
Method Article
Analisi singola particella open-source per Super-risoluzione Microscopia con VirusMapper
In questo articolo
Riepilogo
Questo manoscritto utilizza l'open-source pacchetto software VirusMapper Fiji-based per applicare l'analisi singola particella di immagini di microscopia super-risoluzione al fine di generare modelli precisi di struttura nanoscala.
Abstract
Super-risoluzione microscopia a fluorescenza è attualmente rivoluzionando la ricerca di biologia cellulare. La sua capacità di rompere il limite di risoluzione di circa 300 nm permette di immagini di routine di complessi biologici nanoscala e processi. Questo aumento della risoluzione significa anche che i metodi popolari microscopia elettronica, come l'analisi singola particella, possono essere facilmente applicati a super-risoluzione microscopia a fluorescenza. Combinando questo approccio analitico con imaging ottico super-risoluzione, diventa possibile sfruttare la capacità etichettatura specifica molecola di microscopia a fluorescenza per generare mappe strutturali degli elementi molecolari all'interno di una struttura metastabile. A questo scopo, abbiamo sviluppato un nuovo algoritmo - VirusMapper - confezionato come un plugin ImageJ facile da usare e ad alte prestazioni, e high-throughput. Questo articolo presenta una guida approfondita a questo software, mostra la capacità di individuare nuove caratteristiche strutturali in m biologicacomplessi olecular. Qui, presentiamo come assemblare dati compatibili e fornire un protocollo passo-passo su come utilizzare questo algoritmo di applicare l'analisi singola particella a immagini super-risoluzione.
Introduzione
Super-risoluzione (SR) microscopio ha avuto un impatto importante sulla biologia cellulare, fornendo la possibilità di processi molecolari chiave di immagine con l'etichettatura specifica molecolare cruciale per la loro comprensione. SR consente ora microscopio ottico per avvicinarsi alle risoluzioni (20-150 nm) precedentemente ottenibile solo con microscopia elettronica (EM) pur mantenendo i principali vantaggi di microscopia ottica, come il potenziale di immagine cellule vive 1, 2. Inoltre, la conservazione strutturale trova a livello nanoscala consente l'applicazione di analisi singola particella (SPA) ai dati SR, un concetto ampiamente utilizzato in microscopia elettronica 3. Utilizzando SPA, molte copie altamente conservate della struttura possono essere esposte e mediati insieme per migliorare la risoluzione, precisione, o segnale-rumore dell'oggetto visualizzato. Quando utilizzato in combinazione con SR, SPA è stato dimostrato di essere un potente strumento per l'alta pmappatura recision dei componenti del complesso del poro nucleare 4, 5, 6 centrosomi e virus, come l'HIV 7 e HSV-1 8.
Tuttavia, l'applicazione combinata di routine della SR e SPA è stata contestata da una mancanza di software disponibile. Per questo motivo, abbiamo sviluppato VirusMapper, un plugin per il popolare software di elaborazione delle immagini ImageJ / Figi 9. Questo è il primo pacchetto software liberamente disponibile per SPA generalizzata con immagini di fluorescenza 10 progettati per fornire veloce, facile da usare, multicanale media naive di strutture viste con microscopio SR. Sebbene progettato per i virus, può essere applicato a qualsiasi complesso macromolecolare in cui differenti specie molecolari possono essere esposte, identificati e localizzati.
VirusMapper può essere utilizzato per la produzione di alta precisione molecolaremodelli di qualsiasi struttura nota, consentendo il calcolo di medie dimensioni ed altri parametri. Il disegno algoritmo rende particolarmente utile per separare popolazioni di strutture, che prevede la determinazione di orientamenti distinte o diversi stati morfologici. Inoltre, l'imaging multicanale può essere utilizzato per impiegare un canale di riferimento nei casi in cui la struttura di base è noto, così da consentire la scoperta struttura di riferimento basata. Le istruzioni per il download e l'installazione del software sono forniti su https://bitbucket.org/rhenriqueslab/nanoj-virusmapper . dati di esempio si possono trovare anche lì, e gli utenti sono invitati a praticare utilizzando il software sui dati di esempio prima di tentare di applicarlo alla propria.
Qui, sono descritti i passaggi per utilizzare questo plugin per produrre modelli spa da dati grezzi. Il software prende immagini prime contenenti o singolor strutture a più marcato come input. Ritorna, soggetto ad una serie di parametri che sono adeguati come il software viene eseguito, modelli SPA mostrano le distribuzioni medie dei componenti etichettati all'interno delle strutture imaged.
L'obiettivo di questo protocollo è quello di produrre precisi modelli SPA dando le localizzazioni medi dei componenti all'interno delle strutture impressi secondo la conduttura illustrato in figura 1. Come mostrato in figura 1, il flusso di lavoro software viene utilmente suddiviso in tre fasi. La prima fase è quella di segmentare immagini di grandi dimensioni, con conseguente pile di particelle per ciascun canale. Queste particelle sono le unità che saranno in media per creare modelli e produrre semi per la generazione del modello. La seconda fase è quella di generare immagini di semi, che vengono utilizzati per registrare l'intero insieme di particelle nella fase finale. Questo viene fatto scegliendo un canale di riferimento e manualmente selezionando particelle in questo canale che contribuirà alla seeds. I semi sono scelti in questo canale di riferimento, ma possono essere generati per tutti i canali. Le particelle sono inizialmente riallineate montando una gaussiana 2D in questo canale. Tutte le particelle che sono state selezionate e riallineate sono quindi mediati per produrre un seme. Per ogni struttura comune visto nei dati che deve essere modellata, le particelle devono essere selezionati come semi che chiaramente e accuratamente rappresentano tale struttura. L'interfaccia in questa fase è anche utile per la scansione dei dati per tali strutture.
La fase finale è quello di generare modelli utilizzando il modello di corrispondenza. Questo risultato è ottenuto attraverso la registrazione delle particelle originariamente estratte alle immagini seme generati nella sezione precedente correlazione incrociata. Un sottoinsieme di particelle registrati è mediata insieme, e il processo viene iterato ulteriormente per ridurre modello errore quadratico medio, se desiderato. Questo sottoinsieme è determinato impostando una somiglianza minima contro il seme che deve essere soddisfatto. Durante la creazione del modellos contemporaneamente in più canali, la somiglianza articolazione, o la media delle somiglianze per ciascun canale, viene utilizzato. I modelli risultanti e le particelle registrati che hanno contribuito ad essi possono essere ulteriormente analizzati.
Protocollo
NOTA: Questo protocollo e il video completano la carta originale 10 che descrive il pacchetto software in modo più dettagliato. I lettori sono invitati a rivedere con attenzione per ulteriori indicazioni per quanto riguarda l'utilizzo del software. Ci sono tre fasi principali: estrazione delle particelle, quali segmenti grandi immagini in singole particelle; selezione del seme, in cui le strutture comuni sono identificati nei dati e allineati per produrre semi, che vengono utilizzati nella fase finale; e generazione di modelli, dove template matching basato su questi semi allinea le particelle estratte e le medie un sottoinsieme di produrre modelli SPA.
1. Installazione prima di eseguire il pacchetto software
- Preparare campioni della struttura in esame su un vetrino o nelle condizioni sperimentali pertinenti.
- Immagine i campioni con super-risoluzione microscopia a fluorescenza, come ad esempio l'illuminazione microscopia strutturato (SIM) 11 o stimolideplezione ted emissione (STED) 12 microscopia.
NOTA: I dettagli precisi su come preparare e campioni di immagini dipende molto dalla natura della struttura in fase di studio, in modo da letteratura rilevante dovrebbe essere consultato. Come esempio, il metodo preciso per la preparazione e l'imaging campioni di virus vaccinia, come quelli utilizzati qui, è descritto nella sezione rappresentativa dei risultati. - Creare immagini di più campi di vista che mostra un gran numero di copie separate della struttura o particelle, preferibilmente migliaia. particelle di immagine che sono così separati l'uno dall'altro come possibile e garantire che le immagini siano puliti o altre strutture fluorescenti che non sono di interesse.
- Aprire tutte le immagini che contengono le particelle in Fiji trascinando i file nella barra degli strumenti Fiji o selezionando "File"> "Apri".
- Selezionare "Immagine"> "stack"> "Strumenti"> "concatenate" per concatenare ilimmagini in un unico stack. Quindi, se l'immagine risultante è un HyperStack, trasformarlo in una pila selezionando "Immagine"> "Hyperstacks"> "HyperStack impilare".
NOTA: Lo stack finale dovrebbe avere canali intercalate. Se vi sono due canali, la prima sezione nella catasta deve essere canale 1 dalla prima immagine, la seconda fetta dovrebbe essere il corrispondente canale 2, la terza fetta dovrebbe essere il canale 1 dalla seconda immagine, e così via.
2. Estrarre i Particelle
- Scegli l'immagine di segmentare e selezionare "Estrai strutture virali". Scegliere dove salvare le particelle estratte e visualizzare i "estratto Strutture virali" dialogo (Figura 2).
- Assegnare i parametri di estrazione con le prime stime inserendoli nella finestra "Extract Strutture virali" come segue. Mettere a punto questi parametri dopo l'anteprima del segmentazione di immagini.
- Impostare l'intorpiditoer di canali nel set di dati come il numero di diversi canali di fluorescenza che sono state immagini formate (per esempio, 2).
- Impostare il canale di riferimento da cui le particelle vengono estratti per il rilevamento di picchi nel canale inserendo il numero della scelta del canale. Selezionare il canale più consistente; cioè, il canale in cui la maggior parte delle particelle ha lo stesso aspetto.
NOTA: Se possibile, particelle in questo canale avranno un massimo centrale. - Scegliere se applicare o meno un pre-rilevamento sfocatura gaussiana. Impostare il pre-rilevamento sfocatura gaussiana a 0 per non applicare sfocatura; se si aumenta questo valore, un filtro sfocatura gaussiana con il raggio indicato viene applicato prima rilevazione massimi locali. Utilizzare questa funzione se il canale di riferimento non ha un massimo centrale (ad esempio, forma ad anello); sfocatura induce la comparsa di uno.
NOTA: segmentato regioni di interesse (ROI) non hanno la sfocatura gaussiana applicata a loro, in quanto questa funzione è solo per usod per posizionare ROI nel canale di riferimento. - Impostare il raggio ROI (che sarà impostato attorno ad ogni massimo locale) in pixel. Scegliere un valore tale che tale ROI sono leggermente più grandi delle grandi particelle, come in figura 3. Ad esempio, se le particelle più grandi sembrano avere un diametro di circa 30 pixel (stimato approssimativamente a occhio), quindi impostare il raggio ROI a 20 pixel.
- Impostare il numero di ROI da utilizzare per frame per un primo, relativamente piccolo valore inferiore a 100.
- Impostare la sovrapposizione il massimo ROI. Se le particelle sono ben separati, mantenere questo piccolo; se le particelle sono raggruppati, aumentare questo per consentire le ROI a sovrapporsi.
- Selezionare "Mostra anteprima".
NOTA: Quando è selezionato, appariranno le ROI estratte per il canale di riferimento associato con il frame corrente. - Regolare il raggio ROI, il numero di ROI, e la sovrapposizione massima ROI avere ROI di dimensioni adeguate intorno come molte particelle possibili, Come nella figura 3.
- Selezionare "OK" per eseguire la segmentazione. Chiudere l'immagine e il gestore ROI.
NOTA: Non modificare i nomi dei file dei set di particelle. Questi nomi devono essere in formato "particelle virali - ChannelX" per le seguenti sezioni.
3. Selezionare Seeds
- Selezionare "Genera Seeds", selezionare la cartella in cui vengono salvati i particelle estratte, e visualizzare il "Genera Semi" di dialogo e le finestre (Figura 4).
- Assegnare i parametri di semi-selezione iniziale inserendoli nella finestra "Genera Seeds", come segue.
- Impostare il canale di riferimento che verrà montato allineare e centro tutti i canali. Scegliere un canale in cui la maggior parte particelle hanno lo stesso aspetto e che ha un massimo centrale, se possibile.
- Selezionare le caselle per tutti i canali per i quali un seme deve essere generato.
- Selezionare se i semi devono essere rotatcato di 90 °. Utilizzare questa caratteristica di avere l'allineamento coerente con gli altri modelli
- Scegliere se applicare o meno una sfocatura gaussiana pre-allineamento. Impostare il raggio di sfocatura per ogni canale a 0 per non applicare sfocatura. Aumentare questo valore per applicare un filtro sfocatura gaussiana con il raggio indicato prima riallineamento. Utilizzare questa funzione se le particelle non hanno un massimo centrale; sfocatura induce la comparsa di uno.
NOTA: I semi non hanno la sfocatura applicata, in quanto questa funzione è solo per ottenere l'allineamento coerente. - Scegliere se utilizzare la correzione spostamento per centrare separatamente i semi per i canali non di riferimento, anche se non ruotarle, selezionando o deselezionando le caselle "correzione Shift" per ciascun canale.
NOTA: Utilizzare questo se i canali non sono ben allineati tra loro; può anche essere utile per l'allineamento altri canali esclusivamente in base al riferimento, senza correzione spostamento.
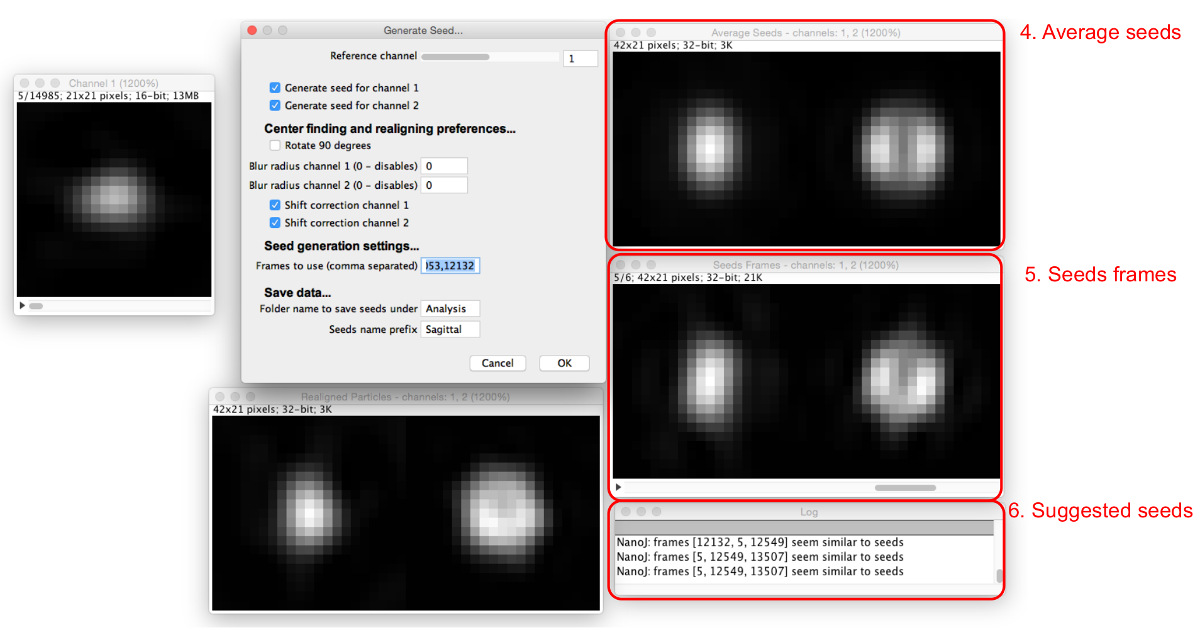
- Scegli particelle da utilizzare come semi. Ricerca attraverso tsi particella sequenza di trovare una particella che assomiglia alla struttura che deve essere modellata e registrare il numero di frame.
- Inserire il numero nella sezione "Frames utilizzare", e appariranno più finestre (Figura 5). Immettere più numeri di frame separati da virgole.
- Visualizzare i fotogrammi di sementi ei semi medi risultanti nelle finestre che appaiono.
NOTA: Il registro suggerirà semi simile alla media. - Regolare il canale di riferimento, il raggio di sfocatura, e le opzioni di correzione di spostamento per ottimizzare il processo di selezione del seme in modo che un numero di fotogrammi seme trovato hanno un aspetto simile. Continuare semi aggiungendo fino a quando i semi media vengono creati in modo soddisfacente che assomigliano la struttura visto nei dati che deve essere modellata.
- Nome i semi e dove saranno salvati e selezionare "OK" per salvare le immagini semi finali per un uso successivo nella generazione del modello.
4. generare modelli
- Selezionare "Genera modelli basati su Seeds", selezionare la cartella in cui salvare le particelle estratte, e visualizzare la finestra "Genera Models" (figura 6).
- Caricare i semi per ogni canale utilizzando le caselle di controllo.
NOTA: I semi verranno salvati in una sottocartella che è stato chiamato nella finestra di dialogo "Genera Seeds". Il nome di default è "Analysis". Fare attenzione a selezionare le medie del seme, non i Frames, che sono anche salvati per riferimento. - Assegnare parametri iniziali generazione modello inserendoli nella finestra di "generare modelli", come segue. Mettere a punto questi parametri in seguito.
- Scegliere se utilizzare un canale di riferimento per l'allineamento, che calcola traslazioni e rotazioni solo dal canale di riferimento e li applica a tutti i canali. Se si utilizza questa opzione, selezionare il canale da utilizzare come riferimento.
