), 287-296 (2019).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nature Structural & Molecular Biology. 18 (1), 91-93 (2011).
- Marina, R. J., et al. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. EMBO Journal. 35 (3), 335-355 (2016).
- Weast, R. C., Weast, R. C., Astle, M. J., Beyer, W. H., et al. . Handbook of chemistry and physics: a ready-reference book of chemical and physical data. 56th edn. , (1975).
- Aydin, S. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides. 72, 4-15 (2015).
- Gan, S. D., Patel, K. R. Enzyme immunoassay and enzyme-linked immunosorbent assay. Journal of Investigative Dermatology. 133 (9), 12 (2013).
- Porstmann, T., Kiessig, S. T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Chao, H. P., et al. Systematic evaluation of RNA-Seq preparation protocol performance. BMC Genomics. 20 (1), 571 (2019).
- Song, Y., et al. A comparative analysis of library prep approaches for sequencing low input translatome samples. BMC Genomics. 19 (1), 696 (2018).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Ku, W. L., et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nature Methods. 16 (4), 323-325 (2019).
- Carter, B., et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nature Communications. 10 (1), 3747 (2019).
- Maskell, D. P., et al. Structural basis for retroviral integration into nucleosomes. Nature. 523 (7560), 366-369 (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten