
Method Article
种测定和定量混合物中使用肽应用的MRM质谱肉认证
摘要
We present a protocol for identifying and quantifying the components in mixtures of species possessing similar proteins. Mass spectrometry detects peptides for identification, and gives relative quantitation by ratios of peak areas. As a tool food for fraud detection, the method can detect 1% horse in beef.
摘要
We describe a simple protocol for identifying and quantifying the two components in binary mixtures of species possessing one or more similar proteins. Central to the method is the identification of 'corresponding proteins' in the species of interest, in other words proteins that are nominally the same but possess species-specific sequence differences. When subject to proteolysis, corresponding proteins will give rise to some peptides which are likewise similar but with species-specific variants. These are 'corresponding peptides'. Species-specific peptides can be used as markers for species determination, while pairs of corresponding peptides permit relative quantitation of two species in a mixture. The peptides are detected using multiple reaction monitoring (MRM) mass spectrometry, a highly specific technique that enables peptide-based species determination even in complex systems. In addition, the ratio of MRM peak areas deriving from corresponding peptides supports relative quantitation. Since corresponding proteins and peptides will, in the main, behave similarly in both processing and in experimental extraction and sample preparation, the relative quantitation should remain comparatively robust. In addition, this approach does not need the standards and calibrations required by absolute quantitation methods. The protocol is described in the context of red meats, which have convenient corresponding proteins in the form of their respective myoglobins. This application is relevant to food fraud detection: the method can detect 1% weight for weight of horse meat in beef. The corresponding protein, corresponding peptide (CPCP) relative quantitation using MRM peak area ratios gives good estimates of the weight for weight composition of a horse plus beef mixture.
引言
The European horse meat scandal of 2013, in which undeclared horse meat was found in a number of supermarket beef products1, highlights the need for testing methods capable of detecting and measuring food fraud in meat. Several technologies have been explored, especially enzyme-linked immunosorbent assay (ELISA) and DNA-based methods2. An alternative route, based on mass spectrometry, targets species-specific peptides which in turn arise from species-specific proteins. Here we outline one such peptide-based approach that offers both identification and relative quantitation of the adulterant species in a meat mixture3.
The protocol is framed in the context of red meats and the desire to determine the presence of one in another at the level of 1% by weight, the level considered by some to represent fraudulent food adulteration as opposed to contamination4. The method relies in the first instance on identifying a protein which is nominally 'the same' in all target meats. Myoglobin, the protein responsible for the red color of meat, is a good candidate since it is abundant, relatively heat tolerant and water soluble, and has been used for species determination of meat previously5,6. The myoglobins for beef (Bos Taurus), pork (Sus scrofa), horse (Equus caballus) and lamb (Ovis aries)3, for instance, are nominally the same, as required, but their sequences are not identical. Such groups of 'similar but different' proteins, like these four myoglobins, can conveniently be described as 'corresponding proteins'. The sequence differences in these four myoglobins are species-specific: for example, the full myoglobin proteins for beef and horse, P02192 and P68082 respectively, each comprise 154 amino acids with 18 sequence differences between the two. Subject to proteolysis using trypsin these proteins produce two sets of peptides, some of which are identical, and some which show one or more species-specific amino acid differences: corresponding proteins therefore give rise to corresponding peptides.
The CPCP approach, therefore, seeks first to identify proteins from two or more species where these proteins exhibit limited species-specific sequence variants. These are corresponding proteins. Following proteolysis, corresponding proteins give rise to peptides, some of which likewise display species-specific sequence variants inherited from the parent protein. These are corresponding peptides. The CPCP approach can be used to compare levels of two corresponding proteins in a mixed species sample by monitoring the levels of corresponding peptides.
The natural technology for the detection of known peptides is multiple reaction monitoring mass spectrometry, or MRM-MS7. Species-specific peptides yield precursor ions, which along with their mass spectrometry fragment ions, are easily itemized in advance by software tools. These lists are then used to instruct the mass spectrometer to record only specific precursor plus fragment ion pairs, called transitions. A particular target peptide is therefore identified not only by its retention time in the chromatography preceding the mass spectrometer, but also by a set of transitions sharing a common precursor ion. This is a highly selective means of detecting known peptides that makes efficient use of the mass spectrometer resource.
Other authors have used mass spectrometry to test for meat adulteration via peptide markers but from disparate proteins8-14. Using the corresponding proteins, corresponding peptides (CPCP) scheme, however, means experimental conditions can be optimized, aiding identification of the species in the mixture from known species-specific transitions. In addition, corresponding proteins and peptides will generally behave similarly in the extraction, proteolysis and detection stages. Since transition peak areas are quantitative and reproducible, ratios of peak areas arising from pairs of corresponding peptides from different species provide a direct estimate of the relative quantities of two meats in a mixture. In contrast, more traditional quantitation routes exploit calibrations based on reference materials to establish absolute quantitation14,15.
Though the protocol is outlined in the context of myoglobin and meat, proteins other than myoglobin could be used for identification and relative quantitation via the CPCP strategy in meat mixtures, though potentially with modifications to the protocol. In addition the strategy is also applicable to binary mixtures of other species sharing one or more corresponding proteins.
The starting point for the protocol is purified 'reference' myoglobin, which for some species can be purchased but which for others must be prepared by conventional size-exclusion chromatography. The procedure for preparing reference myoglobin is not included in the protocol, but is described elsewhere3. Software tools16 are used to list candidate peptides and transitions arising from myoglobins of interest. Each reference myoglobin is subjected to proteolysis and the resultant peptides analyzed by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) to discover which of the candidate precursor ions and transitions are most useful, and to determine the matching peptide retention times. The outcome of this stage is a revised list of target peptides with their transitions, suitable for species determination, and a list of CPCP pairs, suitable for relative quantitation. To test real meats, sample extractions are prepared