
Method Article
工程人造血干细胞和祖细胞中的致癌杂合功能获得突变
摘要
忠实地模拟造血干细胞和祖细胞(HSPC)体细胞突变的新策略对于更好地研究造血干细胞生物学和血液恶性肿瘤是必要的。在这里,描述了通过结合使用CRISPR / Cas9和双rAAV供体转导来模拟HSPC中杂合功能获得突变的方案。
摘要
在其一生中,造血干细胞和祖细胞(HSPC)获得体细胞突变。其中一些突变改变了HSPC的功能特性,如增殖和分化,从而促进血液系统恶性肿瘤的发展。需要对HSPC进行有效和精确的遗传操作,以建模,表征和更好地了解复发性体细胞突变的功能后果。突变可能对基因产生有害影响并导致功能丧失(LOF),或者形成鲜明对比的是,可能增强功能,甚至导致特定基因的新特征,称为功能获得(GOF)。与LOF突变相反,GOF突变几乎完全以杂合方式发生。目前的基因组编辑方案不允许选择性靶向单个等位基因,从而阻碍了模拟杂合子GOF突变的能力。在这里,我们提供了一个详细的协议,说明如何通过结合CRISPR / Cas9介导的同源定向修复和重组AAV6技术来设计人类HSPC中的杂合GOF热点突变,以实现高效的DNA供体模板转移。重要的是,该策略利用双荧光报告系统来跟踪和纯化成功的杂合编辑HSPC。该策略可用于精确研究GOF突变如何影响HSPC功能及其向血液恶性肿瘤的进展。
引言
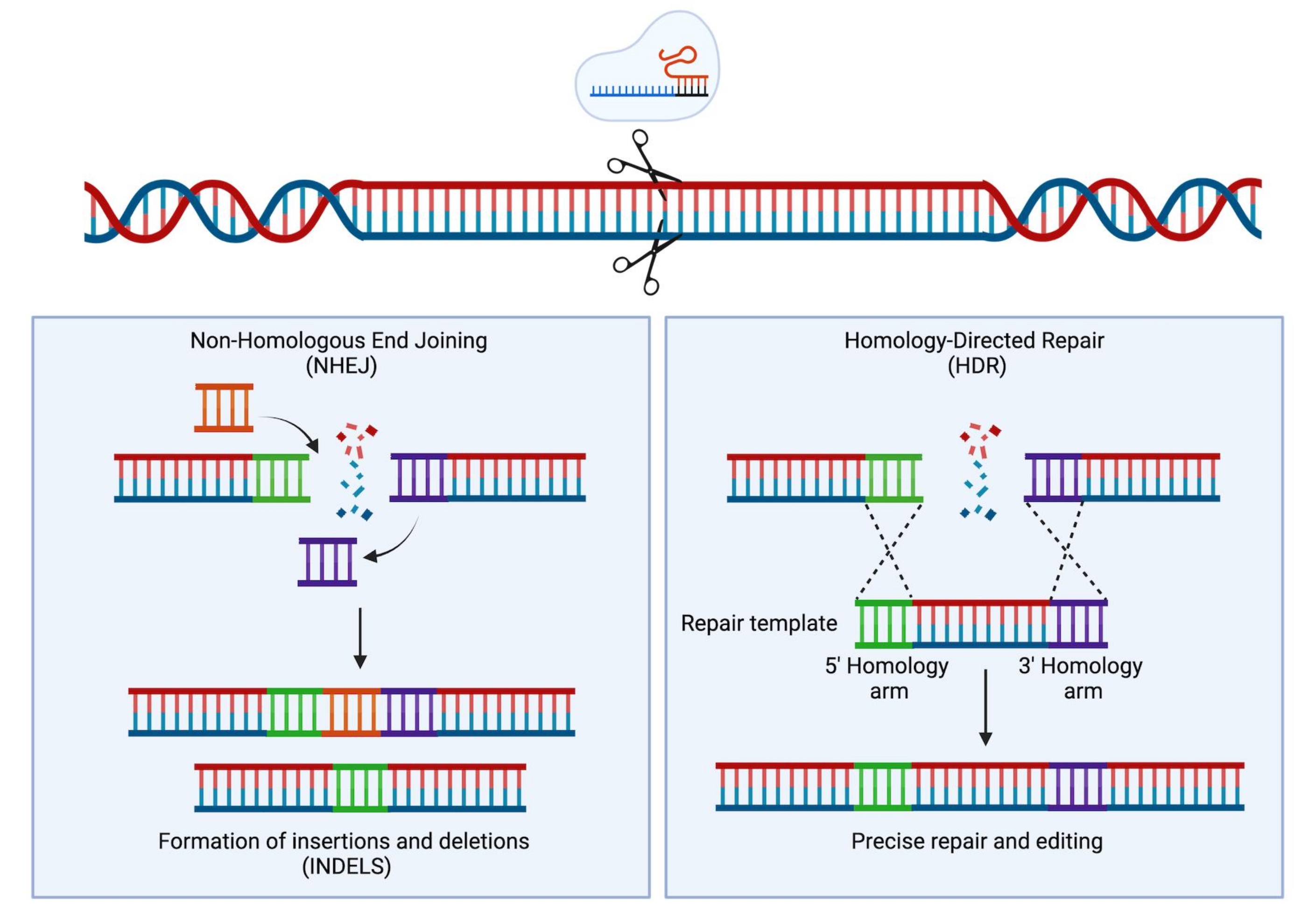
随着成簇规则间隔短回文重复序列(CRISPR)/Cas9技术的发展,科学家的工具包中增加了一种新的极其强大的仪器。该技术允许基因组的精确工程,并且已经证明自己不仅对研究目的非常有用(Hsu等人1中审查),而且最近也已成功转化为临床环境2,3,4。CRISPR/Cas9编辑策略依赖于Cas9蛋白和单向导RNA(sgRNA)的活性5,6,7。在宿主细胞中,Cas9蛋白被引导到DNA中与sgRNA序列互补的特定位点,并将引入DNA双链断裂(DSB)。一旦生成DSB,就会发生两种主要且相互竞争的修复机制:非同源末端连接(NHEJ)和同源定向修复(HDR)。NHEJ是一种容易出错的,主要使用的修复机制导致插入和缺失(插入缺失),而HDR通过使用姐妹染色单体作为修复模板,非常精确,但仅限于细胞周期的S或G2期8。在基因组工程中,HDR 可用于 DNA 的靶向修饰,方法是提供供体模板,该供体模板两侧是与 Cas9 诱导的 DSB 的两个 DNA 末端相同的同源臂(图 1)。用于 HDR 的捐赠者模板类型会极大地影响编辑效率。对于人类HSPC的基因工程,腺相关病毒血清6型(AAV6)最近被描述为递送单链DNA模板的极好载体9,10。
CRISPR / Cas9基因组工程可用于治疗性纠正有害突变11 ,但也可用于将致病突变引入DNA以模拟癌症发展12。血癌,如白血病,通过健康HSPC中体细胞突变的顺序获得 而 发展13,14。早期遗传事件导致克隆增殖优势,导致不确定潜力的克隆造血(CHIP)15,16。突变的进一步获得最终将导致白血病转化和疾病的发展。体细胞突变可以在控制自我更新、存活、增殖和分化的基因中找到17.
