
Method Article
与VirusMapper超分辨率显微镜开源单粒子分析
摘要
这份手稿使用,以便产生纳米级结构的精确模型的单粒子分析应用于超分辨率显微镜图像基于斐济开源软件包VirusMapper。
摘要
超分辨率荧光显微镜正在彻底改变细胞生物学研究。其容量打破的周围300的分辨率极限纳米允许纳米生物复合物和方法的常规成像。增加的分辨率也意味着方法电子显微镜流行,如单粒子分析,可以很容易地应用到超分辨率荧光显微镜。通过结合超分辨率光学成像这种分析方法,可以采取荧光显微镜的特定分子标记能力亚稳结构内产生分子元件的构造图的优点。为此,我们已经开发出一种新的算法 - VirusMapper - 打包为一个易于使用的,高性能,高吞吐量ImageJ的插件。本文介绍了深入的指南,该软件,展示其揭示在生物米新颖的结构特征的能力olecular复合物。这里,我们提出如何组装兼容的数据,并提供关于如何使用该算法单粒子分析适用于超分辨率图像的步骤一步协议。
引言
超分辨率(SR)显微镜已经通过与分子特异性标记,以了解他们至关重要一起提供的能力,以图像的关键分子过程中对细胞生物学产生了重大影响。 SR现在使光镜接近,同时保持光学显微镜的主要好处,如潜在图像活细胞1,2中的分辨率(20-150纳米)以前只有用电子显微镜(EM)实现的。另外,在纳米级水平中发现的结构保守允许单粒子分析(SPA),以SR的数据,在电子显微镜3中广泛使用的一个概念的应用。使用SPA,一个结构的许多高度保守的副本可以被成像和一起平均以提高分辨率,精度或信号与噪声的可视化对象的。当与SR组合使用,SPA已被证明是用于高 - 对一个强大的工具核孔复合体4,5的部件,中心体6,和病毒,如HIV 7和HSV-1 8的recision映射。
然而,SR和SPA的常规联合应用已经缺乏可用的软件的挑战。出于这个原因,我们开发VirusMapper,一个插件来流行的图像处理软件ImageJ的/斐济9。这是第一个免费提供的软件包,用于与被设计来提供快速,方便用户,多通道天真与SR显微镜成像结构的平均荧光图像10广义SPA。虽然设计用于病毒,它可以被应用到任何大分子复合物,其中不同的分子种类可以被成像,识别,和本地化。
VirusMapper可用于生产高精度分子任何已知的结构模型,允许平均尺寸和其它参数的计算。该算法的设计使得它用于分离结构的人群中,提供用于不同取向或不同的形态的状态的确定是特别有用的。此外,多通道成像可用于采用在例的参考信道,其中的底层结构是公知的,从而允许基于引用的结构的发现。上提供了下载和安装该软件的说明https://bitbucket.org/rhenriqueslab/nanoj-virusmapper 。例如数据也可以在那里找到,并建议用户使用上的实例数据的软件试图将其应用到自己之前练习。
在这里,描述了用于使用该插件产生从原始数据SPA模型的步骤。该软件需要包含单ö原始图像ř多重标记的结构作为输入。它返回,受到众多的参数作为软件运行被调整,SPA型号表示成像的结构内的标记的成分的平均分布。
此协议的目标是产生精确SPA模型给出根据图1中所概述的管道成像的结构内的组件的平均本地化。 如图1所示,该软件工作流被有效地划分为三个阶段。第一阶段是将大的图像,从而产生用于每个信道的颗粒堆。这些粒子将被平均,以创建模型和生产用于模型生成种子的单位。第二阶段是产生种子的图像,其被用于记录整个粒子集合中的最后阶段。这是通过选择一个基准信道和手动在该通道中,这将有助于在S中选择的粒子进行电火工品。种子被选择在该参考信道,但可以用于所有信道而产生。颗粒最初由在此通道中安装一个2D高斯重新对准。已被选择和重新排列,所有的颗粒都然后取平均,以产生种子。对于要被建模的数据可以看出每一个公共结构中,颗粒应被选择作为种子,清楚和准确地表示该结构。在这个阶段的接口也用于扫描数据为这样的结构是有用的。
最后一个阶段是通过使用模板匹配,从而生成模型。这是通过最初提取到前面部分由互相关中所产生的晶种图像中的颗粒的登记实现的。注册颗粒的子集一起平均,并且处理被进一步迭代如果需要的话,以减少模型均方误差。该子集是通过设置对种子必须满足一最小相似度来确定。当创建模型同时S IN多个信道,联合相似性,或相似的每个信道的平均,被使用。那对他们作出贡献的所得的模型和注册颗粒然后可进一步分析。
研究方案
注:本协议和视频补充原有纸10在详细描述软件包。鼓励读者关于使用该软件的仔细审查这额外的指导。有三个主要阶段:粒子提取,其中,分段放大图成单独的颗粒;选种,其中常见的结构在数据中识别并对齐,以产生种子,其在最终阶段中使用;和模型生成,其中模板匹配基于这些种子对齐提取的颗粒和平均的子集以产生SPA型号。
1.安装之前运行的软件包
- 在盖玻片上或在相关的实验条件制备所研究的结构的样品。
- 图像的超分辨率荧光显微镜的样品中,如结构化照明显微镜(SIM)11或stimula泰德发射损耗(STED)显微镜12。
注释:如何准备和图像样本在很大程度上取决于所研究的结构的性质,因此相关文献应征询的精确细节。作为一个实例,用于制备和成像牛痘病毒的样品,如这里所使用的精确的方法,在代表性的结果部分中描述。 - 创建的多个视场示出了大量的结构或颗粒,优选数千个单独的副本的图像。被以及彼此分开尽可能并确保图像是无污垢或不感兴趣的其他荧光结构图像的颗粒。
- 打开由文件拖入斐济工具栏或通过选择"文件">"打开"含有斐济颗粒的所有图像。
- 选择"图像">"堆栈">"工具">"串联"来串联图像为单个叠层。然后,如果所得到的图像是一个Hyperstack,通过选择"图像">"Hyperstacks">"Hyperstack到堆叠"把它变成一个堆栈。
注:最终的堆栈应该有夹层通道。如果有两个通道,在所述堆叠中的第一切片应通道1从第一图像,所述第二片应该是对应信道2中,第三片应该是信道1从第二图像,以此类推。
2.提取颗粒
- 选择图像片段,然后选择"提取病毒结构"。选择保存所提取的颗粒,并查看"提取病毒结构" 对话框( 图2)。
- 将它们输入"提取病毒结构"对话框如下分配初步估计提取参数。微调预览图像分割后,这些参数。
- 设置麻木在数据集中为已被成像的不同的荧光通道的数目的通道ER( 例如,2)。
- 设置从哪个颗粒通过检测峰通过输入信道的选择的数量提取在该信道的参考信道。选择最稳定的渠道;即,信道,其中最颗粒具有相同的外观。
注:如果可能的话,在该通道的颗粒将具有一个中央最大。 - 选择是否要申请一个预检测高斯模糊。预检测高斯模糊设置为0申请无残影;如果该值增大时,给定半径的高斯模糊过滤器之前的局部最大值检测施加。使用此功能,如果参考信道不具有中央最大( 例如,环状);模糊诱导一个的外观。
注:(投资回报)的利益分割区域没有应用到他们的高斯模糊,因为此功能只使用D为基准的频道位置的ROI。 - 设置在像素ROI半径(其将围绕每个局部最大值设置)。选择一个值,使得该感兴趣区比最大的颗粒,如在图3中稍大。例如,如果最大的颗粒表现为直径大约30个像素(由眼睛粗略估计),则设置ROI半径至20个像素。
- 设定ROI的数每帧使用至低于100的初始的,相对小的值。
- 设置最大的投资回报率的重叠。如果颗粒完全分离,保留这个小;如果颗粒被聚集,增加此,以使感兴趣区重叠。
- 选择"展前预览"。
注意:当选择此项时,与当前帧相关联的参考信道所提取的ROI将出现。 - 调整ROI半径,ROI的数,和最大ROI重叠以具有的ROI合适大小的周围尽可能多的粒子尽可能,如在图3中 。
- 选择"确定",以运行分割。关闭图像和ROI管理。
注意:不要改变颗粒系列的文件的名称。这些名称必须是格式"病毒颗粒 - channelX"为以下几个部分。
3.选择种子
- 选择"生成种子",选择其中所提取的颗粒被保存的文件夹,并查看"生成种子"对话框和窗( 图4)。
- 将其输入到"生成种子"对话框中,如下分配初始种子选择参数。
- 设置将要装配到对准和中心的所有信道的参考信道。选择其中大部分粒子具有相同的外观,并且具有中央最大,如果可能的信道。
- 选择应该为其生成种子的所有通道的箱子。
- 如果选择的种子应该是ROTAT90°编使用此功能可以与其他机型一致对齐
- 选择是否要申请一个预对准高斯模糊。每个通道的模糊半径设置为0,不应用模糊。增加该值调整之前应用给定半径的高斯模糊滤镜。使用此功能,如果颗粒不具有一个中心最大;模糊诱导一个的外观。
注:种子不会有应用模糊,因为此功能只得到一致的对准。 - 选择是否使用偏移校正分开中心非参考信道的种子,虽然没有旋转它们,通过检查或取消选中"偏移校正"框用于每个信道。
注意:如果信道没有被很好地相互对齐使用此;它也可以是仅用于根据所述参考对准其它信道,而不会偏移校正有用。
- 选择颗粒作为种子使用。搜索到T他粒子序列发现类似于待建模并记录帧数结构的粒子。
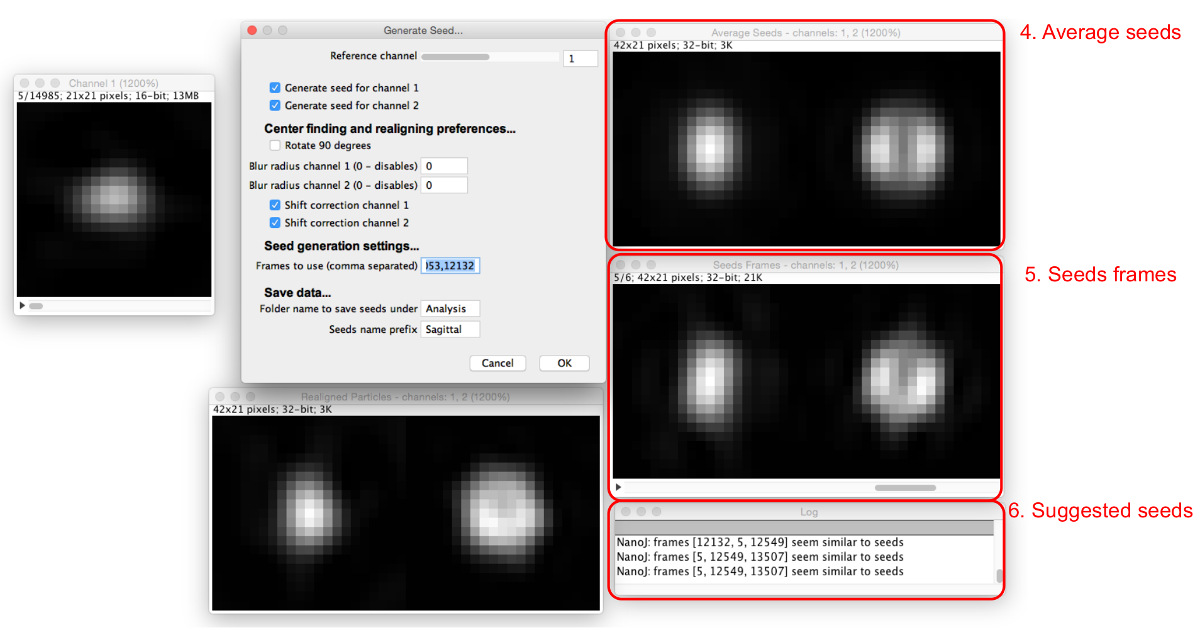
- 在输入号码"框架使用"框中多窗口将出现( 图5)。输入由逗号分隔的多个帧编号。
- 查看种子帧和所述平均所得种子中出现的窗口。
注:日志将建议类似平均种子。 - 调整参考信道,高斯模糊半径,以及偏移校正选项,以便若干发现种子帧的具有相似的外观,以优化种子选择过程。继续添加种子直到平均种子创建的令人满意类似于在待建模的数据看到的结构。
- 命名种子,在那里他们将被保存并选择"OK"保存最终的种子图像在模型生成以后使用。
4.生成模式
- 硒LECT"基于种子模型"中,选择所提取的颗粒被保存的文件夹,并查看"生成模型"对话框( 图6)。
- 通过使用复选框加载用于每个信道的种子。
注:种子将被保存在一个在"生成种子"对话框命名的子文件夹。默认名称是"分析"。小心选择种子的平均值 ,而不是框架,这也被保存以供参考。 - 将其输入到"生成模式"对话框,如下分配初始模型生成的参数。微调这些参数之后。
- 选择是否使用用于对准的参考信道,其只从参考信道计算的平移和旋转,并将其应用于所有通道。如果使用该选项,选择作为参考使用该信道。
注意:此选项只能被选择为专门使用一个信道作为参考,NCE,例如,如果进行基于参考结构的发现。否则,将导致更少的精确模型。这些通道应该如果使用此选项得到很好的对准。 - 选择是否在模板匹配过程中正方形图像强度。这将加重小的差异,因此,创建具有特别细微特征的模型时,使用此。
- 通过使用"最小相似度对种子"滑块或通过输入数字入箱选择针对种子的最小相似性。
注:只有具有相似性的种子比这个截止将使用更大的粒子。 60-80%通常是一个不错的选择开始。 - 迭代的最大数目设置为1,对最优化的种子最小相似度,后来增加它,根据需要。
- 选择框来选择要在模型生成过程的要素。
注:"显示种子"将显示已经加载了boxe种子■在对话框的顶部。 "显示模式"将显示从平均颗粒满足对种子的最低相似的子集创建的模型的所有迭代。 "显示MSE"将显示均方误差(MSE)的图像,突出是最变量模型的领域。 "显示粒子"将显示被用于创建模型的粒子的子集,根据所显示的模型的最高重复注册。
- 选择是否使用用于对准的参考信道,其只从参考信道计算的平移和旋转,并将其应用于所有通道。如果使用该选项,选择作为参考使用该信道。
- 选择"显示预览"生成预览模式并查看结果。
注意:这是该过程的计算最密集的一步。运行时间为一组几千颗粒直径为几十台式PC上的像素的应该是10分钟左右。如果计算时间是一个问题,用户应该首先尝试对数据的较小的子集的算法或者如果可能的话,在步骤2.2.4使用较小的ROI半径。 - 查看生成三维模型和优化参数,特别是针对种子的最小相似性。增加针对种子的最小相似度直到仅有建模形态真实颗粒包括在模型中。
- 通过使用滑块"的迭代的最大数目",或者通过输入一个数到盒子增加迭代的最大数量,并允许模型生成过程进行迭代。为最大迭代使用约10的值。
- 名称型号,然后选择"确定"保存包含最终的模型生成过程的所有迭代模型演进堆栈。
注意:如果对种子的最低相似性是如此之高,没有粒子具有这种相似性,什么都不会被更新。如果插件似乎被冻结,认为最小相似度过高的可能性。
结果
在这里,我们展示的模型痘病毒,牛痘病毒软件。其中最复杂的哺乳动物病毒,牛痘包的350×270×250nm的3砖形颗粒13,14内的约80种不同的蛋白质。三个子结构是通过电子显微镜看到:中央纤芯,其包含双链DNA基因组; 2层蛋白质的结构,称为横向机构,其侧翼的芯;和单个蛋白脂质双层包络15。大尺寸,结构复杂,并顺从重组荧光蛋白标记使牛痘优异的系统来演示VirusMapper工作流程。
使用该软件在此描述,多种对牛痘病毒蛋白的分布进行建模。蛋白质被标记并在COMBI成像,可能如所描述的,以产生在颗粒蛋白质的定位的平均模型使用国家与已知分布作为基准的另一种蛋白质,和该软件。在这个例子中,两种蛋白质被建模,内芯蛋白质L4,和主要侧向主体部件F17。
具有F17的重组痘苗病毒具有标记的GFP和L4具有标记的mCherry 16而使用。纯化的病毒稀释在1毫摩尔Tris,pH为9,并通过将它们包衣,在室温下30分钟结合到洗涤,高性能的盖玻片上。然后将样品在PBS中施加4%甲醛20分钟固定的。盖玻片立即安装到在抗褪色封固剂幻灯片。成像是通过SIM卡在商业SIM显微镜进行。被选择的视场包含数百个病毒和图像用5个相移并用561纳米(3个格转32μm的光栅PE被收购RIOD)和488纳米(32微米光栅周期)激光器。图像使用一个SCMOS相机获取和使用显微镜软件进行处理。基于与所述相同的图像获取的设置成像的多色珠粒滑动通道对齐。之后SIM重建和信道对齐的图像中斐济被打开并连接成单个图像栈。
病毒颗粒是从使用信道L4作为参考和不施加任何高斯模糊的图像中提取的,因为这些颗粒具有中央最大。 15000左右的颗粒在这个实验中提取。
由于牛痘的几何形状,横向机构具有基于病毒取向明显不同的外观。我们可视化两个方向,其中一个或两个横向机构可以区别开来。我们称这些定向额叶和矢状,尊重结构延续。
通过粒子列表中"生成种子"阶段搜索被选定为额叶和矢状方向分开的种子( 图4和5);颗粒显然在一个方向或另一个被选择。在L4信道被用作参考信道,以彼此对准的种子。同样,没有高斯模糊是必要的。选择5个粒子每个方向和进行平均,以产生种子。
基于这些种子每个方向产生模型。既不是参考信道,也不平方强度值使用。最大迭代数最初被设置为1,并且最小相似性被设置为包括在每一种情况下,这给了一个一致的外观对于每个方向大约1000颗粒。然后迭代的最大数量增加至允许模型的收敛。物从而在两个信道的两个方向( 图7)生成的模型。

图1:VirusMapper 工作流程。该插件分为三个主要阶段。病毒颗粒从大的图像中提取,模板图像或种子从数据半手动选择,并最终SPA模型从所述数据通过参照种子生成的。 请点击此处查看该图的放大版本。
{kind=link}

图2:" 提取病毒结构"对话框。当选择"提取病毒Structur ES",该对话框将会出现。该参数应该充满了最佳分割的初步估计。‘展前预览’然后可以选择,使投资回报率进行预览和参数进行微调。 请点击这里查看更大的版本这个数字。
{kind=link}

图3: 设置提取参数。预览将要提取的感兴趣区中,ROI半径,ROI的数,和最大ROI重叠之后被调整以实现这样的情况。的ROI比颗粒稍大,所有的粒子都包含在ROI,并且可以的ROI充分重叠,以允许分离的群集的颗粒。ANK">点击此处查看该图的放大版本。

图4: 生成模板匹配种子。 "生成种子"对话框(1)规定了将被分配的参数。基准粒子序列(2)允许用户通过在所述参考信道扫描的颗粒。当颗粒在颗粒参考序列被观察时,对所有信道重新排列的粒子可以在重新排列粒子预览(3)看到。 请点击此处查看该图的放大版本。
{kind=link}

图5: 添加种子的图片。作为晶种加入到 "帧使用"盒,(5)则显示所有的种子(4)的平均和帧参与。颗粒其类似于目前的平均种子建议在对话框中(6)。 请点击此处查看该图的放大版本。
{kind=link}

图6:" 生成模型"对话框。当选择"基于模型的种子",会出现这个对话框。该参数应该被填充有用于优化模型生成的初始估计,并且计算过程中要显示的模型生成程序的元件应该被选中。 "查看预览"然后可以选择,使模型生成过程以运行和参数进行微调。ftp_upload / 55471 / 55471fig6large.jpg"目标=‘_空白’>点击此处查看该图的放大版本。

图7: 与VirusMapper生成的模型。与具有标记的mCherry的L4核心蛋白并标有EGFP的F17侧向体蛋白的牛痘病毒粒子使用SIM成像。模型,然后用软件生成,如在协议中所述。两个取向,额叶和矢状面,通过横向机构的外观区别。比例尺= 100纳米。 请点击此处查看该图的放大版本。
{kind=link}
讨论
利用这种方法,研究人员都配备SPA和SR显微镜的电源,从而产生的病毒和其他大分子复合物的蛋白质结构的高精度,多通道的二维模型结合起来。然而,一些重要的考虑因素应该被考虑在内。
种子应选择以表示始终可见的结构。因此,原始数据,应检查被选择的种子之前仔细。这是为了防止偏模型非常重要。选择可以通过为包括一定数量的在模型中颗粒的所需的最小相似性阈值的检查来验证。显然,对于种子的选择,这个较高的阈值需要为粒子的给定数量,越该结构是在数据显而易见。
当存在异质性在数据中的模板匹配的概念是特别有用的。所有不同的结构是六锡布尔赫丁应查明并为每种情况创建不同的模型。通过在一个沟道分离异质结构,但同时在第二信道创建模型,图案可以出现,不会一直立即明显。
另一个考虑是知道使用这种算法时是迭代过程将最大限度地随机不对称。例如,建模具有两个对称的最大值的结构的情况下,最大值之间的所有细微的不对称将彼此迭代期间对准,并因此最终模型将是最大程度不对称的。如果这没有反映在结构已知的对称建模,那么这应该加以考虑。目前,为了避免这种最大化的唯一途径是迭代的次数限制为1,虽然发展潜力将是VirusMapper纳入对称轴为模型生成过程。 VirusMapper的任何新版本将是阿瓦伊拉布勒引用的网站上(见材料表 )。用户也将在这里找到一个常见问题回答任何常用查询。
如所描述的软件是适用于能够具有足够分辨率可视化用户希望建模的特征进行成像的任何结构。虽然SPA可以提高分辨率,它显然不会提高在其他方面不可见特征的可见性。这个协议不是,因此,一种方法来提高数据的质量。与任何技术,仔细的样品制备和优化成像策略将提供最干净数据和最佳所得模型。
SR成像模态的选择也很重要,并且在一般情况下,将取决于手边的样品。 VirusMapper已被证实与SIM和STED 10很好地工作,而且还可以提供高品质的定位显微镜数据使用,但应注意在这种情况下服用,稀疏的标签可能会导致类似的不对称性最大化的问题。
目前,VirusMapper为荧光图像的单粒子分析和唯一的通用2D SPA平均软件的唯一免费提供的算法。已经利用了相同的原则4,6其他研究,8使用了专门为每个特定的学习习惯的软件。对于3D数据的重建通用算法已经出版5,18,虽然没有软件提供。
当用作本文中描述所使用的,VirusMapper可用于生产病毒和其它复合物的大分子蛋白质结构的精确,准确和可靠的模型。有了这些模型,研究人员可以利用该结构的平均尺寸的精确测量正在研究数目字,有可能使他们能够达到生物的结论,即否则不会成为可能。
此外,该技术的多通道功能,能够映射络合物内无限数量的蛋白质和部件,并且发现新的蛋白质的组织。检查在纳米结构的变化在不同的生物相关条件,如病毒生命周期的不同阶段,所提供的有价值的见解生物学的潜力。
披露声明
作者什么都没有透露。
致谢
我们要感谢科里纳·比尔,耶日·萨莫勒琼,佩德罗·马特斯·佩雷拉,克里斯托弗·布莱克和凯思林·舍雷尔他们对原有的开发和验证VirusMapper的贡献。我们还要感谢阿尔图尔·亚基莫维奇的手稿的批判性阅读。这项工作是由来自生物技术和生物科学研究理事会的资助(BB / M022374 / 1)(RH);核心资金的MRC实验室分子细胞生物学,伦敦大学学院(JM);欧洲研究理事会(649101-UbiProPox)(JM);和医学研究理事会(MR / K015826 / 1)(RH和JM)。 RG是由工程和物理科学研究理事会(EP / M506448 / 1)资助。
材料
| Name | Company | Catalog Number | Comments |
| Fiji | Open-source image analysis software | ||
| NanoJ-VirusMapper | developed by the Henriques lab | Open source-Fiji plugin (https://bitbucket.org/rhenriqueslab/nanoj-virusmapper) | |
| VectaShield antifade mounting medium | Vector Labs | H-100 | |
| Elyra PS1 | Zeiss | ||
| ZEN BLACK | Zeiss | Image processing software for SIM | |
| High performance coverslip | Zeiss | 474030-9000-000 | |
| TetraSpeck beads | ThermoFisher | T7279 |
参考文献
- Henriques, R., Griffiths, C., Rego, E. H., Mhlanga, M. M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers. 95 (5), 322-331 (2011).
- Gustafsson, N., Culley, S., et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 7, 12471 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 161 (3), 438-449 (2015).
- Szymborska, A., de Marco, A., Daigle, N., Cordes, V. C., Briggs, J. A. G., Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341 (6146), 655-658 (2013).
- Broeken, J., Johnson, H., et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl Fluoresc. 3 (1), 14003 (2015).
- Burns, S., Avena, J., Unruh, J., Yu, Z. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. , (2015).
- Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Nat Acad Sci U S A. 109 (22), 8564-8569 (2012).
- Laine, R. F., Albecka, A., Svan de Linde, ., Rees, E. J., Crump, C. M., Kaminski, C. F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun. 6, 5980 (2015).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Gray, R. D. M., Beerli, C. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci Rep. 6, 29132 (2016).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J of Micros. 198 (2), 82-87 (2000).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Let. 19 (11), 780 (1994).
- Chung, C. -. S., Chen, C. -. H., Ho, M. -. Y., Huang, C. -. Y., Liao, C. -. L., Chang, W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 80 (5), 2127-2140 (2006).
- Moss, B. Poxviridae: the viruses and their replication. Fields virology. , (2010).
- Condit, R. C., Moussatche, N., Traktman, P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 66 (6), 31-124 (2006).
- Schmidt, F. I., Bleck, C. K. E., et al. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 4 (3), 464-476 (2013).
- . Nanoj-virusmapper Available from: https://bitbucket.org/rhenriqueslab/nanoj-virusmapper (2016)
- Fortun, D., Guichard, P., Unser, M. Reconstruction From Multiple Poses in Fluorescence Imaging: Proof of Concept. IEEE J Sel Topics Signal Process. 10 (1), 61-70 (2016).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。