NOTA: Questa opzione deve essere scelto soltanto utilizzare specificamente un canale come DI RIFERIMENTOnce, come se facendo scoperta struttura di riferimento-based. In caso contrario, si tradurrà in modelli meno accurati. I canali devono essere ben allineati, se si utilizza questa opzione. - Scegliere se far quadrare intensità di immagine durante template matching. Ciò accentuerà piccole differenze, quindi utilizza questo quando si crea un modello che ha caratteristiche particolarmente sottili.
- Scegli una somiglianza minima contro seme utilizzando il cursore "somiglianza minima contro seme" o inserendo un numero nella casella.
NOTA: particelle Solo con una somiglianza con i semi maggiori di questo cut-off verrà utilizzato. 60-80% è in genere una buona scelta per iniziare. - Impostare il numero massimo di iterazioni a 1, ottimizzare la somiglianza minima contro il seme, e aumentare in un secondo momento, se necessario.
- Selezionare le caselle di scegliere gli elementi del processo di modello di generazione che apparirà.
NOTA: "Mostra semi" visualizzerà i semi che sono stati caricati boxe con ils in alto della finestra di dialogo. "Mostra modelli" mostrerà tutte le iterazioni dei modelli che vengono creati da una media del sottoinsieme di particelle che incontra la somiglianza minima contro il seme. "Mostra MSE" mostrerà un'immagine di errore quadratico medio (MSE) che evidenzia le aree del modello che sono più variabili. "Mostra particelle" mostreranno il sottoinsieme di particelle che vengono utilizzati per creare i modelli, registrato secondo la massima iterazione del modello che viene visualizzato.
- Scegliere se utilizzare un canale di riferimento per l'allineamento, che calcola traslazioni e rotazioni solo dal canale di riferimento e li applica a tutti i canali. Se si utilizza questa opzione, selezionare il canale da utilizzare come riferimento.
- Selezionare "Mostra anteprima" per generare modelli di anteprima e visualizzare i risultati.
NOTA: Questo è il passo più intensa vista computazionale del processo. Il tempo di esecuzione per un insieme di poche migliaia di particelle con un diametro di poche decine di pixel su un PC desktop dovrebbe essere intorno a 10 min. Se il tempo di calcolo è un problema, gli utenti devono prima provare l'algoritmo su un sottoinsieme più piccolo dei dati o utilizzare un raggio minore ROI nel passaggio 2.2.4, se possibile. - Visualizza il generared modelli e ottimizzare i parametri, in particolare la somiglianza minima contro il seme. Aumentare la somiglianza minima contro seme fino unici veri particelle della morfologia modellato sono inclusi nel modello.
- Aumentare il numero massimo di iterazioni utilizzando il "numero massimo di iterazioni" slider o inserendo un numero nella casella e consentire al processo di generazione modello iterare. Utilizzare un valore di circa 10 per l'iterazione massima.
- Nome modelli e selezionare "OK" per salvare le pile evoluzione del modello che contengono tutte le iterazioni del processo finale di generazione del modello.
NOTA: Se la somiglianza minima contro seme è così alta che nessun particelle hanno questa somiglianza, non sarà aggiornato. Se il plugin sembra essersi fermato, considerare la possibilità che la somiglianza minima è troppo alto.
Risultati
Qui, dimostriamo il software sul modello poxvirus, virus vaccino. Uno dei virus di mammifero più complesse, pacchetti vaccinia circa 80 differenti proteine all'interno di un 350 x 270 x 250 nm 3 particelle a forma di mattone 13, 14. Tre sottostrutture sono distinguibili mediante microscopia elettronica: un nucleo centrale, che contiene il genoma dsDNA; due strutture proteiche, corpi laterali chiamati, che fiancheggiano il nucleo; e un singolo doppio strato proteolipid busta 15. Le grandi dimensioni, struttura complessa, e amenability ricombinante codifica proteina fluorescente fanno vaccinia un ottimo sistema per dimostrare il workflow VirusMapper.
Utilizzando il software come descritto qui, la distribuzione di una varietà di proteine sulla virione vaccinia può essere modellato. Una proteina è stato etichettato e ripreso, eventualmente in combinazione con un'altra proteina di distribuzione nota come riferimento, e il software è stato usato come descritto per produrre modelli medi della localizzazione di tale proteina sulla particella. In questo esempio, due proteine sono state modellate, L4 proteina nucleo interno, e il principale componente F17 corpo laterale.
Un virus vaccinico ricombinante che ha F17 etichettato con GFP e L4 etichettato con mCherry 16 è stato utilizzato. virus purificato è stato diluito in 1 mM Tris, pH 9, e destinato a coprioggetto lavato, ad alte prestazioni rivestendoli per 30 minuti a temperatura ambiente. I campioni sono stati quindi fissati applicando formaldeide al 4% in PBS per 20 minuti. Vetrini sono stati montati su vetrini immediatamente a antifade mezzo di montaggio. Imaging è stata effettuata da SIM su un microscopio SIM commerciale. Un campo di vista è stato selezionato contenente centinaia di virus e le immagini sono state acquisite utilizzando 5 sfasamenti e 3 rotazioni griglia con 561 nm (32 micron reticolo periodo) e 488 nm (32 um reticolo periodo) laser. Immagini sono state acquisite utilizzando una fotocamera sCMOS ed elaborati utilizzando il software microscopio. Canali erano allineati basato su un vetrino multicolore tallone ripreso con le stesse impostazioni di acquisizione delle immagini. Dopo SIM ricostruzione e di allineamento del canale le immagini sono state aperte nelle isole Figi e concatenati in una singola serie di immagini.
particelle virali sono stati estratti dalle immagini utilizzando il canale L4 come riferimento e senza applicare alcuna sfocatura gaussiana, come queste particelle hanno un massimo centrale. Circa 15.000 particelle sono stati estratti in questo esperimento.
A causa della geometria di vaccinia, i corpi laterali hanno un aspetto nettamente diverso in base all'orientamento virus. Abbiamo visualizzato due orientamenti in cui uno o due corpi laterali potrebbero essere distinti. Abbiamo fatto riferimento a questi orientamenti come frontale e sagittale, il rispettotivamente.
Semi separati per frontale e sagittale orientamenti sono stati selezionati dalla ricerca attraverso l'elenco delle particelle nella fase "Generate Seeds" (figure 4 e 5); particelle che erano chiaramente in un senso o nell'altro sono stati scelti. Il canale L4 è stato utilizzato come canale di riferimento per allineare i semi uno con l'altro. Anche in questo caso, nessuna sfocatura gaussiana era necessario. 5 particelle per ciascun orientamento sono stati selezionati e sono stati mediati per produrre i semi.
I modelli sono stati generati per ciascun orientamento sulla base di questi semi. Né un canale di riferimento né valori di intensità quadrati sono stati usati. Il numero massimo di iterazioni è stata impostata inizialmente a 1, e la somiglianza minimo è stato impostato per includere circa 1000 particelle in ogni caso, che ha dato un aspetto coerente per ogni orientamento. Il numero massimo di iterazioni è stata poi aumentata aconsentire la convergenza del modello. I modelli sono stati così generati per i due orientamenti nei due canali (Figura 7).

Figura 1: workflow VirusMapper. Il plugin è organizzata in tre fasi principali. particelle virali sono estratti da immagini di grandi dimensioni, immagini modello o semi vengono selezionati semi-manualmente dai dati, e modelli finali SPA vengono generati dai dati facendo riferimento ai semi. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2: "Estratto Strutture virale" finestra di dialogo. Quando si seleziona "Estrai Viral Structur es", apparirà questa finestra di dialogo. I parametri devono essere riempiti con le prime stime per la segmentazione ottimale. 'Mostra anteprima' può quindi essere selezionato, permettendo ai ROI da visualizzare in anteprima ed i parametri per essere messo a punto. Cliccate qui per visualizzare un più grande versione di questa figura.
{kind=link}

Figura 3: impostazione dei parametri di estrazione. Dopo l'anteprima ROI che saranno estratti, il raggio ROI, numero di ROI, e massima sovrapposizione ROI sono adeguati per raggiungere una situazione come questa. ROI sono leggermente più grandi delle particelle, tutte le particelle sono inclusi in un ROI e ROI possono sovrapporsi in misura sufficiente a consentire particelle cluster di essere separati.ank "> Clicca qui per vedere una versione più grande di questa figura.