通过基因组工程 将 单个突变引入健康的HSPC可以精确地模拟这种逐步白血病生成过程。在骨髓性肿瘤(如急性髓性白血病(AML))中发现的复发性突变数量有限18,19 使这种疾病特别适合使用基因组工程工具进行概括。
体细胞突变可能只出现在一个等位基因(单等位基因/杂合突变)或两个等位基因(双等位基因/纯合突变)上,并且可以对基因的功能产生深远的影响,这可能导致功能丧失(LOF)或功能获得(GOF)。LOF突变导致基因的LOF减少(如果一个等位基因受到影响)或完全(如果两个等位基因都受到影响),而GOF突变导致基因的激活或新功能增加。GOF突变通常是杂合的20。
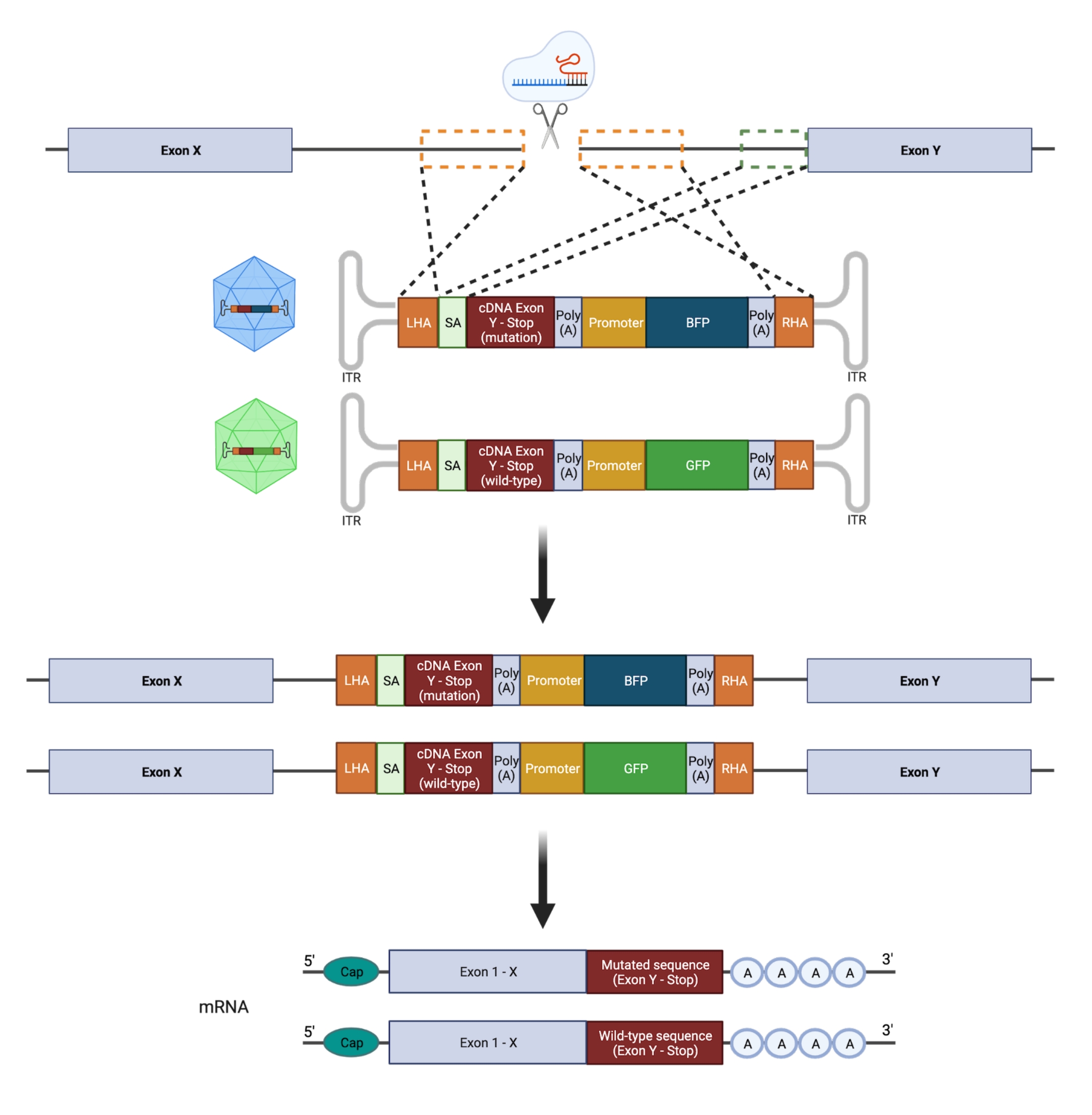
重要的是,合子性(异合子与纯合子)对尝试忠实地模拟突变具有重大意义;因此,仅对基因的一个等位基因进行靶向操作对于设计杂合热点GOF突变是必要的。容易出错的NHEJ导致不同长度的插入缺失21 ,这可能导致不同的,不可预测的生物学后果。然而,由于NHEJ是引入DSB后细胞采用的主要修复程序,因此目前用于操纵HSPC的大多数CRISPR / Cas9平台都不允许精确预测遗传结果22,23。相比之下,引入 CRISPR/Cas9 介导的双链断裂 (DSB),结合使用基于重组腺相关病毒 (rAAV) 载体的 DNA 供体模板通过 HDR 进行 基因组工程,允许在人类 HSPC 中插入等位基因特异性突变11,24。可以同时整合突变体和野生型(WT)序列,并在单个等位基因上结合不同的荧光报告基因,以选择杂合基因型(图2)。该策略可用作精确表征复发性、白血病性、杂合性 GOF 热点突变对 HSPC 功能、疾病起始和进展的影响的有力工具。
本文提供了原代人HSPC中复发性突变杂合GOF突变的有效工程的详细方案。该策略结合了CRISPR / Cas9和双AAV6转导的使用,为杂合GOF突变的预期生成提供WT和突变DNA供体模板。例如,钙网蛋白(CALR)基因中复发性1型突变(52 bp缺失)的工程将显示25。 CALR 外显子 9 中的杂合 GOF 突变经常见于骨髓增殖性疾病,例如原发性血小板增多症 (ET) 和原发性骨髓纤维化 (PMF)26。CALR是一种内质网驻留蛋白,在新合成蛋白质的折叠过程中主要具有质量控制功能。其结构可分为三个主要结构域:参与蛋白质伴侣功能的氨基(N)末端结构域和富含脯氨酸的P结构域,以及参与钙储存和调节的C结构域27,28。 CALR 突变导致 +1 帧偏移,导致新型扩展 C 端的转录和内质网 (ER) 保留信号 (KDEL) 的丢失。突变型 CALR 已被证明可结合血小板生成素 (TPO) 受体,从而导致 TPO 非依赖性信号传导,增殖增加29。

图 1:NHEJ 和 HDR 修复。 在DNA中引入双链断裂后NHEJ和HDR修复机制的简化示意图。 请点击此处查看此图的大图。
{kind=link}

图 2:双等位基因 HDR 编辑策略的示意图。 示意图显示了供体模板与目标等位基因的整合,然后将它们翻译成功能性mRNA。橙色虚线框表示对应于左同源臂 (LHA) 和右同源臂 (RHA) 的区域。每个 HA 的理想大小为 400 bp。绿色虚线框表示对应于 SA 序列的区域。SA 的大小为 150 bp。 请点击此处查看此图的大图。
{kind=link}
研究方案
该协议要求使用健康的供体来源的CD34 + HSPC,并需要当地机构审查委员会(IRB)的伦理批准和签署的知情同意书。该方案中使用的CD34 + HSPC是从足月分娩(妊娠>34周)的脐带血(UCB)中分离出来的。分娩前获得了母亲的知情同意,并从格拉茨医科大学获得了收集UCB的伦理批准(IRB批准:31-322 ex 18/19)。该协议中使用的材料的完整列表可以在 材料表中找到。
1. sgRNA设计与切割效率评估
- 在在线数据库工具(例如,COSMIC,https://cancer.sanger.ac.uk/cosmic)上搜索所需突变的位置和正确的转录本。
- 使用 sgRNA 设计工具选择与突变相邻的 sgRNA(外显子靶向)或前一个内含子(内含子靶向)。在此协议中,使用了Benchling的在线工具。有关用于sgRNA设计的可能在线工具列表,请参阅 材料表 。
- 选择 + (创建) 选项 > DNA 序列 > 导入 DNA 序列 > 从数据库导入。输入所需的基因,例如 CALR,然后选择人类作为物种 > 搜索 > 选择正确的转录本(例如,CALR-201 -ENST00000316448)> 导入。选择要引入DS中断的感兴趣区域(例如,内含子7)。
- 选择屏幕右侧的 CRISPR 选项,然后选择 设计和分析指南。选择 单个导流板,将导板长度保持在20 bp,并使用SpCas9(NGG)进行编辑PAM序列。
- 选择一个目标分数高(在所需位点进行编辑的机会高)和高脱靶分数(在不需要的位点进行编辑的机会低)的指南。选择至少三个指南进行测试,以找到性能最佳的sgRNA。从商业供应商处订购 sgRNA 作为化学修饰的合成 sgRNA。
注意:建议在此阶段订购少量合成sgRNA进行初始筛选。一旦鉴定出表现良好的 sgRNA,继续处理所选 sgRNA 的更大顺序。 - 解冻 2 x 10 5-5 x 105 CD34+ HSPC。 将细胞转移到补充有 1% 抗生素(例如青霉素/链霉素)的 10 mL 预热的 RPMI1640 中。
注意:应使用纯度为>90%的CD34 + HSPC,以获得最佳和最可重复的结果。 - 在室温(RT)下以350× g 离心10分钟。用血细胞计数器计数细胞,并将细胞悬浮在补充有 0.2% 青霉素/链霉素 (P/S)、100 ng/mL 血小板生成素 (TPO)、100 ng/mL 干细胞因子 (SCF)、100 ng/mL FMS 样酪氨酸激酶 3 配体 (FLT3L)、100 ng/mL 白细胞介素-6 (IL-6)、35 nM UM171 和 0.75 μM StemRegenin1 (SR1) 的 SFEM II 培养基中,浓度为 2.5 x 105 个细胞/mL。在37°C / 5%CO2 下孵育48小时 - 72小时。
注意:从现在开始,完整的培养基将被称为HSPC保留培养基。 - 将细胞收集在 15 mL 管中并计数细胞。通过台盼蓝排除检查细胞活力。
- 在开始之前,打开转染系统并选择比色皿选项。为HSPC选择合适的程序和核连接溶液(见材料表)。2 x 10 5 和5 x 106 个细胞可以在一个 100 μL 比色皿中进行核转染。留出少量细胞(例如,1 x 10 5-2 x 105)在培养物中保持48小时以用作WT对照。
- 准备RNP复合物。在 1.5 mL 管中,加入 15 μg Cas9 和 8 μg sgRNA(摩尔比 1:2.5),并在 25 °C 下在加热块中孵育 10 分钟。
- 当RNP复合物孵育时,在室温下以350× g 离心细胞5分钟,并使用移液管弃去上清液。将细胞悬浮在 100 μL 核连接溶液中。
- 将细胞与RNP复合物混合并转移到比色皿中。轻轻拍打比色皿以去除转移过程中可能形成的任何最终气泡。
- 将比色皿插入转染系统的支架中,并使用DZ-100程序对细胞进行电穿孔。
- 电穿孔后,立即加入 400 μL 不含 P/S 的预热 HSPC 保留培养基。
- 用精细转移移液管将细胞转移到含有不含P / S的预热HSPC保留培养基的培养板中。 根据细胞数量,使用适当的培养板(24孔、12孔或6孔板),以达到0.25 x 106 和1 x 106 细胞/ mL之间的密度。
- 将板转移到37°C / 5%CO2的培养箱中。孵育核细胞6-8小时。
- 6-8小时后,除去旧培养基并用补充有P / S的新鲜预热HSPC保留培养基替换.以2.5 x 105 和5 x 105 的浓度悬浮细胞并转移到细胞培养板(24,12或6孔板)。将细胞在37°C / 5%CO2下孵育48小时。
- 收获2 x 10 5个细胞,并在室温下以350 x g 离心5分钟。在开始之前,将加热块设置为65°C和98°C。
- 弃去上清液并将细胞悬浮在 1.5 mL 管中的 1 mL 1x DPBS 中。在室温下以350× g 离心5分钟。
- 弃去上清液并将细胞悬浮在 50 μL DNA 提取溶液中。涡旋15秒。
- 在65°C孵育6分钟。 涡旋15秒,并在98°C孵育2分钟。
- 使用引物通过PCR扩增DSB区域,引物产生约400-600 bp的扩增子,DSB位于中心。使用 0.5-1 μL 提取的 DNA 进行 PCR 反应。
注意:由于DNA提取溶液的组成,DNA浓度无法通过分光光度法准确定量。 - 用DNA分子量标准在100V的1.5%琼脂糖凝胶上运行PCR产物45-60分钟。将凝胶放在蓝光或紫外透射仪上。
- 根据制造商的说明,使用市售试剂盒从凝胶中提取正确大小的DNA条带(参见 材料表)。
- 使用PCR的正向引物或反向引物通过Sanger测序对样品进行测序。对于长度为 400-600 bp 的 PCR 产物,测序需要 75 ng 的 DNA,总体积为 15 μL 的浓度为 5 ng/mL。
- 通过将测序文件上传到旨在通过识别 sgRNA 产生的插入和缺失来计算编辑效率的程序来分析 sgRNA 的编辑效率。选择性能最佳的 sgRNA 进行。
注意:专用的在线工具列在 材料表中。该分析需要转染和WT HSPC的.ab1文件,sgRNA序列和PAM序列。
2. 同源定向修复(HDR)载体构建
- HDR 模板设计
注意:应设计两个 HDR 模板:WT 序列模板和突变序列模板。- 将所需基因的基因组序列和编码序列(CDS)导入适当的分子克隆软件中(专用工具可在 材料表中找到)。从基因组序列文件中,通过在DSB的5'端选择理想的400 bp来设计左同源臂(HA)。将此序列粘贴到新文件中。
- 如果内含子被sgRNA靶向,则需要包括剪接受体(SA)序列。选择目标内含子的最后 150 bp 并将其粘贴到左侧 HA 之后。如果外显子是直接靶向的,则不需要此序列(图3)。
- 从 CDS 文件中,选择感兴趣的 cDNA。如果靶向外显子序列,请确保cDNA从DSB位点的下游立即开始,并包括目的基因的所有后续外显子以及终止密码子。如果靶向内含子,请确保cDNA以下一个外显子的第一个密码子开头。密码子优化cDNA并将其插入SA序列之后。为此,请使用专用的在线工具(材料表)。
- 在感兴趣的cDNA(例如SV40或bGH)之后插入3'聚腺苷酸化(PolyA)信号(表1)。在PolyA之后,插入荧光蛋白的启动子序列(例如,脾病灶形成病毒[SFFV]或多泛素C [UBC], 表1)。
- 插入荧光蛋白的序列(即GFP,BFP或mCherry)。插入第二个但不同的 PolyA 信号。
注意:插入不同的PolyA序列将避免诸如质粒的细菌重组或由于模板中有两个相同序列而导致的序列比对问题等问题。 - 从基因组序列文件中,通过在DSB的3'端选择理想的400 bp来设计正确的HA。在第二个 PolyA 之后插入此序列。
- 创建整个模板的副本并修改感兴趣的cDNA,使其包含所需突变的序列。
- 用不同的荧光蛋白交换荧光蛋白。例如,如果 GFP 用于 WT 模板,则将其与 BFP 或 mCherry 交换为突变模板。构建HDR模板并将其克隆到pAAV-MCS质粒(或其他合适的骨架)中。
注意:组装所需的片段可以通过PCR生产或商业订购,并且需要包含与其相邻片段重叠的序列。如果感兴趣的突变是点突变、小插入或小缺失,则可以通过PCR在含有WT cDNA的HDR模板上进行定点诱变来产生突变的cDNA。 - 使用热冲击法将合格的 大肠杆菌 与组装的产品转化。在开始之前,将细菌放在冰上解冻10分钟。
- 将 2 μL 组装好的产物添加到 50 μL 细菌中。轻轻轻弹混合内容物。
- 将样品放在冰上30分钟。将样品转移到设置为42°C的热块中30秒。
- 将样品在冰上转移5分钟。加入 450 μL 室温 SOC 培养基,并将样品在 37 °C 下孵育 1 小时。
- 将样品铺展到含有氨苄西林的LB琼脂平板上。对于每个样品,散布 100 μL 三种不同稀释度的细菌溶液(未稀释、1:5 和 1:10),以获得具有可采摘的单个菌落的 LB 琼脂平板。将板在37°C孵育过夜。
- 第二天,每个样品选择三个菌落。将菌落转移到15mL管中,盖子上含有4mL补充有氨苄青霉素的LB培养基,并在37°C的摇床中孵育过夜。
- 从每个菌落中分装 500 μL 细菌溶液,并将其储存在冰箱中以备后用。按照制造商的说明进行小型制备以提取质粒DNA。
- 将样品送去进行Sanger测序,以确认质粒的正确组装。使用分布在整个质粒中的足够引物,以确保不间断的序列确认,理想情况下,通过正向和反向读取覆盖每个区域两次。
- 将 200 μL 含有正确组装质粒的细菌溶液加入补充有氨苄青霉素的 200 mL LB 培养基中,并在 37 °C 下在摇床中孵育过夜。
- 按照制造商的说明进行中型或大型制备以提取质粒DNA。将质粒DNA储存在-20°C。
- 重组AAV6制剂
注意:需要执行两个单独的AAV6准备:一个用于带有WT HDR模板的rAAV6,另一个用于带有突变HDR模板的rAAV6。本节介绍仅制备一种病毒所需的步骤。- 解冻HEK293T细胞,将细胞转移到补充有10%FBS,1%P / S和25mM HEPES的10mL预热DMEM中,并在室温下以350 x g 离心5分钟。
- 将细胞悬浮在 1 x 105 个细胞/mL 的浓度下,并转移到合适的烧瓶(例如,175 cm2 烧瓶)中。将烧瓶转移到37°C / 5%CO2的培养箱中。
注意:HEK293T细胞应提前解冻,以使细胞完全恢复并获得足够数量的细胞(产生一种病毒需要11 x 107 个细胞)。建议在至少三次传代后使用细胞,并将细胞保持在20代以下。HEK293T细胞应每周分裂三次。在维持细胞的同时,这些细胞不应超过70%-80%的汇合度。AAV制剂中使用的DMEM也应该已经补充了L-谷氨酰胺和丙酮酸钠。 - 将细胞收集在 50 mL 管中,并在室温下以 350 x g 离心 5 分钟。
- 将细胞悬浮在补充有 10% FBS、1% P/S 和 25 mM HEPES 的 20 mL DMEM 中,并用血细胞计数器计数。使用台盼蓝进行死细胞排除。
- 在 175 cm2 烧瓶中接种 3 x 10 6个细胞,在 20 mL DMEM 中补充有 10% FBS、1% P/S 和 25 mM HEPES。为了生产一种病毒,准备至少四个175cm2烧瓶。将细胞在37°C / 5%CO2下孵育3天。
注意:此步骤最好在星期五执行,因为它允许细胞在周末扩增。 - 收获并计数HEK293T细胞。
- 为了制备其中一种病毒,准备十个 150 毫米培养皿,每个培养皿在 20 mL DMEM 中含有 11 x 106 HEK293T,并补充有 10% FBS、1% P/S 和 25 mM HEPES。
- 将培养皿置于37°C / 5%CO2 的培养箱中24小时。小心丢弃旧培养基,并用补充有 10% FBS、25 mM HEPES 和 1 mM 丁酸钠的 20 mL 无抗生素 DMEM 替换。在继续之前,请检查细胞的汇合度是否不超过80%。
- 准备两个包含转染混合物的 15 mL 管。向试管 1 中加入 5 mL 还原血清培养基、60 μg rAAV6 突变 HDR 质粒和 220 μg pDGM6(辅助质粒)。向试管 2 中加入 5 mL 还原血清培养基和 1120 μL 1 mg/mL 聚乙烯亚胺溶液(PEI,转染试剂)。
- 将管2的内容物添加到管1中并涡旋30秒。在室温下孵育15分钟,以确保DNA正确封装到PEI胶束中。
注意:不要将溶液保存超过20分钟。 - 小心地将 1.1 mL 溶液滴入每个培养皿中,轻轻旋转。将培养皿置于37°C / 5%CO2 的培养箱中48小时。
- 48小时后,向每个培养皿中加入250μL0.5M EDTA,并将培养皿放入培养箱中10分钟。通过洗净培养皿来收获细胞,然后转移到 500 mL 离心管或多个 50 mL 管中。
- 在4°C下以2,000× g 离心10分钟。 弃去上清液。为确保完全除去上清液,在4°C下再次以2,000× g 离心1分钟。
- 丢弃任何剩余的上清液。任何残留的上清液都可能损害病毒纯化。
- 通过涡旋松开颗粒,并根据制造商的说明使用AAV纯化试剂盒(见 材料表)提取病毒。或者,通过碘沙醇梯度超速离心30,31进行提取。
- 将纯化的病毒分装并储存在-80°C。 功能滴定AAV或通过数字微滴PCR(ddPCR)32定量AAV滴度,以确定最佳转导条件。
- 重复此过程以制备rAAV6 WT HDR质粒。
- 通过滴涕聚合酶链反应进行AAV滴定
- 在开始之前,将加热块设置为65°C和98°C。
- 通过将 15 μL DNA 提取溶液添加到 5 μL 病毒中来提取病毒 DNA。涡旋15秒。
- 在65°C孵育6分钟。 涡旋15秒。在98°C孵育2分钟。
- 使用提取的DNA(病毒DNA的1:4稀释)制备无核酸酶(nf)H2O的系列稀释液(1:400,1:40,000,1:160,000,1:640,000)用于ddPCR。
注意:如果不立即进行ddPCR,稀释液可以储存在-20°C。 - 为ddPCR选择三种稀释液。选择使用的稀释液取决于病毒的浓度。优选地,使用三种不同的稀释度(例如,1:40,000、1:160,000 和 1:640,000)以找到在机器检测限内的最佳稀释度。
- 工作时将试剂放在冰上。准备预混液(所有样品将一式两份测量)。对于每个反应,准备以下内容:12.5 μL 用于探针的 ddPCR 超级混合物,不含 dUTP,1.25 μL 黄金时间标准 qPCR 测定、AAV-ITR(参见 材料表)和 6.25 μL nf-H2O。
- 将 5 μL DNA 与 20 μL 预混液混合。对 1:40,000、1:160,000 和 1:640,000 稀释液执行此操作。
- 将墨盒放入墨盒支架中。将 70 μL 液滴生成油添加到标名为油的行中的探针中。注意不要产生任何气泡。
- 在标价为样品的行中加入 20 μL 样品体积。注意不要产生任何气泡。
- 用垫圈密封墨盒并将其放入液滴发生器中。通过按下机器上的启动按钮产生液滴,取下垫圈,然后用多通道移液管小心地将 40 μL 产生的液滴转移到 96 孔板中。缓慢工作以避免破坏液滴和气泡。
- 使用PCR板封口机用穿孔箔(红色条纹朝上)密封96孔板。将板放入热循环仪中。
- 将体积设置为 40 μL,盖子温度设置为 105 °C,升温速率设置为 2 °C/s。如 表2所述运行PCR程序。
- 使用前30分钟打开液滴读取器。在桌面上打开软件。
- 输入板布局,然后在实验选项卡中选择 ABS ,在超级混合选项卡中选择 ddPCR超级混合物 。然后,首先按 PRIME,然后单击软件上的刷新系统以启动 系统 。
- 将板输入阅读器。开始运行,完成后将数据导出为 CSV 文件,以便作为电子表格进行后续分析。
注意:软件生成的数字是每μL的基因组拷贝数(GC)。这些需要乘以稀释因子:GC/μL x 5(预混液中DNA的稀释)x初始稀释度(即40,000或160,000)。

图3:CRISPR/Cas9 HDR敲入过程中内含子和外显子靶向的示意图。 CRISPR/Cas9 HDR敲入的内含子和外显子靶向策略之间的示意图比较。(A)在内含子靶向过程中,在DNA的内含子中引入双链断裂。HDR 模板由 LHA、cDNA 序列和 RHA。内含子靶向还需要存在包含 3' 剪接位点、分支点和聚嘧啶束的切片受体。这允许正确的拼接。绿色虚线框表示对应于 SA 序列的区域。SA的大小为150 bp.(B)外显子靶向依赖于直接在外显子中产生双链断裂。HDR 模板由 LHA、cDNA 序列和 RHA。橙色虚线框表示对应于LHA和RHA的区域。每个 HA 的理想大小为 400 bp。 请点击此处查看此图的大图。
{kind=link}
| 发起人 | ||||
| 名字 | 长度 | 描述 | ||
| SFFV | 492 基点 | 脾病灶形成病毒启动子。强大的哺乳动物促进剂。构成性表达 | ||
| 不列颠哥伦比亚大学 | 400 基点 | 启动子来源于人类泛素C基因。构成性地表达。 | ||
| 巨细胞病毒 | 508 基点 | 启动子来源于巨细胞病毒。它可以包含一个增强子区域。构成性地表达。强大的哺乳动物促进剂。可以静音。 | ||
| EF-1a | 1182 基点 | 人真核翻译伸长因子1α启动子。构成性地表达。强大的哺乳动物促进剂。 | ||
| EFS | 200-300 基点 | EF-1 α 无内含子短形式 | ||
| 全能农业天然气集团 | 584 基点 | 含有CMV早期增强子(C),鸡β肌动蛋白启动子(A)和兔β-珠蛋白基因剪接受体(G)的杂交哺乳动物启动子。构成性地表达。 | ||
| 聚腺苷酸化 (PolyA) 信号 | ||||
| 缩写 | 长度 | 描述 | ||
| SV40 聚甲 | 82-122 基点 | 猿猴病毒40聚腺苷酸化信号 | ||
| bGH PolyA | 224 基点 | 牛生长激素聚腺苷酸化信号 | ||
| rbGlob PolyA | 56 基点 | 兔β珠蛋白聚腺苷酸化信号 | ||
表1:启动子和聚腺苷酸化信号。
| 步 | 温度 | 时间 | 周期 |
| 酶活化 | 95°C | 10 分钟 | 1 倍 |
| 变性 | 94°C | 30 秒 | 42 倍 |
| 退火 | 60°C | 1 分钟 | |
| 外延 | 72°C | 30 秒 | |
| 酶失活 | 98°C | 10 分钟 | 1 倍 |
| 拿 | 4°C | ∞ |
表2:数字微滴PCR程序。
3. HSPC的编辑
- HSPC的转染和转导
- 解冻CD34 + HSPC并将细胞转移到补充有1%P / S的10mL预热RPMI中,在室温下以350 x g 离心10分钟。
- 将细胞悬浮在HSPC保留培养基中至2.5 x 10 5细胞/ mL的浓度,并在37°C /5 %CO2 下孵育48小时 - 72小时。
- 在 15 mL 管中收获细胞。计数细胞并通过台盼蓝排除检查细胞活力。2 x 10 5 和5 x 106 个细胞可以在单个比色皿中进行核转染。根据您计算的细胞数量准备适当数量的比色皿。
- 准备RNP复合物。在 1.5 mL 管中,加入 15 μg Cas9 和 8 μg sgRNA(摩尔比 1:2.5),并在 25 °C 下在加热块中孵育 10 分钟。
- 当RNP复合物孵育时,将细胞以350× g 离心5分钟并弃去上清液。
- 将细胞悬浮在 100 μL 核连接溶液中。将细胞与RNP复合物混合并转移到比色皿中。
- 轻轻拍打比色皿以去除转移过程中可能形成的任何残留气泡。
- 将比色皿插入转染系统的支架中,并按照前面的sgRNA设计部分所述对细胞进行电穿孔。
- 电穿孔后,立即加入 400 μL 不含 P/S 的预热 HSPC 保留培养基,并用精细转移移液管将细胞转移到含有 500 μL 不含 P/S 的预热 HSPC 保留培养基的组织培养板中。 根据细胞数量,使用适当的培养板(24、12 或 6 孔板),以达到 0.25 x 106 和 1 x 106 细胞/mL 之间的密度。将板转移到37°C / 5%CO2的培养箱中。
- 在冰上解冻含有冷冻rAAV的小瓶。通过将每个rAAV6的最佳量移液到细胞悬液中来转导细胞。在电穿孔后20-30分钟内进行转导,以获得高转导效率。
注意:每种病毒的最佳浓度(WT HDR模板为一种病毒,突变HDR模板为另一种单独的病毒)应通过实验确定。通常,5,000-10,000 GC/细胞(例如,5 x 109 GC 用于 1 x 106 细胞)可产生高转导。 - 将细胞悬液与移液器轻轻混合。将转导的细胞在37°C / 5%CO2下孵育6-8小时。6-8小时后,将细胞收集在管中,并在室温下以350× g 离心5分钟。
- 丢弃旧培养基,并更换为补充有P / S的新鲜预热HSPC保留培养基。
- 将细胞悬浮在 2.5 x 10 5-5 x 105 个细胞/mL 之间的浓度,并转移到组织处理的细胞培养板(24、12 或 6 孔板)中。将细胞在37°C / 5%CO2下孵育48小时,然后进行分选。
- 携带杂合GOF突变的工程HSPC的流动分选
- 在 15 mL 管中收获细胞,并在室温下以 350 x g 离心 5 分钟。取出上清液,将细胞悬浮在含有0.1%BSA的1mL DPBS中,并在室温下以350× g 再次离心5分钟。
- 弃去上清液并将细胞悬浮在适当体积的DPBS + 0.1%BSA中,具体取决于细胞数。
注意:建议将细胞悬浮在最小体积为 200 μL 且不超过 1 x 107 个细胞/mL 时,以降低堵塞分选机的风险。 - 将细胞转移到装有盖子的无菌FACS管中。将7-AAD或其他活性染料(取决于它们与荧光蛋白的相容性)添加到细胞悬液中,用于活/死细胞排除。
- 将荧光报告蛋白双重阳性的活细胞分选到含有 200 μL HSPC 保留培养基的收集管中。
注意:应添加由仅用AAV转导的细胞组成的适当对照,以确保在分选过程中正确设门。 - 将分选的细胞在室温下以350 x g 离心5分钟,并将细胞悬浮在HSPC保留培养基中,浓度为2.5 x 105 个细胞/ mL,以进一步扩增培养物或直接将细胞用于功能测定。
4. 确认基因编辑成功
- 基因组DNA提取
- 在开始之前,将加热块设置为65°C和98°C。 收获2 x 10 5基因组编辑和分选纯化的细胞,并以350 x g 离心5分钟。
- 如前所述提取gDNA。直接使用DNA溶液进行PCR或储存在-20°C。
- 进外聚合酶链反应
- 设计两个引物以扩增5'插入位点(图4A):引物向前1靶向左同源臂外的基因组位点;引物反向 2 靶向整合序列。
- 对于由内而出的PCR,每个反应准备以下PCR混合物:
10 μL PCR 预混液(2x;参见 材料表)
1 μL 引物前向 1(10 μM 原液)
1 μL 引物反向 2(10 μM 储备液)
0.5-1.0 μL 提取的 DNA
无核酸酶 H2O 至最终体积为 20 μL
注意:对于多个反应,建议准备一个包含一个额外反应的预混液,以解决移液错误。重要的是包括非模板对照和来自模拟处理样品的模板DNA。 - 使用适当的热循环程序运行PCR反应(请参阅制造商的说明)。
注意:建议首先测试引物对的最佳退火温度。这可以通过计算Tm,然后使用可以执行梯度温度的热循环仪运行PCR来完成。 - 用DNA分子量标准在100V的1.5%琼脂糖凝胶上运行PCR产物45-60分钟(图4B)。将凝胶放在蓝光或紫外透射仪上。从凝胶中切除条带。
- 使用 DNA 凝胶提取试剂盒从条带中提取 DNA。将提取的PCR样品与适当的引物一起发送以进行Sanger测序,以确认所需cDNA在内源性基因位点的正确和无缝整合。

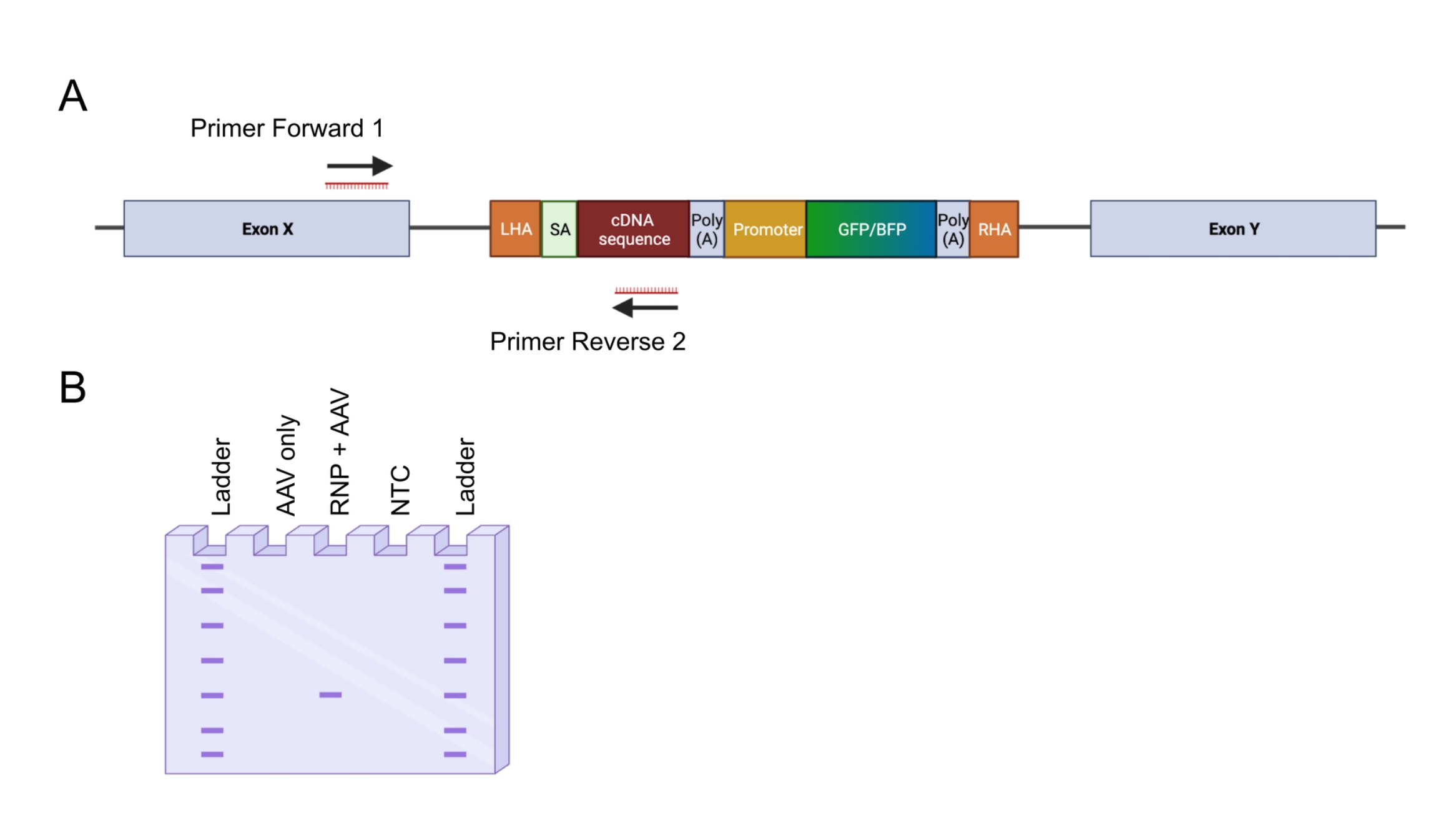
图4:通过由内而外PCR验证基因组整合 。 (A)由内而外PCR策略的示意图。在所描述的策略中,设计了两个引物。引物正向1靶向LHA外的基因组位点,引物反向2靶向密码子优化序列。(B)琼脂糖凝胶电泳的示意图。只有成功编辑的细胞(RNP + AAV)才会在进出PCR过程中产生PCR产物,而未编辑的样品(仅限AAV)不会产生PCR产物。缩写:NTC = 非模板控件。 请点击此处查看此图的大图。
{kind=link}
结果
通过应用上述方案,杂合子1型 CALR 突变可重复地引入脐带血来源的HSPC中。该突变包括外显子 9( CALR 的最后一个外显子)中的 52 bp 缺失,导致 +1 帧偏移,导致一种新的带正电荷的 C 端结构域26,33 的翻译。为了在内源性基因位点引入CALR突变,采用了外显子9上游的内含子靶向策略,因为在Cas9诱导的DSB未通过 HDR 机制 修复 的情况下,这将规避编码序列中任何不必要的变化。在这种特定情况下,由于具有高靶上和低脱靶序列以及有利的同源臂(缺乏序列重复; 图5A)。
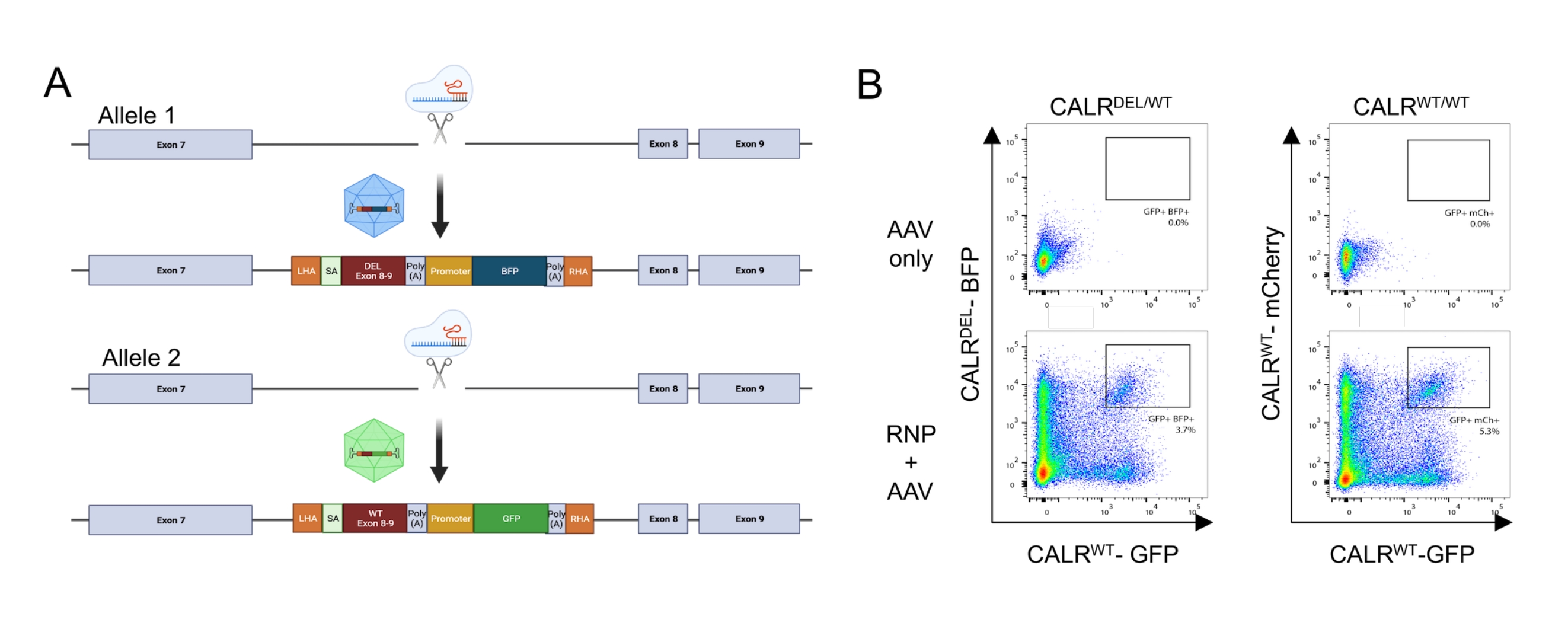
然后将两个供体模板设计并包装在AAV6载体中。为了实现从内源性外显子到整合的cDNA的正确剪接,供体模板包含(i)SA序列,包括3'剪接位点,分支点和聚嘧啶束,(ii)外显子8-9的密码子优化cDNA序列,包含WT(CALRWT)或突变序列(CALRDEL)包括终止密码子,(iii)猿病毒40(SV40)polyA信号,(iv)在单独的内部启动子,脾病灶形成病毒(SFFV)启动子的控制下编码荧光蛋白的序列,然后是(v)牛生长激素(bGH)polyA信号。含有 CALR WT cDNA 的供体模板设计为包含 GFP 盒,而含有 CALRDEL cDNA 序列的供体模板设计为包含 BFP 盒。整个结构的两侧是左右HA(图5A)。
在用RNP复合物转染和用rAAV6病毒转导两天后,通过流式细胞术分析细胞。可以检测到四个主要群体:(i)既不表达GFP也不表达BFP的细胞,代表没有基于HDR的基因组编辑的细胞,(ii)仅对GFP呈阳性的细胞,代表仅整合WT构建体的细胞,(iii)仅对BFP呈阳性的细胞,代表仅整合突变构建体的细胞,以及(iv)GFP和BFP双阳性细胞, 代表整合了WT和突变序列的细胞(图5B)。为了获得携带杂合1型 CALR 突变的HSPC纯群体,通过流式细胞术对双阳性细胞进行分选。其中两个WT序列被敲入的HSPC用作对照细胞(GFP+ mCherry+; 图 5B)。用作对照细胞的有效替代方案是HSPC,其荧光蛋白在安全港位点(即AAVS1;未显示)中的双等位基因整合。对分选的HSPC进行的台盼蓝排除细胞计数表明,超过90%的细胞是活的。
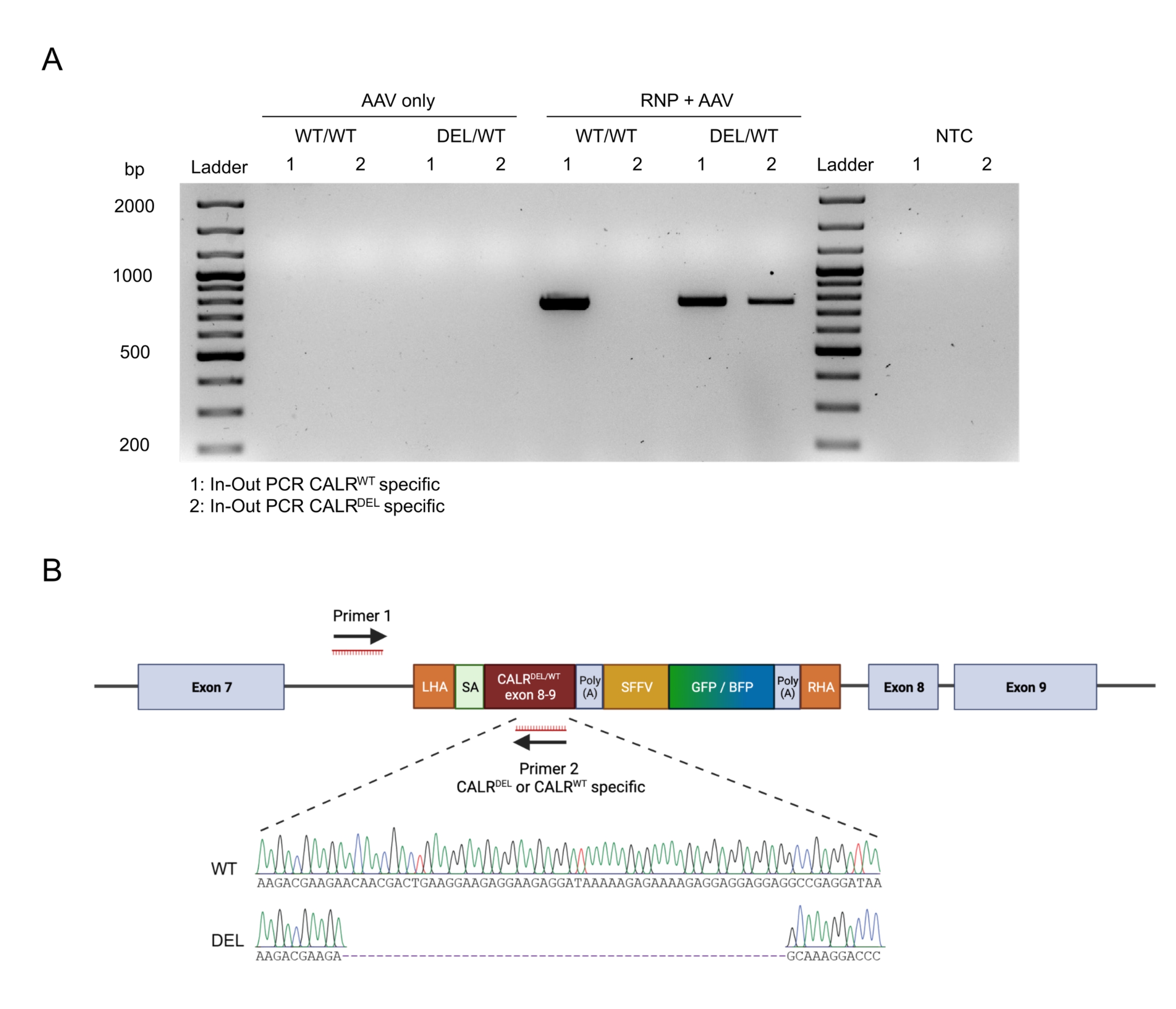
通过应用In-Out PCR策略证实了构建体的无缝靶向整合(图6A)。在这种特定情况下,我们进行了两个单独的输入输出PCR,一个用于敲入CALR WT序列(图6A中凝胶电泳的泳道1),另一个用于敲入的CALRDEL序列(图6A凝胶电泳的泳道2)。对从凝胶条带中提取的DNA进行的Sanger测序证实了WT和突变序列在CALRDEL / WT HSPC中的正确插入(图6B)。

图5:携带杂合CALR突变的HSPC的产生 。 (A)描述杂合 CALR 突变插入的编辑策略的代表性方案。RNP 复合物靶向 CALR 基因的外显子 7 和外显子 8 之间的内含子。两个AAV,一个包含突变外显子8-9和一个BFP,另一个包含WT外显子8-9和一个GFP,将作为供体修复模板,并将促进突变序列在一个等位基因中的整合以及WT序列在其余等位基因中的整合。(B)代表性流式细胞术图,描述HSPC转染和转导后48小时GFP和BFP或GFP和mCherry的表达。 请点击此处查看此图的大图。
{kind=link}

图 6:HSPC 中成功杂合 CALR 突变的验证。 (A) 对从 AAV 对照、CALR WT/WT 和 CALRDEL/WT 中提取的基因组 DNA 进行的进出 PCR 产物的凝胶电泳。使用100 bp DNA分子量标准。缩写:NTC = 非模板控件。(B)从对CALRDEL/WT 进行的进出PCR获得的Sanger测序结果,证实了WT和突变序列的成功整合。 请点击此处查看此图的大图。
{kind=link}
讨论
人类原代HSPC的高效和精确的遗传操作为探索和理解影响正常造血的过程,最重要的是造血细胞的白血病转化提供了很好的机会。
在该协议中,描述了一种设计人类HSPC以表达复发性杂合GOF突变的有效策略。该程序利用CRISPR / Cas9技术和rAAV6载体作为DNA模板的供体,将WT和突变DNA序列精确地插入其内源性基因位点。将工程化的cDNA(WT和突变体)与分离的荧光报告蛋白偶联,可以富集和跟踪具有明确杂合状态的细胞。
与常用的基于慢病毒(LV)的方法相比,该策略具有几个优点。一个主要优点是基于CRISPR/Cas9的系统允许对内源性位点进行精确编辑,从而保存内源性启动子和调控元件。这导致编辑基因在细胞中的表达呈同质性,当使用基于LV的方法时,这一目标几乎无法实现。与左心室载体的基因转移导致基因的半随机整合,优先于转录活性位点34。这可能转化为转移基因的过表达和编辑细胞之间的异质性,最终导致难以研究和分析突变和基因相互作用的作用。第二个优点是所描述的系统作为位点特异性编辑系统,消除了插入诱变的风险35。
双荧光报告策略允许对在两个等位基因上成功编辑的细胞进行精确富集和跟踪,其中一个等位基因整合WT cDNA,另一个等位基因整合突变的cDNA序列。仅表达单个报告基因的细胞仅代表具有相同荧光报告基因的HDR模板的单等位基因整合或双等位基因整合。只有产生单细胞衍生克隆并进行单独分析,才能精确区分这两种情况。然而,HSPC在 体外的增殖能力有限,当长时间培养时,HSPC开始分化为更成熟的后代,并失去其自我更新和植入能力。这使得选择和扩增携带所需杂合突变的单细胞克隆不可行。双荧光蛋白策略的应用和流式细胞术富集携带杂合突变的细胞可以绕过扩展体 外 培养引起的问题。
在这个具体的例子中,成功地证明了HSPC可以有效地设计和分类,以获得携带杂合 CALRDEL/WT突变的纯HSPC群体。
然而,该系统不仅限于工程杂合移码突变,而且还可以很容易地用于创建其他突变类型,包括错义和无义突变。通过应用含有WT的AAV的不同组合或具有不同荧光报告蛋白的突变序列,该系统还可用于引入纯合突变(同时转导两个rAAV都携带突变cDNA,但荧光报告基因不同)甚至校正突变(同时转导两个AAV都携带WT cDNA,但荧光报告基因不同)。此外,值得一提的是,该策略不仅限于引入致癌性GOF突变。事实上,所描述的方案可用于多种替代策略,包括基因敲除、基因替换36,37、转基因(即嵌合抗原受体)的靶向敲入38,甚至用于校正致病突变11,39。
将 CRISPR/Cas9 和 AAV6 与多种荧光报告基因相结合的策略也被证明适用于许多其他细胞类型,包括 T 细胞、浆细胞样树突状细胞、诱导多能干细胞、神经元干细胞和气道干细胞24,38,40,41,42,43,44.该策略可用于生产上嵌合抗原受体(CAR)T细胞。例如,最近发表的CRISPR/Cas9介导的敲除CAR-T细胞中TGFBR2基因的敲除大大增加了它们在抑制性TGF-β丰富的肿瘤微环境中的功能45。这种方法可以提供一步方案,既可以设计T细胞以表达CAR,也可以通过将CAR特异性插入TGFBR2基因的两个等位基因中来敲除TGFBR2基因。此外,这种方法还可用于通过将CAR整合到T细胞受体α常数(TRAC)基因46,47中来产生通用CAR-T细胞。
为了提高可重复性并保证细胞的有效编辑,需要考虑一些重要的因素。确保细胞成功编辑的主要关键点在于(i)sgRNA的选择,(ii)HDR模板的设计,以及(iii)rAAV6的生产。
选择性能良好的sgRNA至关重要,因为它将决定可以整合HDR模板的最大等位基因数量。由于现在有许多软件可用,因此对候选sgRNA的搜索已得到简化。通过选择感兴趣的区域,该软件可以提出一系列具有靶标评分和脱靶评分的sgRNA,分别指示在所需位点和不需要的位点进行编辑的机会。这些分数是根据以前发布的评分模型48,49 计算得出的。虽然这是选择性能良好的sgRNA的良好起点,但sgRNA的性能需要确认,因为它在 计算机中的 预测性能并不总是与 体外有效的sgRNA相对应。因此,强烈建议设计和测试至少三种sgRNA,以增加找到最佳sgRNA的机会。一旦鉴定出真正表现良好的sgRNA,建议继续进行HDR模板的设计。
设计 HDR 模板时应考虑预防措施。左同源臂和右同源臂(分别为LHA和RHA)应分别跨越sgRNA切割位点的上游和下游400 bp,因为较短的HA可能导致HDR频率降低。 可以通过 HDR引入的cDNA的大小取决于AAV的包装能力,大约为4.7 kb。由于HDR模板中有许多必需的元素(LHA,RHA,SA,PolyA,启动子和荧光报告序列),突变或WT cDNA的剩余空间是有限的。如果所需的突变位于基因的3'末端附近或具有整体短CDS的基因中,则这是没有问题的。然而,如果突变位于具有长CDS的基因的转录起始侧(TSS)附近(超过AAV的剩余包装空间),这种描述的方法可能不可行。为了解决这个问题,Bak及其同事最近开发了一种依赖于将HDR模板拆分为两个AAV的策略。该策略依赖于两个独立的HDR介导的整合来获得大基因50的最终无缝整合。
病毒的质量及其滴度是可以成就或破坏细胞基因组工程成功的其他因素。为了获得最佳产量,重要的是不要让HEK293T在培养物中保持完全汇合。理想情况下,HEK293T细胞应在达到70%-80%汇合时分裂。此外,HEK293T不应长时间培养,因为这会降低其产生病毒的能力。新的HEK293T细胞需要在20代后解冻。获得高病毒滴度对于提高实验的效率和可重复性非常重要。低病毒滴度将转化为HSPC转导所需的大量rAAV溶液。作为一般规则,添加到核化细胞中的rAAV溶液不应超过HSPC保留培养基总体积的20%。较高体积的AAV溶液可能导致细胞死亡增加,增殖降低和转导效率受损。因此,在病毒滴度低的情况下,建议进一步浓缩病毒。
总之,该协议提供了一种可重复的方法,通过同时使用CRIPSR / Cas9和rAAV6供体模板以及额外的双荧光报告基因,精确有效地操作人类HSPC。这种方法已被证明是研究正常造血干细胞生物学以及突变对白血病发生的贡献的重要工具。
披露声明
作者没有什么可透露的。
致谢
这项工作得到了奥地利科学基金(FWF;编号P32783和I5021)对A.R.的资助,奥地利内科医学会(约瑟夫·斯柯达奖学金),奥地利血液学和肿瘤学会(OeGHO;临床研究基金)和MEFOgraz。T.K.是白血病和淋巴瘤协会的特别研究员。
材料
| Name | Company | Catalog Number | Comments |
| 175 cm2 Cell Culture Flask, Vent Cap, TC-treated | Corning | 431080 | |
| 150 mm x 25 mm dishes | Corning | 430599 | |
| 293T | DSMZ | ACC 635 | https://www.dsmz.de/collection/catalogue/details/culture/ACC-635 |
| 4D Nucleofector Core Unit | Lonza | - | For nucleofection of human HSPCs use the DZ-100 program. |
| 4D Nucleofector X Unit | Lonza | - | |
| 500 ml Centrifuge Tube | Corning | 431123 | |
| 7-AAD | BD Biosciences | 559925 | |
| AAVpro Purification Kit | Takara | 6666 | |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | 1081058 | |
| Avanti JXN-30 Ultracentrifuge | Beckman Coulter | - | |
| Benchling sgRNA design tool | Online tool for sgRNA design: http://www.benchling.com/crispr | ||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7906-100G | |
| C1000 Touch Thermal Cycler | Bio-Rad | - | |
| Chemically modified synthetic sgRNA | Synthego | Website: https://www.synthego.com/products/crispr-kits/synthetic-sgrna Sequence for the sgRNA targeting intron 7 of CALR: 5’-CGCCTGTAATCCTCGCCCAG-3’ An 80 nucleotide SpCas9 scaffold is added to the 20 nucleotide RNA sequence to complete the sgRNA. Chemical modifications of 2'-O-Methyl are added to the first and last 3 bases and 3' phosphorothioate bonds are added in the first 3 and last 2 bases. *Alternatively chemically modified synthetic sgRNAs can be acquired from IDT (https://eu.idtdna.com/site/order/oligoentry/index/crispr) and Trilink (https://www.trilinkbiotech.com/custom-oligos) | |
| CHOPCHOP sgRNA design tool | Online tool for sgRNA design: http://chopchop.cbu.uib.no | ||
| Costar 24-well Clear TC-treated Multiple Well Plates | Corning | 3526 | |
| CRISPick sgRNA design tool | Online tool for sgRNA design: https://portals.broadinstitute.org/gppx/crispick/public | ||
| CRISPOR sgRNA design tool | Online tool for sgRNA design: http://crispor.tefor.net | ||
| ddPCR 96-Well Plates | Bio-Rad | 12001925 | |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863024 | |
| DG8 Cartridges for QX200/QX100 Droplet Generator | Bio-Rad | 1864008 | |
| DG8 Gaskets for QX200/QX100 Droplet Generator | Bio-Rad | 1863009 | |
| DreamTaq Green PCR Master Mix (2X) | Thermo Scientific | K1081 | |
| Droplet Generation Oil for Probes | Bio-Rad | 1863005 | |
| Dulbecco’s Modified Eagle Medium (DMEM) with high glucose | Sigma-Aldrich | D6429-6X500ML | |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500ML | |
| FACSAria Fusion | BD Biosciences | - | |
| Falcon 5 mL Round Bottom | Corning | 352054 | |
| Fetal Bovine Serum (FBS) Good Forte (heat inactivated), 500 ml | Pan Biotech | P40-47500 | |
| FlowJo 10.8.0 | BD Biosciences | - | |
| GenAgarose L.E. | Inno-train | GX04090 | |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | |
| Gibson Assembly Master Mix | New England Biolabs Inc. (NEB) | E2611L | |
| HEK293T | |||
| HEPES solution | Sigma-Aldrich | H0887-100ML | |
| ICE | Synthego | https://ice.synthego.com | |
| IDT codon optimization tool | IDT | https://www.idtdna.com/pages/tools/codon-optimization-tool | |
| IDT sgRNA design tool | Online tool for sgRNA design: https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM | ||
| LB Broth (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7658-1KG | |
| LB Broth with agar (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7533-1KG | |
| Midori Green Advance | Nippon Genetics | MG04 | |
| Monarch Plasmid Miniprep Kit | NEB | T1010L | |
| Monarch DNA Gel Extraction Kit | NEB | T1020L | |
| NEB 5-alpha Competent E. coli (High Efficiency) | NEB | C2987U | |
| Nuclease-Free Water, 5X100 ml | Ambion | AM9939 | |
| NucleoBond Xtra Midi | Macherey-Nagel | 740410 | |
| Opti-MEM, Reduced Serum Medium, 500 ml | Gibco | 31985070 | |
| P3 Primary Cell 4D-Nucleofetor X Kit L | Lonza | V4XP-3024 | The Lonza Primary P3 solution is supplied as a 2.25 mL P3 Primary Cell Nucleofector Solution and 0.5 mL Supplement 1. To reconstitute, add the Supplement 1 to the P3 Primary Cell Nucleofector Solution and mix. |
| pAAV-MCS2 | Addgene | 46954 | |
| PCR Plate Heat Seal Foil, pierceable | Bio-Rad | 1814040 | |
| pDGM6 | Addgene | 110660 | |
| Penicillin-Streptomycin (P/S) | Gibco | 15140122 | |
| Polyethylenimine (PEI) | Polysciences | 23966 | Add 50 mL of PBS 4.5 pH (made with HCl) to 50 mg of PEI in a tube. Dissolve by placing the tube in a 70°C water bath and vortexing every 10 minutes until the solution is dissolved. After the solution has reached RT, filter sterilze through a 0.22 μm filter, make 1120 μL aliquots, and store at -80°C. |
| Polystyrene Test Tube, with Snap Cap | |||
| Primers | Eurofins | - | Primers were ordered from Eurofins (eurofinsgenomics.eu) as unmodified salt free custom oligos. The primers were designed by using PRIMER-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) Primer 1 Fwd: AAGTGATCCGTTCGCCATGAC; Primer 2 Rev CALR WT specific: ACGTCCTCTTCCTCGTCCTC; Primer 2 Rev CALR DEL specific: CCAACCCTGGAGACACGCTTC |
| PrimeTime qPCR Primer Assay | IDT | - | PrimeTime qPCR Probe Assays (1 probe/2 primers) that can be ordered from IDT (https://eu.idtdna.com/site/order/qpcr/assayentry). Scale: Std - qPCR Assay 500 reactions; Primer 1 Forward (5'-3') : GGAACCCCTAGTGATGGAGTT; Primer 2 Reverse (5'-3'): CGGCCTCAGTGAGCGA; Probe (5'-3'): CACTCCCTCTCTGCGCGCTCG; 5' Dye/3' Quencher: FAM/ZEN/IBFQ; Primer to probe ratio: 3.6 |
| PX1 PCR Plate Sealer | Bio-Rad | 1814000 | |
| QuantaSoft Software | Bio-Rad | ||
| Quick Extract DNA Extraction Solution | Lucigen | QE0905T | |
| QX200 Droplet Generator | Bio-Rad | 1864002 | |
| QX200 Droplet Reader | Bio-Rad | ||
| Recombinant human Flt3-ligand | Peprotech | 300-19 | |
| Recombinant human IL-6 | Peprotech | 200-06 | |
| Recombinant Human SCF | Peprotech | 300-07 | |
| Recombinant Human TPO | Peprotech | 300-18 | |
| RPMI 1640 | Sigma-Aldrich | R8758-6X500ML | |
| SnapGene | Dotmatics | Molecular cloning software https://www.snapgene.com *Alternatively also Benchling (https://www.benchling.com) and Geneious (https://www.geneious.com) can be used. | |
| Soc outgrowth medium | NEB | B9020S | |
| Sodium-butyrate | Sigma-Aldrich | B5887-1G | |
| Stem Regenin 1 (SR1) | Biogems | 1224999 | |
| StemSpan SFEM II | STEMCELL Technologies | 9655 | |
| TAE Buffer (Tris-acetate-EDTA) 50X | Thermo Scientific | B49 | |
| TIDE | http://shinyapps.datacurators.nl/tide/ | ||
| Trypan blue 0.4% | Sigma-Aldrich | T8154-100ML | |
| TrypLE (with phenol red), 500 ml | Thermo Scientific | 16605-028 | |
| UltraPure 0.5: EDTA, pH 8.0, 100 ml | Thermo Scientific | 15575-038 | |
| UM171 | STEMCELL Technologies | 72914 | |
| Vector Builder codon optimization tool | Vector Builder | https://en.vectorbuilder.com/tool/codon-optimization.html |
参考文献
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Gillmore, J. D., et al. CRISPR-Cas9 in vivo gene editing transthyretin amyloidosis. New England Journal of Medicine. 385 (6), 493-502 (2021).
- Frangoul, H., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine. 384 (3), 252-260 (2021).
- Stadtmauer, E. A., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 367 (6481), (2020).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Veldwijk, M. R., et al. Pseudotyped recombinant adeno-associated viral vectors mediate efficient gene transfer into primary human CD34+ peripheral blood progenitor cells. Cytotherapy. 12 (1), 107-112 (2010).
- Song, L., et al. High-efficiency transduction of primary human hematopoietic stem cells and erythroid lineage-restricted expression by optimized AAV6 serotype vectors in vitro and in a murine xenograft model in vivo. PLoS One. 8 (3), 58757 (2013).
- Dever, D. P., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 539 (7629), 384-389 (2016).
- Drost, J., et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521 (7550), 43-47 (2015).
- Jan, M., et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine. 4 (149), (2012).
- Corces-Zimmerman, M. R., Hong, W. J., Weissman, I. L., Medeiros, B. C., Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America. 111 (7), 2548-2553 (2014).
- Jaiswal, S., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine. 377 (2), 111-121 (2017).
- Genovese, G., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New England Journal of Medicine. 371 (26), 2477-2487 (2014).
- Papaemmanuil, E., et al. Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine. 374 (23), 2209-2221 (2016).
- Cancer Genone Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 368 (22), 2059-2074 (2013).
- Ball, M., List, A. F., Padron, E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 128 (20), 2381-2387 (2016).
- Varmus, H. E. The molecular genetics of cellular oncogenes. Annual Review of Genetics. 18, 553-612 (2003).
- Cox, D. B. T., Platt, R. J., Zhang, F. Therapeutic genome editing: Prospects and challenges. Nature Medicine. 21 (2), 121-131 (2015).
- Tothova, Z., et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell. 21 (4), 547-555 (2017).
- Mandal, P. K., et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 15 (5), 643-652 (2014).
- Bak, R. O., Dever, D. P., Porteus, M. H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols. 13 (2), 358-376 (2018).
- Foßelteder, J., et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia. , (2023).
- Nangalia, J., et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New England Journal of Medicine. 369 (25), 2391-2405 (2013).
- Merlinsky, T. R., Levine, R. L., Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clinical Cancer Research. 25 (10), 2956-2962 (2019).
- Belčič Mikič, T., Pajič, T., Zver, S., Sever, M. The contemporary approach to CALR-positive myeloproliferative neoplasms. International Journal of Molecular Sciences. 22 (7), 3371 (2021).
- How, J., Hobbs, G. S., Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 134 (25), 2242-2248 (2019).
- Grieger, J. C., Choi, V. W., Samulski, R. J. Production and characterization of adeno-associated viral vectors. Nature Protocols. 1 (3), 1412-1428 (2006).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 6 (6), 973-985 (1999).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2011).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Bulcha, J. T., Wang, Y., Ma, H., Tai, P. W. L., Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduction and Targeted Therapy. 6 (1), 1-24 (2021).
- Montini, E., et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. The Journal of Clinical Investigation. 119 (4), 964-975 (2009).
- Vaidyanathan, S., et al. Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Molecular Therapy. 30 (1), 223-237 (2022).
- Cromer, M. K., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nature Medicine. 27 (4), 677-687 (2021).
- Wiebking, V., et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica. 106 (3), 847-858 (2021).
- Wilkinson, A. C., et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nature Communications. 12 (1), 1-9 (2021).
- Dever, D. P., et al. CRISPR/Cas9 genome engineering in engraftable human brain-derived neural stem cells. iScience. 15, 524-535 (2019).
- Laustsen, A., et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nature Communications. 9 (1), 1-14 (2018).
- Bak, R. O., et al. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife. 6, 27873 (2017).
- Nakauchi, Y., et al. The cell type-specific 5hmC landscape and dynamics of healthy human hematopoiesis and TET2-mutant preleukemia. Blood Cancer Discovery. 3 (4), 346-367 (2022).
- Vaidyanathan, S., et al. selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 26 (2), 161-171 (2020).
- Tang, N., et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 5 (4), 133977 (2020).
- Georgiadis, C., et al. Long terminal repeat CRISPR-CAR-coupled "universal" T cells mediate potent anti-leukemic effects. Molecular Therapy. 26 (5), 1215-1227 (2018).
- Ren, J., et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical Cancer Research. 23 (9), 2255-2266 (2017).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Doench, J. G., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Bak, R. O., Porteus, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Reports. 20 (3), 750-756 (2017).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。