then subjected to proteolysis to generate peptides both from myoglobin and other extraneous proteins. The myoglobin-based peptides are then monitored by LC-ESI-MS/MS based on their listed transitions. The species present in a mixture are identified by the transition peaks associated with marker peptides. Estimates of the relative amounts of two meats in a binary mixture are calculated using ratios of transition peak areas. A set of test mixtures of pairs of meats will allow the ratio of peak areas for a given pair of transitions to be checked and calibrated against actual mixtures.
研究方案
1.蛋白水解和参考Myoglobins分析
- 参考myoglobins蛋白水解
- 制备纯化的参考myoglobins的溶液(范围0.2 -在25mM碳酸氢铵0.5毫克/毫升)3。
- 转移1ml等份各样品至2ml离心管中。
- 通过在95℃下30分钟在热块加热样品热变性提取的蛋白质。冷却样品约15分钟,直到它达到室温。添加30毫克尿素(终浓度0.5 M),以增强消化,然后混匀。
- 胰蛋白酶水解
- 制备在25mM碳酸氢铵胰蛋白酶的1毫克/毫升溶液,并根据需要在冰上储存。添加胰蛋白酶的足够体积使得最终的酶的活性是420 BAEE(N-苯甲酰基-L-精氨酸乙酯盐酸盐)单位/萃取mg蛋白质,然后通过温和涡旋混合并允许在3至proteolyze过夜7°C。
- 进行十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)17以证明蛋白水解的完整性。
- 蛋白水解后的样品脱盐
- 稀释样品1:2体积:体积水。
- 激活的聚合物反相(RP)柱,加入1毫升甲醇填充有30毫克的RP材料,然后通过添加1毫升%甲酸平衡墨盒。
- 加载到样品在重力作用下的墨盒。
- 与在重力作用下1毫升5%甲醇/ 1%甲酸洗涤。
- 在重力作用下与5微升二甲基亚砜(DMSO)中预先填充2ml微量管;:用1毫升乙腈/水(0.1%甲酸酸酯Ⅴ90:10 v)的洗脱肽。
- 使用离心蒸发器120分钟,在50℃真空下除去溶剂,然后再溶解残余物中加入250μl乙腈/水(3:97体积:体积; 0.1%甲酸)。
- 转让该溶液至低体积自动取样管形瓶中。
注:样品可以储存在4℃直到准备液相色谱质谱(LC / MS)分析。
- 对于MRM过渡名单产生
- 定位为从的UniProt数据库的不同的肉类的肌红蛋白的序列。
- 输入序列肌红蛋白进入肽和过渡预测软件的"目标"框( 如地平线)。如果需要,肽以揭示其片段列表悬停。
- 点击"设置",然后选择"肽设置"。输入为消化的喜好(即胰蛋白酶),并错过了分裂的次数(0)。输入所需的选择附加的参数,特别是,所述肽长度(6 - 25),N-末端排除(0)和假定的氨基酸修饰(无)。
- 点击"设置",然后选择"转换设置"。选择喜好用于LC / MS分析仪器类型。
- 点击"导出",然后选择"转换列表"创建包含生成的MRM转变和参数的电子表格。
- 分析通过LC / MS
- 设置二元梯度的一个系统(水(A)和乙腈(B)中,每个含有0.1%甲酸V:V)进行高效液相色谱(HPLC)用自动取样器,C18芯壳HPLC柱(10厘米×2.1毫米连接到与MRM检测阳性电喷雾模式操作的三重四极质谱仪2.6微米粒径)。
- 在数据收集软件( 例如 ,分析师),选择"文件"和"新建",在弹出框中点击"获取方法",然后点击"确定"。
注意:这将打开仪器方法编辑器,它包含了连接的设备,使一个新的LC / MS方法的设置的列表。 - 点击"二元泵"和我NPUT的流量值(300微升/分钟)并在22分钟在表中的梯度时间,设置3%B的二进制梯度分布到30%B,在23分钟为一个5分钟增加至100%B洗出之前返回到初始条件和再平衡再6分钟。
- 点击"自动进样器",然后插入进样量(5微升)。启用"洗针周期"并输入"洗涤时间"(30秒),然后选择"刷新端口"。
- 点击"柱温控制器"和"柱温箱属性"设置'左温度"和"合适的温度"(40°C)。
- 点击"质谱仪",然后单击"编辑参数"输入气源条件。选择"扫描类型"为"MRM(MRM)"和"极性"为"积极"。转到"期间摘要",并进入"持续时间",总时间为LC分析的ð平衡(35分钟)。
- 在表中点击右键,选择"去聚势(DP)'和'碰撞能量(CP)"将这些列添加到表中。输入Q1,Q3,时间(毫秒),身份证,DP和CE值的所有转变,对于单肉种,在过渡列表(见步骤1.4.5)创建。
注:时间(毫秒)指的是停留时间,质谱仪花费扫描每个过渡时,求和其中不应超过3秒。 - 保存采集方法文件(文件扩展.dam)。
注意:步骤1.5.2 - 1.5.8需要重复为每个肉物种。这将在下面的分析准备创建在屏幕模式下每个品种的肉的单一方法文件。 - 在数据采集软件,点击"捕获"并选择"。平衡"。在打开的对话框中,选择所需的采集方法开始仪器平衡。
- 放入AR样品瓶ACK在自动采样器。
- 点击"文件",选择"新建",然后"批量收购"。在"样品"选项卡中选择"添加设置",然后"添加样本"。将要分析的样品数量,然后点击"确定"。在"收购"框中选择将用于从下拉菜单中进行分析的方法文件。
- 在表中,选择"板代码",然后从下拉菜单中选择相应纸盒配置。点击鼠标左键在"板代码"列标题,然后点击右键并选择"向下填充"。在"瓶位置"输入中的行中的自动取样器各样品的位置。
- 在"数据文件"收购中输入文件名,在列标题,然后左击后点击右键并选择"向下填充"。在"样品名称"插入待分析各样品的身份。除收购批处理文件(文件Ëxtension .dab)。
- 点击"提交"选项卡,然后选中需要的LC / MS要分析的样本。点击"提交"。点击"捕获"和"开始采样"开始进行分析。
注意:每个采集方法将扫描跨过色谱的整个长度的MRM离子为单个肉物种。对于MRM购置质谱仪设置,根据设备型号及肽有所不同。 - 查看使用数据查看软件所生成的数据文件。点击西昌(提取离子),并在下拉列表中突出了一个单一的前体(Q1)中的所有片段(Q3值)下降。一个新的面板将打开只显示所选择的过渡。
- 记录并发跃迁基的保留时间(R t)的 ,因为这些对应于单个肽。
- 重复以峰分配给每个各自的肽为每组变换的前两个步骤的肉种。
- 记录标记的肽是适合于提供物种鉴定( 例如 ,肽HPGDFGADAQGAMTK,前体M / Z = 752,R T = 12.0分钟,对马),其保留时间一起,并注意其形成对应的适合于相对定量成对。
注意:例如,马标记肽(前体M / Z = 752)具有一个相应的牛肉肽,HPSDFGADAQAAMSK(前体M / Z = 767,R T = 13.2分钟)。 - 为了建立一个单一的动态方法包括所有的肉种,在该数据查看软件,依次在每个肉物种,打开XIC过渡数据的每个前体(在1.5.8分配给特定的肽)。
- 放大在通过左击选定的保留时间峰丛并拖动光标集群下方。找出最激烈的转变(由峰标签上单击鼠标右键)。
- 手动记录转换和保留时间在电子表格中。
- 要输入参数的LC / MS软件的新动态方法,点击"质谱仪",然后单击"编辑参数"输入气源条件。选择"扫描类型"为"MRM(MRM)"和"极性"为"积极"。
- 转到"期间摘要"并输入持续时间(设置为总时间为LC分析和平衡)。在表中点击右键,选择"去聚势(DP)'和'碰撞能量(CP)"将这些列添加到表中。
注:"时间"栏现指每次转换的预期保留时间(min)。 - 在LC / MS数据采集软件的"编辑参数"部分中,选中"计划MRM"框。输入Q1,Q3,时间(min),身份证,在电子表格(1.5.21)创造的转变DP和CE值并保存收购法(FILË扩展.dam)。
注意:此方法通常会降低MRM数转换到4最激烈的每个肽和扫描只在整个每个肽峰的保留时间窗口,从而提高了灵敏度和数据的质量。一个"动态"的方法是"引导滞留时间窗口"的方法,有时也被称为调度。
2.校准样品的制备和分析
- 肉混合物提取
- 使用先前冷冻然后研磨成粉末肉,通过称重肉的各自的量(约300毫克的总质量)为15个毫升的塑料离心管中制备一系列肉混合物。
- 加入4毫升提取缓冲液(0.15氯化1M氢+ 0.15M的磷酸盐缓冲液6.5)的。涡旋30秒。提取物对实验室摇动器在室温下以250次/分钟2小时。
注:周期/分指的是振荡运动。 - 传送2毫升的提取到2ml微量离心管中。离心在4℃下5分钟,在17000×g的。
- 转移上清液200μl的等份(保留少量用于蛋白质测定,见2.2)于2ml离心管并干燥使用离心蒸发器(预先设定的程序:50℃,没有排气和120分钟的持续时间)。
- 蛋白检测
- 转移所保留的上清液7微升等份(见2.1.4)中一式三份加入到96孔板的孔中。
- 转移一系列一式三份蛋白质标准的7微升等份,范围0 - 1.0毫克/毫升牛血清白蛋白(BSA),以同样的96孔板中。
- 加入200μl考马斯加蛋白质检测试剂的每个孔中。
- 直观比较样品孔与蛋白质标准的颜色来检查样品中的校准标准的范围内。如有必要,重复用稀释的样品,使其成为在范围内。
- 离开板ST和3分钟。
- 爆裂已经用皮下注射针形成的任何气泡。
- 分析上使用标准的端点协议在595波长下的板读取器的板。
- 确定使用从蛋白质的标准校准数据样品的蛋白质浓度。
注意:这是必需的胰蛋白酶在胰蛋白酶消化所用的量的计算。
- 肉混合物的蛋白水解
- 再溶解从步骤2.1.4干燥残留物于1ml的25mM碳酸氢铵溶液。在rotamixer拌匀。
- 按照步骤1.1.3协议1.3.7。
- 分析通过LC / MS
- 设置LC / MS如先前(步骤1.5.1)。
- 创建如先前概述了新的采集批次(步骤1.5.9 - 1.5.14),选择在步骤在1.5.24中创建的采集方法,它使用一种动态LC / MS方法结合所有的肉种,并获取用于数据该diges特德肉类样品。
- 显示在数据查看软件的完整图谱。显示西昌的依次设置每个过渡。视觉上确认每个簇包含的钟形峰的必要数量以预期的保留时间,从而确认所选择的肽的存在。
- 执行使用数据查看软件通过双击"建立定量方法"导航栏中集成峰面积为每个感兴趣的过渡定量。
- 在"选择示例"窗格中选择"数据文件"和"样品"进行分析,以产生"分析物"表。
- 点击"一体化"选项卡上显示的第一个过渡(分析物)的进行整合。
- 点击"分析物"框中显示下拉转换的列表。依次选择每个过渡来显示它和视觉确认正确的峰值被选择用于集成。到modify或强制整合,右键单击并拖动光标在目标峰(这将在绿色高亮显示)。点击"选择峰"按钮,然后单击"应用"。
- 保存工作区作为方法文件(.qmf)。
注意:这将创建样品峰面积的后续计量定量方法文件。 - 双击"定量向导"导航栏中。在"选择样品"窗口中选择一个"数据文件",那么一个或多个"提供样品"创造"定量设置"。选择"下一步",以显示"选择设置和查询"框中。与保留默认值,选择"下一步",以显示"选择方法"。从下拉"方法"框中,选择在步骤2.4.8创建的"积分法"文件,然后选择"完成"。
注意:这将创建一个"结果表",其中包括肉类产生的混合物转变峰地区。 - 保存"结果表"(文件扩展名.rdb),导出为文本文件(.txt),并在电子表格中打开它来查看数据。
- 从相应的肽,着眼于其中两个片段含有这些情况下一种肉类的另一个的百分比(由过渡峰面积)对所测量的百分比的曲线图图(W / W)在两个肉为选定的MRM所选过渡相同数量的氨基酸从C-末端计数。
注:具有相同的碎片网站相同的片段给予最佳的效果。 - 检查从上面2.4.11的地块。无论是视觉上或使用绘图包中的趋势线的工具,识别一组地块它们都是线性渐变相似的。使用这些CPCP加上片段组合中的任何一个或多个为真实的肉样品中的校准。
注:显出非同一般的梯度作图可能表明,要么肽或片段抑制与信号STRE随之减少NGTH。非线性曲线可指示峰值检测差或其他问题。
3.肉类样品
- 从靶肉样品中提取蛋白质
- 从哪里用抹刀将样品适用,消费无关的非肉类材料。例如,从冷冻烤宽面条刮掉酱和意大利面食。
- 重20克肉入金属烧杯中。
- 加入100毫升0.15氯化物1M氢/ 0.15M的磷酸钾缓冲液,在pH 6.5。
- 通过在高速均化器混合肉1分钟提取蛋白质。
- 2.3.2 - 步骤2.1.4按照协议。
- 通过LC / MS分析样品
- 重复步骤2.4.2使用动态LC / MS方法来采集数据。
- 确定从每个肉肌红蛋白的肽作为在步骤2.4.3进行。
- 对于定量,使用定量软件对峰面积整合为每个感兴趣的过渡,如在步骤2.4.9概述。
- 对于混合品种的鉴定,记录这些标记肽满足商定的标准过渡的数量和信噪为那些过渡。
- 对于定量,使用来自步骤2.4.12商定集成化转变的峰面积,并利用百分比由过渡峰面积,由两个物种中的混合物计算的肌红蛋白的百分比。
- 从肉可能肌红蛋白水平的文献18使用前的知识来估计相对w / w的量存在于样品以两种肉类。
结果
在一个单一的动态模式的MRM试验每个编程过渡被单独地记录(如每秒检测器计数,厘泊)在特定的保留时间窗口。因此,从在一个实验中收集到的所有数据,峰值强度为每个转变可以单独萃取。那么唯一的有限信号是为该过渡设置的保留时间窗口。窗口的外面,该信号值被定义为零。为任何一种过渡的信号,例如,752→1269从马(肽单一同位素质量1,501.66道尔顿,母体离子M / Z 751.84道尔顿,充电状态= 2,片段离子ÿ13)典型地具有仅与测量噪声和不竞争从其他过渡峰认为也许可以从其他物种。因此,输出是一组清洁的峰,每过渡之一,在一个共同的保留时间为那些过渡共享一个共同的前体离子。
图1示出的1%w / w的混合物输出为组四个跃迁752→(1269,706,248,1366)马牛肉。由于显示四个跳变与马相关联,并且是在纯牛肉,羊肉或猪肉样品不存在,这些峰表示马的存在。取决于鲁棒性的标准,一组两个或多个转换的每个超过一些指定的信号到噪声电平建立鉴定。因此该图中建立的马中的1%的混合物的存在下w / w的在牛肉马。
偶尔,在检测到单个孤立的过渡。这表明前体离子和单个片段的机会的比赛,可能来自外来蛋白,与那些从系统预期和程序性进入质谱仪。峰的奇异性质,其在一个意想不到的保留时间发生,是在Si可以被忽略意外过渡gnature。
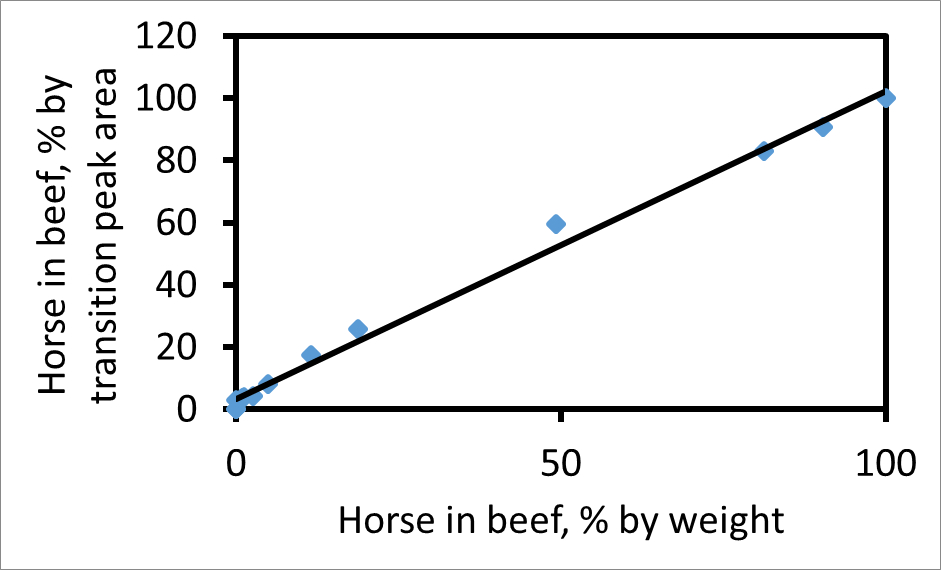
每个转变峰下的面积可以单独计算。根据一个合适的片段,马牛肉过渡峰面积的比率,例如,752→1269(马),以767→1299(牛肉),将正比于在混合物中的实际的肉类的比率。 图2示出了由这两个过渡与在牛肉马的混合物的重量百分比为重量的马的峰面积百分率的曲线图。如果百分比过渡峰面积相匹配的重量百分比为重量肉则斜率为1。在该图的斜率为1.03,这表明,这些过渡和CPCP对中,过渡峰面积得到的相对量的可靠测量在混合物中这两个肉类。如果在样品中的马肉是两倍富含肌红蛋白作为牛肉然后,与其他因素不变,的斜率线将是大于一。

图1. 的MRM跃迁强度与保留时间为1%w / w的在牛肉马该转换是752→(1269,706,248,1366),在橙,黑,蓝,绿,分别示出。标记肽是HPGDFGADAQGAMTK。四个过渡片段可以表示分别ý13,Y 7,Y 2和Y 14,其中y n表示从肽C-末端在正氨基酸计数。噪声信号变化从23到53以上的四个转变。另外一个红色线表示的0%马,100%牛肉进行比较的752→1269的过渡。只显示的保留时间非零区域。该图已被从Watson等 3修改。ES / ftp_upload / 54420 / 54420fig1large.jpg"目标="_空白">点击此处查看该图的放大版本。

图2. 情节牛肉马,至于重量百分比重量,相对于牛肉的百分数转变峰区马。该地块使用对肽牛肉(767)和马(752),并为在y 13碎片离子。如果A表示的峰面积那么纵坐标是100 A H /(A H + A B)。最佳拟合线(R 2 = 0.99)的斜率为1.03。这个数字已经从Watson等 3修改。 请点击此处查看本图的放大版本。
{kind=link}
讨论
合适的靶蛋白的选择是重要的。一个好的目标蛋白质需要在目标物质相应的形式,充分物种依赖性序列变异,种属特异性,与生物体内存在着大量访问。评估已经历处理混合物(例如,热处理),具有相对地不受该处理的序列的蛋白质是理想的。肌红蛋白是红肉,包括熟食红肉一个很好的候选人,但并不是唯一的可能性。一旦目标蛋白质决定,该协议的最关键的部分是蛋白质水解。从不同的肌红蛋白的蛋白质很可能需要一个替代蛋白水解协议。
如所描述的协议包括基于参考纯化的蛋白质的片段。这旨在发现保留时间窗口和合适的前体和碎片离子。这部分是非常有益的,但不是必需的。
尽管来自两个物种的兴趣相应肽对即使不实验中列出,有时,一个序列差异对消化轮廓严重后果的情况。例如,所述肽对VLGFHG(牛肉)和ELGFQG(马)得到异常的定量结果(表现为一个梯度小于一在图2中)。这是因为,后面的肽源于相对抑制KE裂解,引起马的混合物中的水平的低估。因此最好的开始与不同的氨基酸相应的肽被避免。常来自两个对应的肽片段具有相同的氨基酸序列,并表现良好,但是这并非总是如此,需要方法开发期间进行检查。种属鉴定是敏感的这些问题比相对定量少得多。该协议已被证明四红肉第3条 。附加肉种类可以包括,但过渡峰形的质量可能如果太多标记肽共洗脱恶化,有效地减少了停留时间和最终降解相对定量估计。改进的仪器,已经上市,将改善这一点。一个相关的问题是,并非所有的肉都有不同myoglobins。例如,马,驴和斑马myoglobins是相同的,因此严格来说该方法只能够在牛肉检测马,驴或斑马。在某些情况下,即使myoglobins不相同,一些关键的肽即可。例如,一些羊肉肌红蛋白衍生肽标记也出现在山羊。
面对此以及任何其他基于蛋白质的定量方法的复杂性在于,在蛋白质水平必须假定在所有物种恒定如果蛋白质或肽水平是平凡等同于混合物中的肉类的水平。对于肌红蛋白和四个红M吃这不是普遍如此。在一般的水平是物种依赖性的,猪肉呈现四个的最低水平。此外,肌红蛋白水平分割肉和动物的年龄而异。所以虽然过渡峰面积的比率可靠地映射到肌红蛋白的比率时,映射到实际的肉类的比率是对有关在该混合物的肉类的可能的来源的假设的估计图。
在这项工作中所概述的方法的不同之处的一些其他发表的贡献的方式。更典型的途径是使用蛋白质组学方法来识别各种不同的物种依赖性标记的肽,在这种情况下,对于不同物种的标记物具有彼此8-12,14,19没有特别的关系。相比之下,我们选择共同所有感兴趣的物种的蛋白质多达物种依赖性序列变体3。除了是中央到我们的相对定量的策略,这具有的优点是样品制备策略可以被优化。此外,这种相应的蛋白质可以预料到的行为类似,例如,在提取或样品如烹调或罐头的商业加工。种属鉴定,然后通过检测不同标记肽的正常进行,而通过检测通常拥有一个或两个序列的差异密切相关肽的CPCP办法物种鉴定的收益。最后,蛋白的定量在另一个重量的一种物种的估计的百分比通过分别基于已知标准7,14,15各蛋白质的绝对定量可能常规进行。然而,使用CPCP方法也没有必要进行校准的方法。相反,相对水平通过比较来自两个物种两个相应肽的信号强度,完全绕过绝对测量载台估计。由于最终目标是重量在ANO一个物种的百分比疗法,一个相对定量,则CPCP既更直接和超过比较两个绝对定量测量更简单。这些特点转化为短的实验时间,预计将用精致的协议,大约两小时,使得该技术可用作食品欺诈检测领域快速的监视工具。
披露声明
The authors have nothing to disclose.
致谢
We acknowledge financial support from Institute of Food research BBSRC Core Strategic Grant funds, BBSRC Project BB/J004545/1.
材料
| Name | Company | Catalog Number | Comments |
| Uniprot database | www.uniprot.org | Freely accessible database of protein sequences | |
| Skyline software | www.skyline.gs.washington.edu | Free software to download that enables the creation of targeted methods for proteomic studies, peptide and fragment prediction | |
| Ammonium bicarbonate | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | O9830 | |
| Methanol, HPLC grade | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 10674922 | |
| Acetonitrile, HPLC grade | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 10010010 | |
| Urea | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | U5378 | |
| Trypsin(from bovine pancreas treated with TPCK) | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | T1426 | |
| Formic acid | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | F0507 | |
| Coomassie Plus Protein Assay Reagent | Thermo Fisher Scientific www.thermofisher.com | 1856210 | |
| Protein standard | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | P0914 | |
| Ultra Turrax homogeniser T25 | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 13190693 | |
| Edmund and Buhler KS10 lab shaker | |||
| Heraeus Fresco 17 Centrifuge | Thermo Fisher Scientific www.thermoscientific.com | 75002420 | |
| Vacuum centrifuge RC 1022 | Jouan | ||
| Plate Reader | |||
| Strata-X 33u polymeric reversed-phase cartridges 60 mg/3 ml tubes | Phenomenex, Macclesfield, UK | 8B-S100-UBJ | |
| 4000 QTrap triple-quadrupole mass spectrometer | AB Sciex, Warrington, UK www.sciex.com | ||
| 1200 rapid resolution LC system | Agilent, Stockport, UK | ||
| XB C18 reversed-phase capillary column (100 mm x 2.1 mm, 2.6 µm particle size) | Phenomenex, Macclesfield, UK www.phenomenex.com | ||
| Analyst 1.6.2 software | AB Sciex, Warrington, UK www.sciex.com | QTrap data acquisition and analysis, including peak area integration | |
| Autosampler vials |
参考文献
- O'Mahony, P. J. Finding horse meat in beef products-a global problem. QJM-An Int. J. Med. 106, 595-597 (2013).
- Sentandreu, M. A., Sentandreu, E. Authenticity of meat products: Tools against fraud. Food Res. Int. 60, 19-29 (2014).
- Watson, A. D., Gunning, Y., Rigby, N. M., Philo, M., Kemsley, E. K. Meat Authentication via Multiple Reaction Monitoring Mass Spectrometry of Myoglobin Peptides. Anal. Chem. 87, 10315-10322 (2015).
- Food Standards Agency. . Report of the investigation by the Food Standards Agency into incidents of adulteration of comminuted beef products with horse meat and DNA. , (2013).
- Taylor, A. J., Linforth, R., Weir, O., Hutton, T., Green, B. Potential of electrospray mass-spectrometry for meat pigment identification. Meat Science. 33, 75-83 (1993).
- Ponce-Alquicira, E., Taylor, A. J. Extraction and ESI-CID-MS/MS analysis of myoglobins from different meat species. Food Chem. 69, 81-86 (2000).
- Gallien, S., Duriez, E., Domon, B. Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 46, 298-312 (2011).
- Orduna, A. R., Husby, E., Yang, C. T., Ghosh, D., Beaudry, F. Assessment of meat authenticity using bioinformatics, targeted peptide biomarkers and high-resolution mass spectrometry. Food Addit. Contam. Part A-Chem. 32, 1709-1717 (2015).
- Claydon, A. J., Grundy, H. H., Charlton, A. J., Romero, M. R. Identification of novel peptides for horse meat speciation in highly processed foodstuffs. Food Addit. Contam. Part A-Chem. 32, 1718-1729 (2015).
- von Bargen, C., Brockmeyer, J., Humpf, H. U. Meat Authentication: A New HPLC-MS/MS Based Method for the Fast and Sensitive Detection of Horse and Pork in Highly Processed Food. J. Agric. Food Chem. 62, 9428-9435 (2014).
- von Bargen, C., Dojahn, J., Waidelich, D., Humpf, H. U., Brockmeyer, J. New Sensitive High-Performance Liquid Chromatography Tandem Mass Spectrometry Method for the Detection of Horse and Pork in Halal Beef. J. Agric. Food Chem. 61, 11986-11994 (2013).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Authentication of processed meat products by peptidomic analysis using rapid ambient mass spectrometry. Food Chem. 187, 297-304 (2015).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Rapid detection of Peptide markers for authentication purposes in raw and cooked meat using ambient liquid extraction surface analysis mass spectrometry. Anal. Chem. 86, 10257-10265 (2014).
- Sentandreu, M. A., Fraser, P. D., Halket, J., Patel, R., Bramley, P. M. A Proteomic-Based Approach for Detection of Chicken in Meat Mixes. J. Proteome Res. 9, 3374-3383 (2010).
- Elliott, M. H., Smith, D. S., Parker, C. E., Borchers, C. Current trends in quantitative proteomics. J. Mass Spectrom. 44, 1637-1660 (2009).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26, 966-968 (2010).
- Keeton, J. T., Ellerbeck, S. M., Nunez de Gonzalez, M. T., Devine, C., Dikeman, M. . Encyclopedia of Meat Sciences. 1, 235-243 (2014).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Rapid Detection of Peptide Markers for Authentication Purposes in Raw and Cooked Meat Using Ambient Liquid Extraction Surface Analysis Mass Spectrometry. Anal. Chem. 86, 10257-10265 (2014).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。