Figura 4: Generazione di modelli semi di corrispondenza. La finestra di dialogo "Genera Seeds" (1) stabilisce i parametri da assegnare. La sequenza particelle di riferimento (2) consente all'utente di eseguire la scansione attraverso particelle nel canale di riferimento. Quando una particella viene visualizzato nella sequenza particelle di riferimento, le particelle riallineate per tutti i canali possono essere visualizzati nelle anteprime particelle riallineate (3). Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 5: Aggiunta di immagini seme. Come i semi vengono aggiunti alla "Frames utilizzare", vengono visualizzate la media di tutti i semi (4) e le cornici coinvolti (5). Le particelle che sono simili agli attuali semi medi sono suggerite nella finestra di dialogo (6). Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 6: "generare modelli" di dialogo. Quando si seleziona "Genera modelli basati su Seeds," apparirà questa finestra di dialogo. I parametri devono essere riempiti con stime iniziali per la generazione ottimale modello, e gli elementi della procedura di generazione di modello per essere visualizzati durante il calcolo deve essere selezionato. può quindi essere selezionato "Mostra anteprima", permettendo al processo di modello di generazione per l'esecuzione ed i parametri per essere messo a punto.ftp_upload / 55471 / 55471fig6large.jpg" target = '_ blank'> Clicca qui per vedere una versione più grande di questa figura.

Figura 7: modelli generati con VirusMapper. virioni vaccinia con la proteina core L4 contrassegnate con mCherry e la proteina corpo F17 laterale contrassegnate con EGFP sono stati ripresi utilizzando SIM. Modelli sono stati poi generati con il software, come descritto nel protocollo. Due orientamenti, frontale e sagittale, si distinguono per l'aspetto dei corpi laterali. bar scala = 100 nm. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}
Discussione
Con questo metodo, i ricercatori sono in grado di combinare la potenza di SPA e microscopia SR al fine di generare alta precisione, modelli 2D multicanale dell'architettura proteine di virus e altri complessi macromolecolari. Tuttavia, alcune importanti considerazioni dovrebbero essere prese in considerazione.
I semi dovrebbero essere scelti per rappresentare una struttura che è costantemente visibile. Così, i dati grezzi devono essere controllati con attenzione prima che i semi sono scelti. Questo è importante per prevenire modelli polarizzati. Scelte possono essere convalidate dall'esame delle soglie minime somiglianza necessari per includere un certo numero di particelle nei modelli. Chiaramente, per una scelta di sementi, maggiore è questa soglia deve essere per un dato numero di particelle, tanto più che la struttura è evidente nei dati.
Il concetto di template matching è particolarmente utile quando v'è eterogeneità nei dati. Tutte le strutture differenti che sono VIbile dovrebbe essere identificato e diversi modelli creati per ogni singolo caso. Separando strutture eterogenee in un canale ma contemporaneamente la creazione di modelli in un secondo canale, i modelli possono emergere che non sarebbe stato immediatamente evidente.
Un'altra considerazione da tenere presente quando si utilizza questo algoritmo è che la procedura di iterazione massimizzerà asimmetria stocastico. Ad esempio, quando si modellano una struttura con due massimi simmetrica, tutte lievi asimmetrie tra i massimi saranno allineati con l'altro durante l'iterazione, e il modello finale saranno pertanto massimamente asimmetrico. Se questo non riflette una simmetria noto nella struttura essendo modellata, allora questo dovrebbe essere preso in considerazione. Attualmente, l'unico modo per evitare questo massimizzazione è quello di limitare il numero di iterazioni da 1, anche se un potenziale di sviluppo sarebbe per VirusMapper incorporare assi di simmetria nel processo di generazione del modello. Eventuali nuove versioni di VirusMapper saranno Available sul sito web di riferimento (vedi Materiali Tavolo). Gli utenti potranno anche trovare una FAQ qui per rispondere a qualsiasi domanda comuni.
Il software descritto è applicabile a qualsiasi struttura che può essere ripreso con una risoluzione sufficiente per visualizzare le funzioni che l'utente desidera modellare. Sebbene SPA può migliorare la risoluzione, chiaramente non migliorerà la visibilità delle caratteristiche che non sono altrimenti visibili. Questo protocollo non è, quindi, un metodo per migliorare la qualità dei dati. Come con qualsiasi tecnica, preparazione del campione attenta e l'ottimizzazione della strategia di imaging fornirà i dati più pulite e migliori modelli risultanti.
La scelta della modalità di imaging SR è importante e, in generale, dipenderà dal campione a mano. VirusMapper è stato validato per lavorare bene con SIM e STED 10, e può essere utilizzato anche con i dati di localizzazione microscopia di alta qualità, ma la cura dovrebbe essere presa in questo caso,come l'etichettatura sparse potrebbe causare problemi simili a quelli di asimmetria di massimizzazione.
Attualmente, VirusMapper è l'algoritmo unico liberamente disponibili per l'analisi singola particella di immagini di fluorescenza e l'unico normale software media 2D SPA. Altri studi che hanno fatto uso degli stessi principi 4, 6, 8 hanno usato software personalizzato specializzato per ogni studio particolare. Algoritmi di impiego generale per la ricostruzione di dati 3D sono stati pubblicati 5, 18, anche se nessun software è stato fornito.
Quando viene utilizzato come descritto in questo articolo, VirusMapper può essere usato per produrre modelli precisi, accurati e robusti dell'architettura proteina macromolecolare di virus e altri complessi. Con questi modelli, i ricercatori possono effettuare misurazioni precise delle dimensioni medie del structUres in fase di studio, potenzialmente permettendo loro di raggiungere conclusioni biologiche che non sarebbe altrimenti stato possibile.
Inoltre, con le funzionalità multi-canale di questa tecnica, è possibile mappare un numero illimitato di proteine e componenti all'interno di complessi e di scoprire l'organizzazione delle proteine romanzo. Esaminando cambiamenti nella struttura nanometrica in diverse condizioni biologicamente rilevanti, come ad esempio diverse fasi di un ciclo di vita del virus, ha il potenziale per offrire informazioni preziose nella biologia.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Vorremmo ringraziare Corina Beerli, Jerzy Samolej, Pedro Matos Pereira, Christopher Bleck, e Kathrin Scherer per il loro contributo allo sviluppo originale e la validazione di VirusMapper. Vorremmo anche ringraziare Artur Yakimovich per la sua lettura critica del manoscritto. Questo lavoro è stato finanziato da sovvenzioni dal Biotecnologie e Scienze Biologiche Research Council (BB / M022374 / 1) (RH); finanziamento di base al Laboratorio MRC per biologia cellulare molecolare, University College di Londra (JM); Consiglio europeo della ricerca (649.101-UbiProPox) (JM); e il Medical Research Council (MR / K015826 / 1) (RH e JM). RG è finanziato dal Dipartimento di Ingegneria e Scienze Fisiche Research Council (EP / M506448 / 1).
Materiali
| Name | Company | Catalog Number | Comments |
| Fiji | Open-source image analysis software | ||
| NanoJ-VirusMapper | developed by the Henriques lab | Open source-Fiji plugin (https://bitbucket.org/rhenriqueslab/nanoj-virusmapper) | |
| VectaShield antifade mounting medium | Vector Labs | H-100 | |
| Elyra PS1 | Zeiss | ||
| ZEN BLACK | Zeiss | Image processing software for SIM | |
| High performance coverslip | Zeiss | 474030-9000-000 | |
| TetraSpeck beads | ThermoFisher | T7279 |
Riferimenti
- Henriques, R., Griffiths, C., Rego, E. H., Mhlanga, M. M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers. 95 (5), 322-331 (2011).
- Gustafsson, N., Culley, S., et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 7, 12471 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 161 (3), 438-449 (2015).
- Szymborska, A., de Marco, A., Daigle, N., Cordes, V. C., Briggs, J. A. G., Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341 (6146), 655-658 (2013).
- Broeken, J., Johnson, H., et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl Fluoresc. 3 (1), 14003 (2015).
- Burns, S., Avena, J., Unruh, J., Yu, Z. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. , (2015).
- Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Nat Acad Sci U S A. 109 (22), 8564-8569 (2012).
- Laine, R. F., Albecka, A., Svan de Linde, ., Rees, E. J., Crump, C. M., Kaminski, C. F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun. 6, 5980 (2015).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Gray, R. D. M., Beerli, C. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci Rep. 6, 29132 (2016).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J of Micros. 198 (2), 82-87 (2000).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Let. 19 (11), 780 (1994).
- Chung, C. -. S., Chen, C. -. H., Ho, M. -. Y., Huang, C. -. Y., Liao, C. -. L., Chang, W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 80 (5), 2127-2140 (2006).
- Moss, B. Poxviridae: the viruses and their replication. Fields virology. , (2010).
- Condit, R. C., Moussatche, N., Traktman, P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 66 (6), 31-124 (2006).
- Schmidt, F. I., Bleck, C. K. E., et al. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 4 (3), 464-476 (2013).
- . Nanoj-virusmapper Available from: https://bitbucket.org/rhenriqueslab/nanoj-virusmapper (2016)
- Fortun, D., Guichard, P., Unser, M. Reconstruction From Multiple Poses in Fluorescence Imaging: Proof of Concept. IEEE J Sel Topics Signal Process. 10 (1), 61-70 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati