
Method Article
Análisis de una sola partícula de código abierto para el Super-resolución Microscopía con VirusMapper
En este artículo
Resumen
Este manuscrito utiliza el paquete de software de fuente abierta VirusMapper basada en Fiji de aplicar el análisis de una sola partícula de imágenes de microscopía de super-resolución con el fin de generar modelos precisos de estructura a nanoescala.
Resumen
Super-resolución de la microscopía de fluorescencia está revolucionando la investigación en biología celular. Su capacidad para romper el límite de resolución de alrededor de 300 nm permite la formación de imágenes de rutina de los complejos y los procesos biológicos a nanoescala. Este aumento de la resolución también significa que los métodos populares en microscopía electrónica, como el análisis de una sola partícula, se pueden aplicar fácilmente a la microscopía de fluorescencia super-resolución. Mediante la combinación de este enfoque analítico con imágenes ópticas super-resolución, se hace posible para tomar ventaja de la capacidad de etiquetado de la molécula específica de la microscopía de fluorescencia para generar mapas estructurales de elementos moleculares dentro de una estructura metaestable. Con este fin, hemos desarrollado un nuevo algoritmo - VirusMapper - empaquetado como un plugin de ImageJ fácil de usar y de alto rendimiento y de alto rendimiento. Este artículo presenta una guía en profundidad para este software, mostrando su capacidad para descubrir características estructurales novedosas en m biológicacomplejos MOLECULAR. A continuación, presentamos cómo ensamblar datos compatibles y proporcionar un protocolo de paso a paso sobre cómo utilizar este algoritmo para aplicar el análisis de una sola partícula de las imágenes super-resolución.
Introducción
Super-resolución (SR) microscopía ha tenido un impacto importante en la biología celular, proporcionando la capacidad de los procesos moleculares clave de la imagen mediante el marcaje específico molecular crucial para la comprensión de ellos. SR permite ahora el microscopio óptico para acercarse a las resoluciones (20-150 nm) antes sólo alcanzables con la microscopía electrónica (EM), mientras que la retención de los principales beneficios de la microscopía de luz, tales como el potencial de las células vivas imagen 1, 2. Además, la conservación estructural encontrado en el nivel de nanoescala permite la aplicación de análisis de una sola partícula (SPA) a los datos de SR, un concepto que se utiliza ampliamente en la microscopía electrónica 3. Uso de SPA, muchas copias altamente conservadas de una estructura se pueden obtener imágenes y se promedian en conjunto para mejorar la resolución, precisión, o relación señal-ruido del objeto visualizado. Cuando se utiliza en combinación con SR, SPA ha demostrado ser una herramienta poderosa para la alta pmapeo recision de componentes del complejo de poro nuclear 4, 5, 6 centrosomas, y virus, como el VIH 7 y HSV-1 8.
Sin embargo, la aplicación combinada de rutina de SR y SPA ha sido cuestionada por la falta de software disponible. Por esta razón, hemos desarrollado VirusMapper, un plugin para el popular software de procesamiento de imágenes ImageJ / Fiji 9. Este es el primer paquete de software de libre acceso para SPA generalizada con 10 imágenes de fluorescencia diseñados para proporcionar rápida fácil de usar, con un promedio de múltiples canales ingenua de estructuras proyectadas con el microscopio SR. Aunque está diseñado para virus, se puede aplicar a cualquier complejo macromolecular en el que las especies moleculares diferentes se pueden obtener imágenes, identificados y localizados.
VirusMapper se puede utilizar para producir alta precisión molecularmodelos de cualquier estructura conocida, lo que permite el cálculo de dimensiones medias y otros parámetros. El diseño algoritmo hace que sea particularmente útil para la separación de poblaciones de estructuras, proporcionando para la determinación de las orientaciones distintas o diferentes estados morfológicos. Además, las imágenes de múltiples canales se puede utilizar para emplear un canal de referencia en los casos en que la estructura subyacente es bien conocido, permitiendo de este modo para el descubrimiento de la estructura a base de referencia. Las instrucciones para descargar e instalar el software se proporcionan en https://bitbucket.org/rhenriqueslab/nanoj-virusmapper . Ejemplo de datos también se pueden encontrar allí, y se aconseja a los usuarios a practicar el uso del software en el ejemplo de datos antes de intentar aplicarlo a su propia cuenta.
Aquí, se describen los pasos para utilizar este plugin para producir modelos de SPA de datos en bruto. El software toma imágenes en bruto que contienen una sola or estructuras multi-etiquetado como entrada. Devuelve, sujeto a una serie de parámetros que se ajustan como el software se ejecuta, los modelos de hidromasaje Se muestran las distribuciones medias de los componentes etiquetados dentro de las estructuras de imágenes.
El objetivo de este protocolo es para producir modelos de SPA precisas que dan las localizaciones medias de los componentes dentro de las estructuras fotografiadas de acuerdo con la tubería indica en la Figura 1. Como se muestra en la Figura 1, el flujo de trabajo de software se divide de forma útil en tres etapas. La primera etapa consiste en segmento de grandes imágenes, resultando en pilas de partículas para cada canal. Estas partículas son las unidades que se promedian para crear modelos y para producir semillas para la generación de modelo. La segunda etapa es para generar imágenes de semillas, que se utilizan para registrar la totalidad del conjunto de partículas en la etapa final. Esto se hace mediante la elección de un canal de referencia y seleccionar manualmente las partículas en este canal que va a contribuir a la seeds. Las semillas se eligen en este canal de referencia, pero pueden ser generados para todos los canales. Las partículas son realineados inicialmente por el ajuste de una gaussiana 2D en este canal. Todas las partículas que han sido seleccionados y realineados son entonces promediadas para producir una semilla. Para cada estructura común que se observa en los datos que debe ser modelado, las partículas deben ser seleccionados como semillas que representan claramente y con precisión esa estructura. La interfaz en esta etapa también es útil para la exploración de los datos para tales estructuras.
La etapa final es la generación de modelos usando coincidencia de plantilla. Esto se logra mediante el registro de las partículas extraídas originalmente a las imágenes de semillas generadas en la sección anterior por correlación cruzada. Un subconjunto de partículas registrados se promedia juntos, y el proceso se itera aún más a reducir modelo de error cuadrático medio, si se desea. Este subconjunto se determina mediante el establecimiento de una similitud mínima frente a la semilla que debe ser satisfecha. Al crear el modeloS simultáneamente en múltiples canales, la similitud conjunta, o la media de las similitudes para cada canal, se utiliza. Los modelos resultantes y las partículas registrados que contribuyeron a ellos, entonces pueden analizarse más.
Protocolo
NOTA: Este protocolo y vídeo complementan el documento original 10 que describe el paquete de software con más detalle. Se invita al lector a revisar esto con cuidado para obtener orientación adicional con respecto al uso del software. Hay tres etapas principales: la extracción de partícula, que los segmentos de imágenes de gran tamaño en partículas individuales; selección de semillas, donde se identifican estructuras comunes en los datos y alineados para producir semillas, que se usan en la etapa final; y la generación de modelo, donde la plantilla coincidente basa en estas semillas alinea las partículas y los promedios extraídos un subconjunto para producir los modelos de spa.
1. Configuración antes de ejecutar el paquete de software
- Preparar muestras de la estructura en estudio sobre un cubreobjetos o en las condiciones experimentales relevantes.
- Image las muestras con microscopía de fluorescencia de super-resolución, tales como estructurado microscopía de iluminación (SIM) 11 o STIMULAagotamiento ted emisión (STED) 12 microscopía.
NOTA: Los detalles precisos de cómo preparar y muestras de imagen depende en gran medida de la naturaleza de la estructura en estudio, por lo que la literatura relevante debe ser consultado. Como un ejemplo, el método preciso para la preparación y obtención de imágenes de muestras de virus vaccinia, tales como los que se usan aquí, se describe en la sección de resultados representativo. - Crear imágenes de múltiples campos de visión que muestra un gran número de copias separadas de la estructura o partículas, preferiblemente miles. partículas de imagen que están así separados entre sí como sea posible y aseguran que las imágenes son libres de suciedad u otras estructuras fluorescentes que no son de interés.
- Abrir todas las imágenes que contienen las partículas en Fiji arrastrando los archivos en la barra de herramientas de Fiji o seleccionando "Archivo"> "Abrir".
- Seleccione "Imagen"> "pila"> "Herramientas"> "concatenar" para concatenar ellas imágenes en una sola pila. Entonces, si la imagen resultante es un HyperStack, convertirlo en una pila al seleccionar "Imagen"> "Hyperstacks"> "HyperStack apilar".
NOTA: La pila final debe tener canales intercalados. Si hay dos canales, la primera rebanada de la pila debe ser el canal 1 de la primera imagen, la segunda rebanada debe ser el correspondiente canal 2, la tercera rebanada debe ser el canal 1 de la segunda imagen, y así sucesivamente.
2. Extrae las partículas
- Elija la imagen para segmentar y seleccionar "Extraer estructuras virales". Elegir dónde guardar las partículas extraídas y ver los "Extracto Estructuras viral" de diálogo (Figura 2).
- Asignar parámetros de extracción con las estimaciones iniciales introduciéndolos en el cuadro de diálogo "Extraer estructuras virales" de la siguiente manera. Afinar estos parámetros después de la vista previa de la segmentación de imágenes.
- Ajuste el entumecidaer de canales en el conjunto de datos como el número de diferentes canales de fluorescencia que han sido la imagen (por ejemplo, 2).
- Ajuste el canal de referencia desde el cual las partículas se extraen por la detección de picos en ese canal introduciendo el número de la elección del canal. Seleccionar el canal más consistente; es decir, el canal en el que la mayoría de las partículas tienen la misma apariencia.
NOTA: Si es posible, las partículas en este canal tendrán un máximo central. - Elegir si desea o no aplicar un desenfoque gaussiano pre-detección. Ajuste el desenfoque gaussiano pre-detección de 0 a aplicar ninguna falta de definición; si se incrementa este valor, un filtro de desenfoque gaussiano de la radio dado se aplica antes de la detección máximos locales. Utilice esta función si el canal de referencia no tiene un máximo central (por ejemplo, forma de anillo); desdibujamiento induce la aparición de uno.
NOTA: segmentado regiones de interés (ROI) no tienen el desenfoque gaussiano aplicado a ellos, ya que esta característica es sólo para usod para colocar ROIs en el canal de referencia. - Ajuste el radio de ROI (que se encuentra alrededor de cada máximo local) en píxeles. Elija un valor tal que que las ROIs son ligeramente más grandes que las partículas más grandes, tales como en la Figura 3. Por ejemplo, si las partículas más grandes parecen tener un diámetro de alrededor de 30 pixeles (estimado más o menos por ojo), a continuación, establecer el radio ROI a 20 píxeles.
- Ajuste el número de regiones de interés para usar por trama para un valor relativamente pequeño inicial, por debajo de 100.
- Establecer el máximo retorno de la inversión se superponen. Si las partículas están bien separados, mantener esta pequeña; Si se agrupan las partículas, aumentar esta opción para activar las regiones de interés a superponerse.
- Seleccione "Mostrar vista previa".
NOTA: Cuando se selecciona esta opción, aparecerán los ROIs extraídos para el canal de referencia asociado con el cuadro actual. - Ajuste el radio de ROI, el número de regiones de interés, y la superposición ROI máximo tener ROIs de tamaño adecuado alrededor de tantas partículas como sea posible, Como en la Figura 3.
- Seleccione "OK" para ejecutar la segmentación. Cerrar la imagen y el gestor de retorno de la inversión.
NOTA: No cambie los nombres de los archivos de los conjuntos de partículas. Estos nombres deben estar en el formato "partículas virales - channelx" para las siguientes secciones.
3. Seleccione Semillas
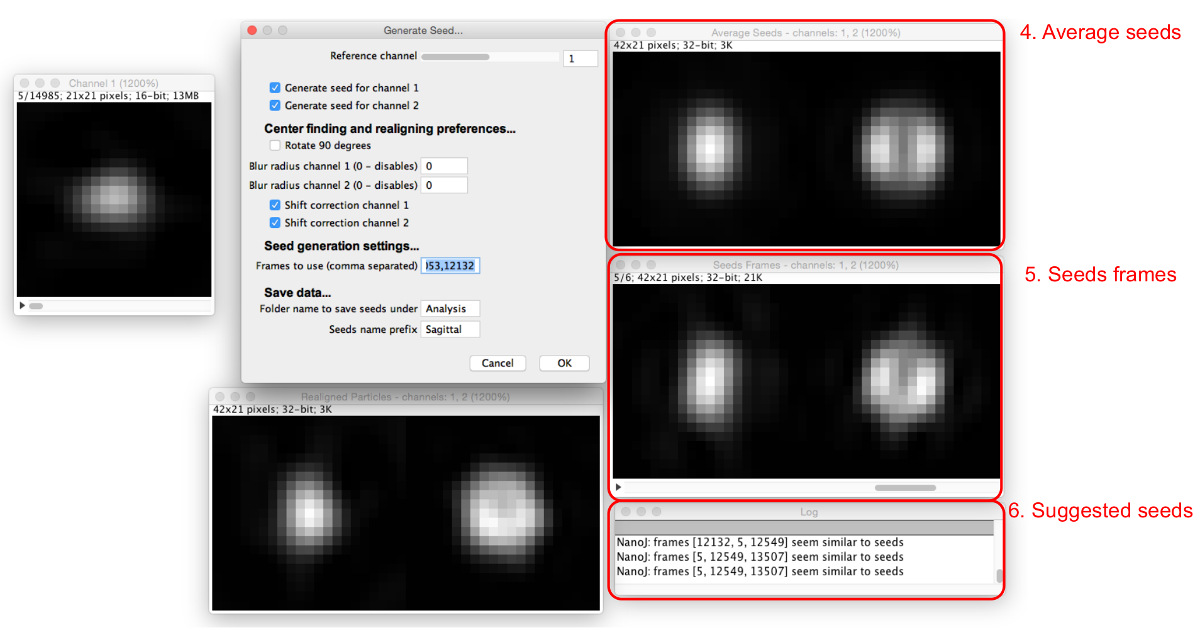
- Seleccionar "Generar Seeds", seleccionar la carpeta donde se guardan las partículas extraídas, y ver el cuadro de diálogo y ventanas "Generar Seeds" (Figura 4).
- Asignar parámetros iniciales de selección de las semillas mediante la introducción de ellos en el cuadro de diálogo "Generar semillas", como sigue.
- Ajuste el canal de referencia que será equipado para alinear y centro de todos los canales. Elegir un canal en el que la mayoría de las partículas tienen la misma apariencia y que tiene un máximo central, si es posible.
- Seleccione las casillas para todos los canales en los que se debe generar una semilla.
- Seleccione si las semillas deben ser Rotated por 90 °. Utilice esta función para tener la alineación consistente con otros modelos
- Elegir si desea o no aplicar un desenfoque gaussiano de pre-alineación. Ajuste el radio de desenfoque para cada canal a 0 para aplicar ninguna falta de definición. Aumentar este valor a aplicar un filtro de desenfoque gaussiano de la radio dado antes de realineación. Utilice esta función si las partículas no tienen un máximo central; desdibujamiento induce la aparición de uno.
NOTA: Las semillas no tienen el desenfoque aplicado, ya que esta característica es sólo para conseguir una alineación consistente. - Seleccione si desea utilizar la corrección de desplazamiento para centrar por separado semillas para los canales no son de referencia, aunque no girarlas, activando o desactivando las casillas de "corrección" Shift para cada canal.
NOTA: Utilice esta si los canales no están bien alineados entre sí; también puede ser útil para alinear otros canales únicamente en función de la referencia, sin corrección de desplazamiento.
- Elija partículas para usar como semillas. Buscar a través de tél de partículas secuencia de encontrar una partícula que se asemeja a la estructura que debe ser modelado y registrar el número de cuadro.
- Introduzca el número de los "marcos de utilizar", y aparecerán más ventanas (Figura 5). Introducir varios números de bastidor separados por comas.
- Ver los marcos de semillas y las semillas resultantes promedio en las ventanas que aparecen.
NOTA: El registro le sugerirá semillas similares a la media. - Ajuste el canal de referencia, el radio de desenfoque gaussiano, y las opciones de corrección de desplazamiento para optimizar el proceso de selección de la semilla de modo que un número de fotogramas de semillas encontradas tienen una apariencia similar. Continuar semillas añadiendo hasta que se creen semillas promedio que satisfactoriamente se asemejan a la estructura se ve en los datos que debe ser modelado.
- Nombre las semillas y en el que se guardarán y seleccionar "OK" para guardar las imágenes finales de semillas para su posterior uso en la generación de modelos.
4. Generar modelos
- SElect "Generar modelos basados en semillas", seleccionar la carpeta donde se guardan las partículas extraídas, y ver el cuadro de diálogo "generar modelos" (Figura 6).
- Cargar las semillas para cada canal mediante el uso de las casillas de verificación.
NOTA: Las semillas se guardarán en una subcarpeta que fue nombrado en el diálogo "Generar Semillas". El nombre por defecto es "Análisis". Velará por seleccionar los promedios de semillas, y no los marcos, que también se guardan para referencia. - Asignar parámetros iniciales de generación de modelo introduciéndolos en el diálogo "Generar modelos", de la siguiente manera. Afinar estos parámetros más tarde.
- Seleccione si desea utilizar un canal de referencia para la alineación, que calcula traslaciones y rotaciones sólo desde el canal de referencia y las aplica a todos los canales. Si se utiliza esta opción, seleccione el canal que se utiliza como referencia.
NOTA: Esta opción sólo se debe elegir a utilizar específicamente un canal como referena vez, como si al hacerlo descubrimiento estructura basada en referencia. De lo contrario, dará lugar a modelos menos exactos. Los canales deben estar bien alineados si se utiliza esta opción. - Seleccione si la cuadratura intensidades de imagen durante la comparación de plantillas. Esto acentuar pequeñas diferencias, a fin de utilizar este cuando se crea un modelo que tiene características particularmente sutiles.
- Elija una similitud mínima frente a la semilla mediante el control deslizante "Similitud mínima contra semilla" o introduciendo un número en el cuadro.
NOTA: sólo partículas con una similitud con las semillas mayores que este punto de corte se utilizará. 60-80% suele ser una buena opción para empezar. - Ajuste el número máximo de iteraciones para 1, optimizar la similitud mínima contra semilla, y aumentar más tarde, según sea necesario.
- Seleccione las casillas para elegir los elementos del proceso de generación de modelos que aparecerá.
NOTA: "Mostrar semillas" mostrará las semillas que han sido cargados con los boxes en la parte superior del cuadro de diálogo. "Mostrar modelos" mostrará todas las iteraciones de los modelos que se crean a partir de un promedio del subconjunto de partículas que se encuentra con la similitud mínima contra semilla. "Mostrar MSE" mostrará una imagen del error cuadrático medio (MSE) que pone de relieve las áreas del modelo que son más variable. "Mostrar partículas" se mostrará el subconjunto de partículas que se utilizan para crear los modelos, registrada de acuerdo con el más alto iteración del modelo que se muestra.
- Seleccione si desea utilizar un canal de referencia para la alineación, que calcula traslaciones y rotaciones sólo desde el canal de referencia y las aplica a todos los canales. Si se utiliza esta opción, seleccione el canal que se utiliza como referencia.
- Seleccione "Mostrar vista previa" para generar modelos de vista previa y ver los resultados.
NOTA: Este es el paso más intensivos computacionalmente del proceso. El tiempo de ejecución para un conjunto de unos pocos miles de partículas con diámetros de unas pocas decenas de píxeles en un PC de escritorio debe estar alrededor de 10 min. Si el tiempo de cálculo es un problema, los usuarios deben tratar primero el algoritmo en un pequeño subconjunto de los datos o el uso de un radio de retorno de la inversión más pequeña en el paso 2.2.4, si es posible. - Ver el generarmodelos D y optimizar los parámetros, especialmente la similitud mínima contra semilla. Aumentar la similitud mínima contra la cabeza hasta que sólo las partículas reales de la morfología modelada se incluyen en el modelo.
- Aumentar el número máximo de iteraciones utilizando el "número máximo de iteraciones" control deslizante o introduciendo un número en el cuadro y permitir que el proceso de generación de modelo para iterar. Utilice un valor en torno a 10 para la iteración máxima.
- Nombre modelos y seleccione "OK" para guardar las pilas modelo de evolución que contienen todas las iteraciones del proceso final de generación del modelo.
NOTA: Si la similitud mínima frente a la semilla es tan alta que no hay partículas tienen esta similitud, nada se actualizará. Si el plugin parece estar congelado, considere la posibilidad de que la similitud mínima es demasiado alta.
Resultados
Aquí, demostramos el software en el modelo de virus de la viruela, virus de la vacuna. Uno de los virus de mamíferos, paquetes de vaccinia más complejos alrededor de 80 proteínas diferentes dentro de un 350 x 270 x 250 nm 3 de partículas en forma de ladrillo 13, 14. Tres subestructuras son discernibles por microscopía electrónica: un núcleo central, que contiene el genoma de ADN de doble cadena; dos estructuras proteináceas, llamados cuerpos laterales, que flanquean el núcleo; y una sola bicapa proteolipídica envelope 15. El gran tamaño, estructura compleja, y IMPREGNABILIDAD etiquetado proteína fluorescente recombinante hacen vaccinia un excelente sistema para demostrar el flujo de trabajo VirusMapper.
Usando el software tal como se describe aquí, la distribución de una variedad de proteínas en el virión de vaccinia puede ser modelado. Una proteína se marcó y se obtuvieron imágenes, posiblemente en combise utilizó nación con otra proteína de distribución conocida como referencia, y el software como se ha descrito para producir modelos promedio de la localización de esa proteína en la partícula. En este ejemplo, dos proteínas se modelaron, la L4 proteína núcleo interno, y el principal cuerpo lateral componente F17.
Un virus vaccinia recombinante que tiene F17 etiquetado con GFP y L4 etiquetado con se utilizó mCherry 16. virus purificada se diluyó en Tris 1 mM, pH 9, y obligado a cubreobjetos lavados, de alto rendimiento mediante el recubrimiento de ellos durante 30 min a temperatura ambiente. Las muestras fueron fijadas mediante la aplicación de formaldehído al 4% en PBS durante 20 min. Los cubreobjetos se montaron sobre portaobjetos inmediatamente en antifade medio de montaje. Imaging se llevó a cabo por SIM en un microscopio SIM comercial. Se seleccionó un campo de visión que contiene cientos de virus y las imágenes fueron adquiridas utilizando 5 cambios de fase y 3 rotaciones cuadrícula con 561 nm (32 micras PE rejillaRIOD) y 488 nm (32 m período) láseres de rejilla. Las imágenes fueron adquiridas usando una cámara sCMOS y procesados utilizando el software microscopio. Los canales fueron alineadas sobre la base de una diapositiva perla de varios colores fotografiado con la misma configuración de adquisición de imágenes. Después de la reconstrucción y la alineación del canal imágenes SIM se abrieron en Fiji y concatenan en una sola pila de imágenes.
Las partículas virales se extrajeron de las imágenes utilizando el canal de L4 como la referencia y sin aplicar ninguna desenfoque gaussiano, ya que estas partículas tienen un máximo central. Alrededor de 15.000 partículas se extrajeron en este experimento.
Debido a la geometría de vaccinia, los cuerpos laterales tienen un aspecto claramente diferente basado en la orientación virus. Visualizamos dos orientaciones en las que uno o dos cuerpos laterales podrían ser distinguidos. Nos referimos a estas orientaciones como frontal y sagital, el respetovamente.
Semillas separadas para las orientaciones frontal y sagital se seleccionaron buscando a través de la lista de partícula en la fase de "Generar Seeds" (Figuras 4 y 5); partículas que eran claramente en una orientación o la otra fueron elegidos. El canal L4 se utilizó como el canal de referencia para alinear las semillas entre sí. Una vez más, no desenfoque gaussiano era necesario. Se seleccionaron 5 partículas para cada orientación y se promediaron para producir las semillas.
Los modelos fueron generados para cada orientación sobre la base de estas semillas. Se han usado Ni un canal de referencia ni valores de intensidad al cuadrado. El número máximo de iteraciones se fijó inicialmente a 1, y la similitud mínimo se fijó para incluir alrededor de 1.000 partículas en cada caso, lo que dio una apariencia consistente para cada orientación. a continuación, se incrementó el número máximo de iteraciones parapermitir la convergencia del modelo. Los modelos se generan así para las dos orientaciones en los dos canales (Figura 7).

Figura 1: Flujo de trabajo VirusMapper. El plugin se organiza en tres etapas principales. Las partículas virales se extraen de las imágenes grandes, imágenes de la plantilla o semillas se seleccionan semi-manualmente de los datos, y modelos finales de SPA se generan a partir de los datos haciendo referencia a las semillas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: "Extracto de Estructuras viral" de diálogo. Al seleccionar "Extraer viral Structur ES", aparecerá este cuadro de diálogo. Los parámetros deben llenarse con las estimaciones iniciales para la segmentación óptima. 'Mostrar vista previa', entonces se puede seleccionar, permitiendo que las regiones de interés que pueden ver y los parámetros a ser ajustado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Configuración de los parámetros de extracción. Después de la vista previa de la ROIs que se extrae, el radio de ROI, el número de regiones de interés, y el máximo solapamiento ROI se ajustan para conseguir una situación como esta. ROIs son ligeramente más grandes que las partículas, todas las partículas se incluyen en un retorno de la inversión, y ROIs pueden solaparse suficientemente para permitir que las partículas agrupadas a ser separados.ank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Generación de semillas de comparación de plantillas. El cuadro de diálogo "Generar Semillas" (1) establece los parámetros para ser asignadas. La secuencia de partículas de referencia (2) permite al usuario escanear a través de las partículas en el canal de referencia. Cuando una partícula se ve en la secuencia de partículas de referencia, las partículas realineados para todos los canales se pueden ver en las vistas previas de partículas realineados (3). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Adición de imágenes de semillas. A medida que se añaden semillas a la "Marcos utilizar", el promedio de todas las semillas (4) y los marcos se trate (5) se muestran. Las partículas que son similares a las semillas promedio actuales se sugieren en el cuadro de diálogo (6). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: "generar modelos" de diálogo. Al seleccionar "Generar modelos basados en semillas", aparecerá este cuadro de diálogo. Los parámetros deben rellenarse con estimaciones iniciales para la generación de modelo óptimo, y los elementos del procedimiento de generación de modelos que se muestran durante el cálculo debe ser seleccionado. "Mostrar vista previa", entonces se puede seleccionar, permitiendo que el proceso de generación de modelos para correr y los parámetros a ser ajustado.ftp_upload / 55471 / 55471fig6large.jpg" target = '_ blank'> Haga clic aquí para ver una versión más grande de esta figura.

Figura 7: Los modelos generados con VirusMapper. viriones Vaccinia con la proteína del núcleo L4 etiquetadas con mCherry y la proteína cuerpo F17 lateral etiquetadas con EGFP fueron imágenes utilizando SIM. Los modelos se generan entonces con el software, como se describe en el protocolo. Dos orientaciones, frontal y sagital, se distinguen por la aparición de los cuerpos laterales. Barra de escala = 100 nm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Con este método, los investigadores están equipados para combinar el poder de SPA y microscopía SR a fin de generar alta precisión, modelos 2D multi-canal de la arquitectura de proteínas de virus y otros complejos macromoleculares. Sin embargo, algunas consideraciones importantes deben tenerse en cuenta.
Las semillas deben ser elegidos para representar una estructura que se observa constantemente. Por lo tanto, los datos en bruto deben ser examinadas cuidadosamente antes se escogen las semillas. Esto es importante para la prevención de los modelos sesgados. Las opciones pueden ser validados por el examen de los umbrales de similitud mínimos necesarios para incluir un cierto número de partículas en los modelos. Claramente, para una selección de la semilla, la más alta de este umbral tiene que ser para un número dado de partículas, más que la estructura es evidente en los datos.
El concepto de emparejamiento de plantillas es particularmente útil cuando hay heterogeneidad en los datos. Todas las estructuras diferentes que son visible debe ser identificado y creado diferentes modelos para cada caso. Mediante la separación de estructuras heterogéneas en un canal, pero al mismo tiempo la creación de modelos en un segundo canal, los patrones pueden surgir que no habría sido evidente de inmediato.
Otra consideración a tener en cuenta cuando se utiliza este algoritmo es que el procedimiento de iteración maximizará la asimetría estocástico. Por ejemplo, cuando se modela una estructura con dos máximos simétrica, todos ligeras asimetrías entre los máximos estarán alineados entre sí durante la iteración, y el modelo final serán así máximamente asimétrica. Si esto no refleja una simetría conocido en la estructura que está siendo modelado, entonces esto debe ser tomado en cuenta. Actualmente, la única manera de evitar este maximización es limitar el número de iteraciones a 1, aunque un desarrollo potencial sería para VirusMapper para incorporar ejes de simetría en el proceso de generación de modelo. Cualquier nuevas versiones de VirusMapper estarán dislable en el sitio web de referencia (véase la Tabla de Materiales). Los usuarios también encontrarán un FAQ para responder a cualquier pregunta comunes.
El software tal como se describe es aplicable a cualquier estructura que se pueden obtener imágenes con una resolución suficiente para visualizar las características que el usuario desea modelar. Aunque SPA puede mejorar la resolución, está claro que no va a mejorar la visibilidad de las características que de otra manera no son visibles. Este protocolo no es, por lo tanto, un método para mejorar la calidad de los datos. Al igual que con cualquier técnica, la preparación de muestras cuidado y optimización de la estrategia de imagen proporcionará los datos más limpios y los mejores modelos resultantes.
La elección de la modalidad de imagen SR también es importante y, en general, dependerá de la muestra en cuestión. VirusMapper ha sido validado para trabajar bien con SIM y STED 10, y también puede ser utilizado con los datos de microscopía de localización de alta calidad, pero se debe tener cuidado en este caso,como el etiquetado escasa podría causar problemas similares a los de la maximización de la asimetría.
Actualmente, VirusMapper es el algoritmo solamente libremente disponibles para el análisis de una sola partícula de imágenes de fluorescencia y el único de propósito general software de promediado 2D SPA. Otros estudios que han hecho uso de los mismos principios 4, 6, 8 han utilizado software a medida especializado para cada estudio en particular. Algoritmos de propósito general para la reconstrucción de datos en 3D se han publicado 5, 18, aunque no se proporcionó ningún software.
Cuando se utiliza como se describe en este artículo, VirusMapper se puede utilizar para producir modelos precisos, exactos y robustos de la arquitectura de la proteína macromolecular de virus y otros complejos. Con estos modelos, los investigadores pueden realizar mediciones precisas de las dimensiones medias de la estructuraUres en estudio, lo que potencialmente lo que les permite llegar a conclusiones biológicas que no habría sido posible de otra manera.
Además, con las capacidades de múltiples canales de esta técnica, es posible mapear un número ilimitado de proteínas y componentes dentro de los complejos y para descubrir nuevos organización de proteínas. Examinar los cambios en la estructura a nanoescala en diferentes condiciones biológicamente relevantes, como las diferentes etapas del ciclo de vida del virus, tiene el potencial de ofrecer información valiosa sobre la biología.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría dar las gracias a Corina Beerli, Jerzy Samolej, Pedro Matos Pereira, Christopher Bleck, y Kathrin Scherer por sus contribuciones al desarrollo original y validación de VirusMapper. También nos gustaría dar las gracias a Artur Yakimovich por su lectura crítica del manuscrito. Este trabajo fue financiado por subvenciones de la Biotecnología y Ciencias Biológicas de Investigación (BB / M022374 / 1) (RH); financiación básica al Laboratorio MRC de Biología Molecular Cell, University College London (JM); el Consejo Europeo de Investigación (649101-UbiProPox) (JM); y el Consejo de Investigación Médica (MR / K015826 / 1) (RH y JM). RG es financiado por la Ingeniería y Ciencias Físicas de Investigación (EP / M506448 / 1).
Materiales
| Name | Company | Catalog Number | Comments |
| Fiji | Open-source image analysis software | ||
| NanoJ-VirusMapper | developed by the Henriques lab | Open source-Fiji plugin (https://bitbucket.org/rhenriqueslab/nanoj-virusmapper) | |
| VectaShield antifade mounting medium | Vector Labs | H-100 | |
| Elyra PS1 | Zeiss | ||
| ZEN BLACK | Zeiss | Image processing software for SIM | |
| High performance coverslip | Zeiss | 474030-9000-000 | |
| TetraSpeck beads | ThermoFisher | T7279 |
Referencias
- Henriques, R., Griffiths, C., Rego, E. H., Mhlanga, M. M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers. 95 (5), 322-331 (2011).
- Gustafsson, N., Culley, S., et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 7, 12471 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 161 (3), 438-449 (2015).
- Szymborska, A., de Marco, A., Daigle, N., Cordes, V. C., Briggs, J. A. G., Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341 (6146), 655-658 (2013).
- Broeken, J., Johnson, H., et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl Fluoresc. 3 (1), 14003 (2015).
- Burns, S., Avena, J., Unruh, J., Yu, Z. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. , (2015).
- Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Nat Acad Sci U S A. 109 (22), 8564-8569 (2012).
- Laine, R. F., Albecka, A., Svan de Linde, ., Rees, E. J., Crump, C. M., Kaminski, C. F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun. 6, 5980 (2015).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Gray, R. D. M., Beerli, C. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci Rep. 6, 29132 (2016).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J of Micros. 198 (2), 82-87 (2000).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Let. 19 (11), 780 (1994).
- Chung, C. -. S., Chen, C. -. H., Ho, M. -. Y., Huang, C. -. Y., Liao, C. -. L., Chang, W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 80 (5), 2127-2140 (2006).
- Moss, B. Poxviridae: the viruses and their replication. Fields virology. , (2010).
- Condit, R. C., Moussatche, N., Traktman, P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 66 (6), 31-124 (2006).
- Schmidt, F. I., Bleck, C. K. E., et al. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 4 (3), 464-476 (2013).
- . Nanoj-virusmapper Available from: https://bitbucket.org/rhenriqueslab/nanoj-virusmapper (2016)
- Fortun, D., Guichard, P., Unser, M. Reconstruction From Multiple Poses in Fluorescence Imaging: Proof of Concept. IEEE J Sel Topics Signal Process. 10 (1), 61-70 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados