
Method Article
Engenharia de Mutações de Ganho de Função Heterozigotas Oncogênicas em Células-Tronco Hematopoiéticas Humanas e Progenitoras
Neste Artigo
Resumo
Novas estratégias para modelar fielmente mutações somáticas em células-tronco hematopoiéticas e progenitoras (HSPCs) são necessárias para melhor estudar a biologia das células-tronco hematopoiéticas e as neoplasias hematológicas. Aqui, um protocolo para modelar mutações heterozigotas de ganho de função em HSPCs combinando o uso de CRISPR/Cas9 e transdução dupla de doador de rAAV é descrito.
Resumo
Ao longo de sua vida, as células-tronco hematopoiéticas e progenitoras (HSPCs) adquirem mutações somáticas. Algumas dessas mutações alteram as propriedades funcionais do HSPC, como proliferação e diferenciação, promovendo assim o desenvolvimento de neoplasias hematológicas. A manipulação genética eficiente e precisa dos HSPCs é necessária para modelar, caracterizar e entender melhor as consequências funcionais das mutações somáticas recorrentes. As mutações podem ter um efeito deletério em um gene e resultar em perda de função (LOF) ou, em contraste gritante, podem melhorar a função ou mesmo levar a novas características de um gene em particular, denominado ganho de função (GOF). Em contraste com as mutações LOF, as mutações GOF ocorrem quase exclusivamente de forma heterozigótica. Os protocolos atuais de edição de genoma não permitem o direcionamento seletivo de alelos individuais, dificultando a capacidade de modelar mutações GOF heterozigotas. Aqui, fornecemos um protocolo detalhado sobre como projetar mutações de hotspot GOF heterozigotas em HSPCs humanos, combinando reparo dirigido por homologia mediado por CRISPR / Cas9 e tecnologia AAV6 recombinante para transferência eficiente de molde de doador de DNA. É importante ressaltar que essa estratégia faz uso de um sistema de repórter fluorescente duplo para permitir o rastreamento e a purificação de HSPCs editados de forma heterozigótica bem-sucedida. Essa estratégia pode ser empregada para investigar com precisão como as mutações GOF afetam a função do HSPC e sua progressão para neoplasias hematológicas.
Introdução
Com o desenvolvimento da tecnologia de repetições palindrômicas curtas agrupadas regularmente interespaçadas (CRISPR)/Cas9, um novo e extremamente poderoso instrumento foi adicionado ao kit de ferramentas dos cientistas. Essa tecnologia permite a engenharia precisa do genoma e tem se mostrado extremamente útil não apenas para fins de pesquisa (revisada em Hsu et al.1), mas mais recentemente também tem sido traduzida com sucesso para o cenário clínico 2,3,4. As estratégias de edição CRISPR/Cas9 dependem da atividade de uma proteína Cas9 e de um RNA de guia único (sgRNA)5,6,7. Na célula hospedeira, a proteína Cas9 é guiada para um local específico no DNA que é complementar à sequência de sgRNA e introduzirá uma quebra de fita dupla de DNA (DSB). Uma vez que um DSB é gerado, existem dois mecanismos de reparo principais e concorrentes que podem ocorrer: junção final não homóloga (NHEJ) e reparo dirigido por homologia (HDR). O NHEJ é um mecanismo de reparo predominantemente usado e propenso a erros, levando a inserções e deleções (indels), enquanto o HDR, ao usar a cromátide irmã como modelo de reparo, é muito preciso, mas limitado à fase S ou G2 do ciclo celular8. Na engenharia do genoma, o HDR pode ser utilizado para a modificação direcionada do DNA, fornecendo um molde doador que é ladeado por braços de homologia idênticos a ambas as extremidades do DNA do DSB induzido por Cas9 (Figura 1). O tipo de modelo de doador usado para HDR pode afetar muito a eficiência da edição. Para a engenharia genética em HSPCs humanos, o sorotipo 6 do vírus adenoassociado (AAV6) tem sido recentemente descrito como um excelente veículo para a entrega de modelos de DNA de fita simples 9,10.
A engenharia genômica CRISPR/Cas9 pode ser empregada terapeuticamente para corrigir mutações deletérias11, mas também pode ser utilizada para introduzir mutações patogênicas no DNA para modelar o desenvolvimento do câncer12. O câncer de sangue, como a leucemia, desenvolve-se por meio da aquisição sequencial de mutações somáticas em HSPCs saudáveis13,14. Eventos genéticos precoces levam a uma vantagem proliferativa clonal, resultando em hematopoiese clonal de potencial indeterminado (CHIP)15,16. A aquisição adicional de mutações acabará por levar à transformação leucêmica e ao desenvolvimento da doença. Mutações somáticas podem ser encontradas em genes que controlam a auto-renovação, sobrevivência, proliferação e diferenciação17.
A introdução de mutações individuais por meio da engenharia do genoma em HSPCs saudáveis permite modelar com precisão esse processo leucemogênico gradual. O número limitado de mutações recorrentes encontradas em neoplasias mieloides como a leucemia mieloide aguda (LMA)18,19 torna essa doença particularmente passível de ser recapitulada por meio de ferramentas de engenharia genômica.
As mutações somáticas podem surgir em apenas um alelo (mutações monoalélicas/heterozigotas) ou em ambos os alelos (mutações bialélicas/homozigotas) e podem ter efeitos profundos na função do gene, o que poderia causar uma perda de função (LOF) ou um ganho de função (GOF). As mutações LOF levam a um LOF reduzido (se um alelo for afetado) ou completo (se ambos os alelos forem afetados) do gene, enquanto as mutações GOF levam a uma ativação aumentada ou a uma nova função do gene. As mutações GOF são tipicamente heterozigotas20.
É importante ressaltar que a zigosidade (hetero vs. homozigoto) tem grandes implicações para a tentativa de modelar fielmente uma mutação; portanto, a manipulação direcionada de apenas um alelo de um gene é necessária para projetar mutações GOF de hotspot heterozigoto. O NHEJ propenso a erros leva a indels de diferentes comprimentos21 que podem levar a consequências biológicas variadas e imprevisíveis. No entanto, como o NHEJ é o programa de reparo predominante empregado pelas células após a introdução de um DSB, a maioria das plataformas CRISPR/Cas9 atualmente utilizadas para manipular HSPCs não permite prever com precisão o desfecho genético22,23. Em contraste, a introdução de uma quebra de fita dupla mediada por CRISPR/Cas9 (DSBs), combinada com o uso de modelos de doadores de DNA baseados em vetores baseados em vírus adenoassociados recombinantes (rAAV) para engenharia genômica via HDR, permite a inserção alelo-específica de mutações em HSPCs humanos11,24. A integração simultânea de uma sequência mutante e uma do tipo selvagem (WT), juntamente com repórteres fluorescentes distintos nos alelos individuais, pode ser realizada para selecionar um genótipo heterozigoto (Figura 2). Essa estratégia pode ser aproveitada como uma ferramenta poderosa para caracterizar com precisão os efeitos de mutações recorrentes, leucêmicas e heterozigóticas do hotspot GOF na função, início da doença e progressão do HSPC.
Neste artigo, um protocolo detalhado para a engenharia eficiente de mutações GOF heterozigotas recorrentemente mutadas em HSPCs humanos primários é fornecido. Essa estratégia combina o uso de CRISPR/Cas9 e uma transdução dupla de AAV6 para fornecer modelos de doadores de WT e DNA mutante para a geração prospectiva da mutação GOF heterozigótica. Como exemplo, a engenharia das mutações recorrentes tipo 1 (deleção de 52 pb) no gene da calreticulina (CALR) será mostrada25. A mutação GOF heterozigótica no éxon 9 da CALR é recorrentemente encontrada em distúrbios mieloproliferativos, como trombocitemia essencial (TE) e mielofibrose primária (FMP)26. A CALR é uma proteína residente do retículo endoplasmático que tem principalmente uma função de controle de qualidade no processo de dobramento de proteínas recém-sintetizadas. Sua estrutura pode ser dividida em três domínios principais: um domínio amino (N)-terminal e um domínio P rico em prolina, que estão envolvidos na função chaperona da proteína, e um domínio C, que está envolvido no armazenamento e regulação do cálcio27,28. As mutações da CALR causam um desvio de quadro +1, levando à transcrição de uma nova extremidade C-terminal estendida e à perda do sinal de retenção do retículo endoplasmático (RE) (KDEL). Demonstrou-se que a CALR mutante se liga ao receptor de trombopoietina (TPO), levando à sinalização independente da TPO com aumento da proliferação29.

Figura 1: Reparo de NHEJ e HDR. Representação esquemática simplificada dos mecanismos de reparo NHEJ e HDR após a introdução de uma quebra de fita dupla no DNA. Por favor, clique aqui para ver uma versão maior desta figura.
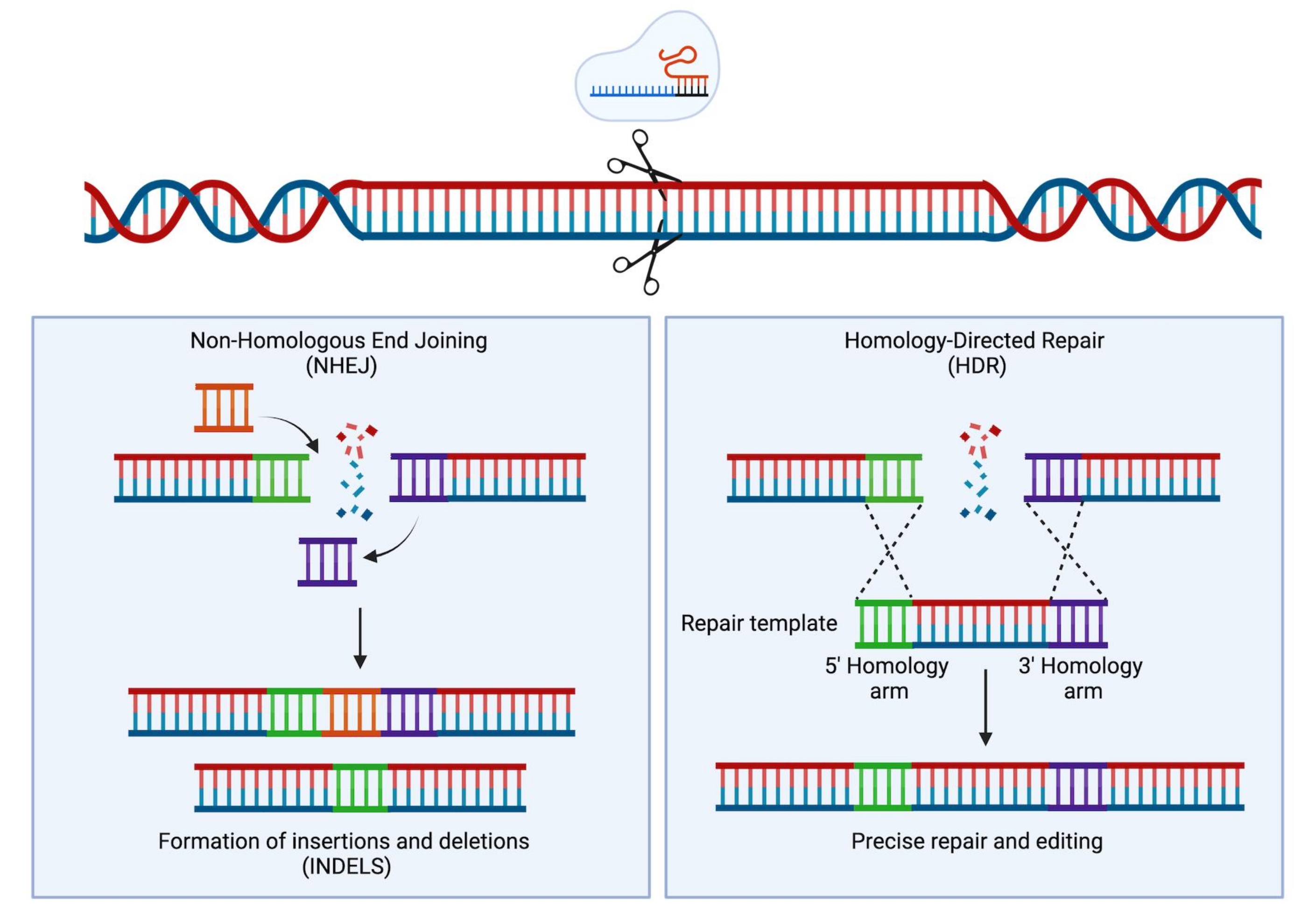{kind=link}

Figura 2: Visão geral esquemática da estratégia de edição HDR bialélica. Representação esquemática mostrando a integração dos modelos doadores nos alelos alvo, seguida de sua tradução em mRNAs funcionais. As caixas pontilhadas laranja indicam as regiões correspondentes ao braço de homologia esquerdo (LHA) e ao braço de homologia direito (RHA). O tamanho ideal dos HAs é de 400 pb cada. A caixa pontilhada verde representa a região correspondente à sequência SA. O tamanho do SA é de 150 pb. Clique aqui para ver uma versão maior desta figura.
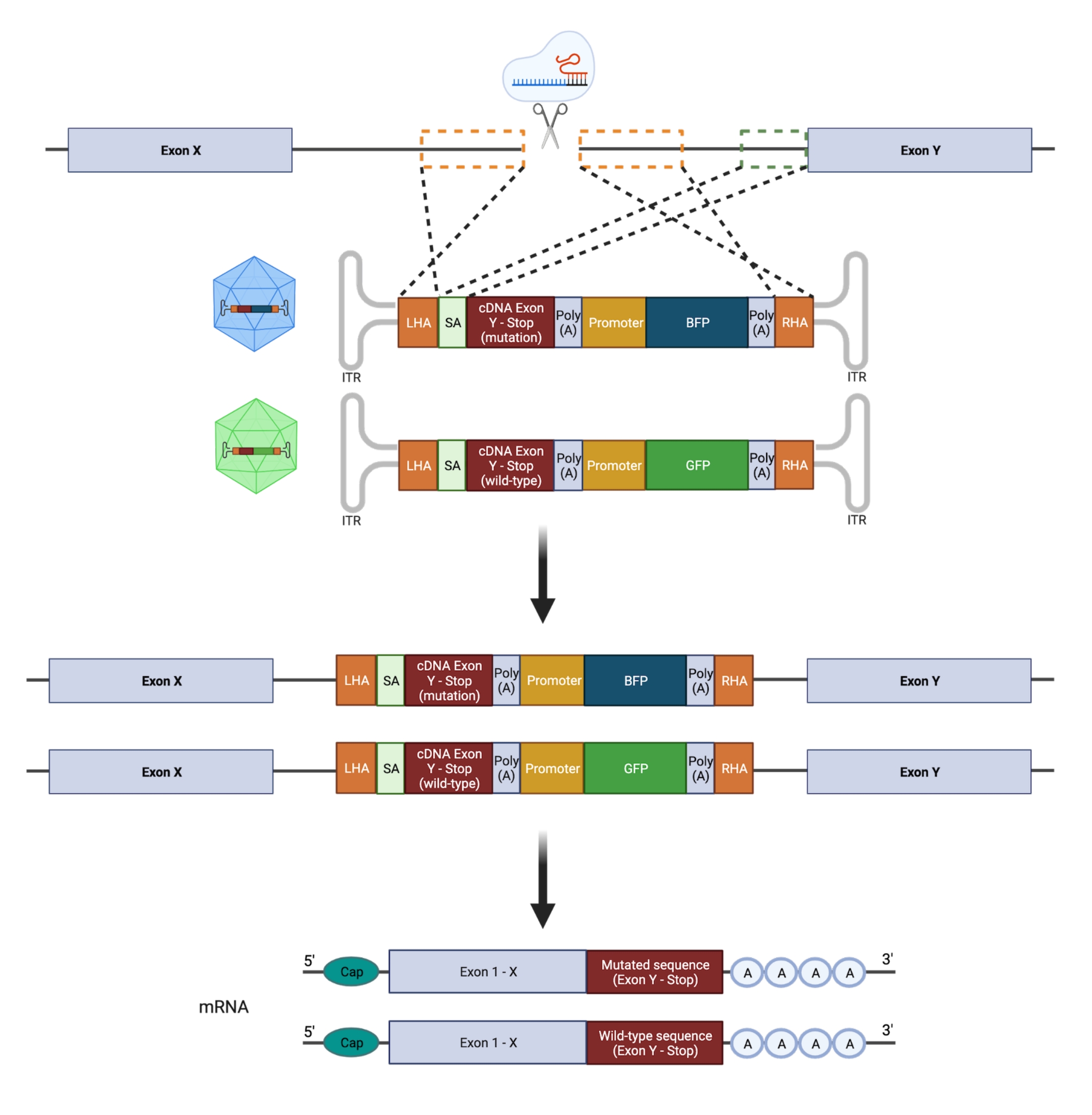{kind=link}
Protocolo
Este protocolo requer o uso de HSPCs CD34+ derivados de doadores saudáveis e requer aprovação ética dos conselhos de revisão institucional locais (IRB) e consentimento informado assinado. Os HSPCs CD34+ utilizados neste protocolo foram isolados do sangue do cordão umbilical (UCB) de partos a termo (>34 semanas de gestação). O consentimento informado foi obtido das mães antes do parto, e a aprovação ética para a coleta de UCB foi obtida (aprovação IRB: 31-322 ex 18/19) da Medical University of Graz. Uma lista completa dos materiais utilizados neste protocolo pode ser encontrada na Tabela de Materiais.
1. Projeto de sgRNA e avaliação de eficiências de corte
- Procure a localização da mutação desejada e a transcrição correta em uma ferramenta de banco de dados on-line (por exemplo, COSMIC, https://cancer.sanger.ac.uk/cosmic).
- Selecione um sgRNA adjacente à mutação (segmentação exônica) ou no íntron anterior (segmentação intrônica) usando uma ferramenta de design de sgRNA. Neste protocolo, a ferramenta online da Benchling é usada. Consulte A Tabela de Materiais para obter uma lista de possíveis ferramentas on-line para o design de sgRNA.
- Selecione a opção + (criar) > Sequência de DNA > Importar sequências de DNA > Importar de bancos de dados. Insira o gene desejado, por exemplo, CALR e selecione humano como a espécie > Pesquisar > Selecione o transcrito correto (por exemplo, CALR-201 -ENST00000316448) > Importar. Selecione a região de interesse na qual introduzir a quebra DS (por exemplo, íntron 7).
- Selecione a opção CRISPR no lado direito da tela e selecione Projetar e analisar guias. Selecione uma única guia, mantenha o comprimento da guia em 20 pb e a sequência PAM para edição com SpCas9 (NGG).
- Escolha um guia com uma alta pontuação no alvo (alta chance de edição no locus desejado) e uma alta pontuação fora do alvo (baixa chance de editar em loci indesejados). Escolha pelo menos três guias para testar, a fim de encontrar o sgRNA de melhor desempenho. Encomende o sgRNA como um sgRNA sintético quimicamente modificado de um fornecedor comercial.
NOTA: É aconselhável nesta fase encomendar uma pequena quantidade de sgRNAs sintéticos para a tela inicial. Uma vez que um sgRNA de bom desempenho tenha sido identificado, prossiga com uma ordem maior do sgRNA selecionado. - Descongelar 2 x 10 5-5 x 105 CD34+ HSPCs. Transfira as células para 10 mL de RPMI1640 pré-aquecido suplementado com antibióticos a 1% (por exemplo, penicilina/estreptomicina).
NOTA: HSPCs CD34+ com uma pureza >90% devem ser usados para alcançar os melhores e mais reprodutíveis resultados. - Centrífuga a 350 x g à temperatura ambiente (RT) durante 10 min. Contar as células com um hemocitômetro e suspender as células em meio SFEM II suplementado com 0,2% de penicilina/estreptomicina (P/S), 100 ng/mL de trombopoietina (TPO), 100 ng/mL de fator de células-tronco (SCF), 100 ng/mL de ligante de tirosina 3 semelhante à FMS (FLT3L), 100 ng/mL de interleucina-6 (IL-6), 35 nM UM171 e 0,75 μM de StemRegenin1 (SR1) até uma concentração de 2,5 x 105 células/mL. Incubar a 37 °C/5% CO2 durante 48 h - 72 h.
NOTA: O meio completo será referido a partir de agora como o meio de retenção do HSPC. - Colete as células em um tubo de 15 mL e conte as células. Verifique a viabilidade celular por exclusão azul de tripano.
- Antes de começar, ligue o sistema de transfecção e selecione a opção para as cuvetes. Selecione o programa apropriado e a solução de nucleofeção para HSPCs (consulte Tabela de Materiais). Entre 2 x 10 5 e5 x 106 células podem ser nucleofectadas em uma cubeta de 100 μL. Reserve um pequeno número de células (por exemplo, 1 x 10 5-2 x 105) para manter em cultura por 48 h para ser usado como controle WT.
- Preparar o complexo RNP. Num tubo de 1,5 ml, adicionar 15 μg de Cas9 e 8 μg de sgRNA (razão molar 1:2,5) e incubar a 25 °C durante 10 min num bloco de aquecimento.
- Enquanto o complexo RNP está incubando, centrifugar as células a 350 x g em RT por 5 min e descartar o sobrenadante usando uma pipeta. Suspender as células em 100 μL de solução de nucleofeção.
- Misture as células com o complexo RNP e transfira para a cubeta. Bata suavemente na cubeta para remover eventuais bolhas de ar que possam ter se formado durante a transferência.
- Insira a cubeta no suporte do sistema de transfecção e eletroporate as células com o programa DZ-100.
- Imediatamente após a eletroporação, adicionar 400 μL de meio de retenção HSPC pré-aquecido sem P/S.
- Transfira as células com uma pipeta de transferência fina para uma placa de cultura contendo meio de retenção HSPC pré-aquecido sem P/S. Dependendo do número de células, use uma placa de cultura apropriada (placa de 24, 12 ou 6 poços) para atingir uma densidade entre 0,25 x 10 6 e 1 x 106 células/mL.
- Transferir a placa para a incubadora a 37 °C/5% de CO2. Incubar as células nucleofecadas por 6-8 h.
- Após 6-8 h, remova o meio antigo e substitua por um meio de retenção de HSPC pré-aquecido fresco suplementado com P/S. Suspenda as células a uma concentração entre 2,5 x 10 5 e 5 x 105 e transfira para uma placa de cultura celular (placa de 24, 12 ou 6 poços). Incubar as células por 48 h a 37°C/5% de CO2.
- Colha 2 x 10 5 células e centrifugar a 350 x g a RT por5 min. Antes de começar, ajuste os blocos de aquecimento a 65 °C e 98 °C.
- Descarte o sobrenadante e suspenda as células em 1 mL de 1x DPBS em um tubo de 1,5 mL. Centrífuga a 350 x g a RT durante 5 min.
- Rejeitar o sobrenadante e suspender as células em 50 μL de solução de extracção de ADN. Vórtice por 15 s.
- Incubar durante 6 min a 65 °C. Vórtice por 15 s e incubar por 2 min a 98 °C.
- Amplificar a região do DSB por PCR usando primers que geram um amplificador de cerca de 400-600 pb com o DSB no centro. Use 0,5-1 μL de DNA extraído para a reação de PCR.
NOTA: Devido à composição da solução de extração de DNA, as concentrações de DNA não podem ser quantificadas com precisão por espectrofotometria. - Execute o produto de PCR com uma escada de DNA em um gel de agarose a 1,5% a 100 V por 45-60 min. Coloque o gel em um transiluminador de luz azul ou UV.
- Extraia a banda de DNA do tamanho correto do gel usando um kit comercialmente disponível de acordo com as instruções do fabricante (consulte a Tabela de Materiais).
- Sequencie as amostras por sequenciamento de Sanger usando o primer para frente ou o primer reverso da PCR. Para produtos de PCR de 400-600 pb de comprimento, são necessários 75 ng de DNA para sequenciamento com uma concentração de 5 ng/mL em um volume total de 15 μL.
- Analise a eficiência de edição dos sgRNAs carregando os arquivos de sequenciamento para um programa projetado para calcular a eficiência de edição, identificando a inserção e as exclusões produzidas pelo sgRNA. Selecione o sgRNA de melhor desempenho para prosseguir.
NOTA: As ferramentas on-line dedicadas estão listadas na Tabela de Materiais. Essa análise requer os arquivos .ab1 dos HSPCs transfectados e WT, a sequência de sgRNA e a sequência PAM.
2. Construção vetorial de reparo dirigido por homologia (HDR)
- Design de modelo HDR
Observação : dois modelos HDR devem ser projetados: um modelo para a sequência WT e um modelo para a sequência mutada.- Importe a sequência genômica e a sequência de codificação (CDS) do gene desejado para um software apropriado para clonagem molecular (ferramentas dedicadas podem ser encontradas listadas na Tabela de Materiais). A partir do arquivo de sequência genômica, projete o braço de homologia esquerda (AH) selecionando idealmente 400 pb na extremidade 5' do DSB. Cole essa sequência em um novo arquivo.
- Se um íntron é direcionado com o sgRNA, uma sequência de aceptor de emenda (SA) precisa ser incluída. Selecione os últimos 150 pb do íntron de destino e cole-o após o AH esquerdo. Se o éxon for direcionado diretamente, essa sequência não será necessária (Figura 3).
- No arquivo CDS, selecione o cDNA de interesse. No caso de uma sequência exônica ser direcionada, certifique-se de que o cDNA comece imediatamente a jusante do local DSB e inclua todos os seguintes éxons do gene de interesse, bem como o códon de parada. No caso de um íntron ser direcionado, certifique-se de que o cDNA comece com o primeiro códon do éxon seguinte. Códon-otimizar o cDNA e inseri-lo após a sequência SA. Use ferramentas on-line dedicadas para essa finalidade (Tabela de Materiais).
- Insira um sinal de poliadenilação (PolyA) de 3' após o cDNA de interesse (por exemplo, SV40 ou bGH) (Tabela 1). Após a PolyA, insira uma sequência promotora (por exemplo, vírus formador de foco do baço [SFFV] ou poliubiquitina C [UBC], Tabela 1) para a proteína fluorescente.
- Insira a sequência para a proteína fluorescente (ou seja, GFP, PBF ou mCherry). Insira um segundo sinal, mas diferente, PolyA.
NOTA: A inserção de uma sequência PolyA diferente evitará problemas como a recombinação bacteriana do plasmídeo ou problemas com o alinhamento da sequência devido a duas sequências idênticas no modelo. - A partir do arquivo de sequência genômica, projete o HA correto selecionando idealmente 400 pb na extremidade 3' do DSB. Insira essa sequência após o segundo PolyA.
- Crie uma cópia de todo o modelo e modifique o cDNA de interesse para que ele contenha a sequência da mutação desejada.
- Troque a proteína fluorescente por uma proteína fluorescente diferente. Por exemplo, se a GFP foi usada para o modelo WT, troque-a com BFP ou mCherry para o modelo mutante. Construa e clone os modelos HDR no plasmídeo pAAV-MCS (ou outros backbones adequados).
NOTA: Os fragmentos necessários para a montagem podem ser produzidos por PCR ou encomendados comercialmente e precisam conter sequências sobrepostas com seus fragmentos vizinhos. Se a mutação de interesse for uma mutação pontual, uma pequena inserção ou uma pequena deleção, o cDNA mutado pode ser produzido por PCR realizando uma mutagênese direcionada ao local no molde HDR contendo o cDNA WT. - Transformar a Escherichia coli competente com o produto montado utilizando o método do choque térmico. Antes de começar, coloque as bactérias para descongelar no gelo por 10 min.
- Adicionar 2 μL dos produtos montados a 50 μL de bactérias. Misture o conteúdo com um movimento suave.
- Coloque as amostras no gelo por 30 min. Transfira as amostras para um termobloco ajustado a 42 °C durante 30 s.
- Transfira as amostras no gelo por 5 min. Adicionar 450 μL de meio SOC à temperatura ambiente e incubar as amostras a 37 °C durante 1 h.
- Espalhe as amostras em placas de ágar LB contendo ampicilina. Para cada amostra, espalhar 100 μL de três diluições diferentes da solução bacteriana (não diluída, 1:5 e 1:10), a fim de obter placas de ágar LB com colónias únicas que possam ser colhidas. Incubar as placas durante a noite a 37 °C.
- No dia seguinte, escolha três colônias por amostra. Transferir as colônias para tubos de 15 mL com tampas contendo 4 mL de LB em meio suplementado com ampicilina e incubar durante a noite em agitador a 37 °C.
- Aliquot 500 μL da solução bacteriana de cada colónia e guardá-la no frigorífico para utilização posterior. Execute uma mini-preparação para extrair o DNA plasmídico de acordo com as instruções do fabricante.
- Envie as amostras para sequenciamento de Sanger para confirmar a montagem correta dos plasmídeos. Use iniciadores suficientes distribuídos por todo o plasmídeo para garantir a confirmação ininterrupta da sequência, idealmente cobrindo cada região duas vezes com leituras para frente e para trás.
- Adicionar 200 μL da solução bacteriana contendo o plasmídeo correctamente montado a 200 ml de meio LB suplementado com ampicilina e incubar num agitador durante a noite a 37 °C.
- Execute uma preparação midi ou maxi para extrair o DNA do plasmídeo de acordo com as instruções do fabricante. Armazenar o DNA plasmidial a -20 °C.
- Preparação recombinante de AAV6
NOTA: Duas preparações separadas de AAV6 precisarão ser executadas: uma para o rAAV6 com o modelo WT HDR e outra para o rAAV6 com o modelo HDR mutado. Esta seção descreve as etapas necessárias para a preparação de apenas um vírus.- Descongele as células HEK293T, transfira as células para 10 mL de DMEM pré-aquecido suplementado com 10% de FBS, 1% P/S e 25 mM de HEPES, e centrifugar a 350 x g em RT por 5 min.
- Suspender as células a uma concentração de 1 x 105 células/ml e transferir para um balão adequado (por exemplo, um balão de 175 cm2 ). Transferir o balão para uma incubadora a 37 °C/5% de CO2.
NOTA: As células HEK293T devem ser descongeladas com antecedência, a fim de permitir que as células se recuperem totalmente e obtenham um número suficiente de células (11 x 107 células são necessárias para a produção de um vírus). Recomenda-se usar as células após um mínimo de três passagens e manter as células abaixo de 20 passagens. As células HEK293T devem ser divididas três vezes por semana. Ao manter as células, estas não devem exceder 70% a 80% de confluência. O DMEM utilizado na preparação do AAV também já deve ser suplementado com L-glutamina e piruvato de sódio. - Recolher as células num tubo de 50 ml e centrífuga a 350 x g a RT durante 5 min.
- Suspender as células em 20 mL de DMEM suplementado com FBS a 10%, P/S a 1% e HEPES a 25 mM e contar com hemocitômetro. Use azul de tripano para exclusão de células mortas.
- Sementes 3 x 106 células em 20 mL de DMEM suplementadas com 10% de FBS, 1% P/S e 25 mM de HEPES em um frasco de 175 cm2 . Para a produção de um vírus, preparar pelo menos quatro balões de 175 cm2 . Incubar as células a 37 °C/5% de CO2 durante 3 dias.
NOTA: Esta etapa é melhor executada em uma sexta-feira, pois permite que as células se expandam durante o fim de semana. - Colha e conte as células HEK293T.
- Para a preparação de um dos vírus, prepare dez pratos de 150 mm, cada um contendo 11 x 106 HEK293T, em 20 mL de DMEM suplementado com 10% FBS, 1% P/S e 25 mM HEPES.
- Colocar os pratos na incubadora a 37 °C/5% de CO2 durante 24 h. Descarte cuidadosamente o meio antigo e substitua por 20 mL de DMEM livre de antibióticos, suplementado com FBS a 10%, HEPES de 25 mM e butirato de sódio a 1 mM. Antes de prosseguir, verifique se a confluência das células não está acima de 80%.
- Prepare dois tubos de 15 mL que conterão as misturas de transfecção. Ao Tubo 1, adicione 5 mL de meio sérico reduzido, 60 μg de plasmídeo HDR mutado rAAV6 e 220 μg de pDGM6 (plasmídeo auxiliar). Ao Tubo 2, adicionar 5 mL de meio sérico reduzido e 1120 μL de 1 mg/mL de solução de polietilenimina (PEI, reagente de transfecção).
- Adicione o conteúdo do tubo 2 ao tubo 1 e vórtice por 30 s. Incubar por 15 min no RT para garantir o encapsulamento adequado do DNA nas micelas PEI.
NOTA: Não mantenha a solução por mais de 20 minutos. - Adicione cuidadosamente 1,1 mL da solução a cada prato e distribua rodopiando suavemente. Colocar os pratos na incubadora a 37 °C/5% de CO2 durante 48 h.
- Após 48 h, adicionar 250 μL de 0,5 M EDTA a cada prato e colocar os pratos na incubadora por 10 min. Colha as células lavando-as do prato e transfira para um tubo de centrífuga de 500 mL ou vários tubos de 50 mL.
- Centrífuga a 2.000 x g durante 10 min a 4 °C. Descarte o sobrenadante. Para garantir a remoção completa do sobrenadante, centrifugar novamente a 2.000 x g durante 1 min a 4 °C.
- Descarte qualquer sobrenadante restante. Qualquer sobrenadante residual pode prejudicar a purificação do vírus.
- Solte o pellet por vórtice e extraia o vírus usando um kit de purificação de AAV (consulte Tabela de Materiais) de acordo com as instruções do fabricante. Alternativamente, realizar a extração por ultracentrifugação gradiente de iodixanol30,31.
- Aliquotar o vírus purificado e armazenar a -80 °C. Titular o AAV funcionalmente ou quantificar o título de AAV por PCR de gotículas digitais (ddPCR)32 para determinar as condições ideais de transdução.
- Repita este processo para a preparação do plasmídeo rAAV6 WT HDR.
- Titulação de AAV por ddPCR
- Antes de começar, ajuste os blocos de aquecimento a 65 °C e 98 °C.
- Extrair o ADN viral adicionando 15 μL de solução de extracção de ADN a 5 μL do vírus. Vórtice por 15 s.
- Incubar durante 6 min a 65 °C. Vórtice por 15 s. Incubar durante 2 min a 98 °C.
- Use o DNA extraído (que é uma diluição 1:4 do DNA viral) para preparar diluições seriadas (1:400, 1:40.000, 1:160.000, 1:640.000) com nuclease livre (nf) H2O para o ddPCR.
NOTA: As diluições podem ser armazenadas a -20 °C se a ddPCR não for imediatamente realizada. - Selecione três das diluições para o ddPCR. A escolha da diluição a ser usada depende da concentração do vírus. De preferência, use três diluições diferentes (por exemplo, 1:40.000, 1:160.000 e 1:640.000) para encontrar a melhor diluição que esteja dentro do limite de detecção da máquina.
- Mantenha os reagentes no gelo enquanto trabalha. Prepare uma mistura mestra (todas as amostras serão medidas em duplicatas). Para cada reação, prepare o seguinte: 12,5 μL de ddPCR Supermix para Sondas, sem dUTP, 1,25 μL de PrimeTime Std qPCR Assay, AAV-ITR (ver Tabela de Materiais) e 6,25 μL nf-H2O.
- Misture 5 μL de DNA com 20 μL da master mix. Execute isso para as diluições de 1:40.000, 1:160.000 e 1:640.000.
- Coloque um cartucho no suporte do cartucho. Adicione 70 μL de óleo de geração de gotículas às sondas na linha denominada como Óleo. Tenha cuidado para não gerar bolhas.
- Adicionar 20 μL do volume da amostra na linha denominada como Amostra. Tenha cuidado para não gerar bolhas.
- Sele o cartucho com a junta e coloque-o no gerador de gotículas. Gere as gotículas pressionando o botão de partida na máquina, remova a junta e transfira cuidadosamente, com uma pipeta multicanal, 40 μL das gotículas geradas para uma placa de 96 poços. Trabalhe lentamente para evitar a destruição das gotículas e bolhas de ar.
- Sele a placa de 96 poços com folha de perfuração (a faixa vermelha voltada para cima) usando o selador de placa de PCR. Coloque a placa em um termociclador.
- Ajuste o volume para 40 μL, a temperatura da tampa para 105 °C e as taxas de rampa para 2 °C/s. Execute o programa de PCR conforme descrito na Tabela 2.
- Ligue o leitor de gotículas 30 minutos antes do uso. Abra o software na área de trabalho.
- Insira o layout da placa e selecione ABS na guia experimento e ddPCR Supermix na guia Supermix . Em seguida, primeiro pressione PRIME, seguido de clicar em FLUSH SYSTEM no software para iniciar o sistema.
- Insira a placa no leitor. Inicie a execução e, quando terminar, exporte os dados como um arquivo CSV para análise subsequente como uma planilha.
NOTA: Os números gerados pelo software são cópias do genoma (GC) por μL. Estes precisam ser multiplicados pelos fatores de diluição: GC/μL x 5 (diluição do DNA na mistura mestra) x diluição inicial (ou seja, 40.000 ou 160.000).

Figura 3: Visão geral esquemática da segmentação intrônica e exônica durante o knock-in CRISPR/Cas9 HDR. Comparação esquemática entre estratégias de direcionamento intrônico e exônico para knock-in CRISPR/Cas9 HDR. (A) Durante a segmentação intrônica, uma quebra de fita dupla é introduzida em um íntron do DNA. O modelo HDR é composto por um LHA, sequência de cDNA e RHA. A segmentação intrônica requer, adicionalmente, a presença de um aceptor de fatia contendo o local da emenda de 3', o ponto de ramificação e o trato de polipirimidina. Isso permite o splicing correto. A caixa pontilhada verde representa a região correspondente à sequência SA. O tamanho do SA é de 150 pb. (B) A segmentação exônica depende da produção de uma quebra de fita dupla diretamente no éxon. O modelo HDR é composto por um LHA, sequência de cDNA e RHA. As caixas pontilhadas laranja indicam as regiões correspondentes ao LHA e RHA. O tamanho ideal dos HAs é de 400 pb cada. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
| Promotores | ||||
| Nome | Comprimento | Descrição | ||
| SFFV | 492 pb | Promotor de vírus formador de foco do baço. Forte promotor de mamíferos. Constitutivamente expresso | ||
| UBC | 400 pb | Promotor derivado do gene da ubiquitina C humana. Constitutivamente expresso. | ||
| CMV | 508 pb | Promotor derivado do citomegalovírus. Pode conter uma região potenciadora. Constitutivamente expresso. Forte promotor de mamíferos. Pode ser silenciado. | ||
| EF-1a | 1182 pb | Fator de alongamento eucariótico humano 1 alfa promotor. Constitutivamente expresso. Forte promotor de mamíferos. | ||
| EFS | 200-300 pb | Forma abreviada sem ítron alfa EF-1 | ||
| CAG | 584 pb | Promotor híbrido de mamíferos contendo o potenciador precoce do CMV (C), o promotor de beta-actina de galinha (A) e o aceptor de emenda para o gene da beta-globina de coelho (G). Constitutivamente expresso. | ||
| Sinais de poliadenilação (PolyA) | ||||
| Abreviação | Comprimento | Descrição | ||
| SV40 PoliA | 82-122 pb | Vírus símio 40 sinal de poliadenilação | ||
| bGH PoliA | 224 pb | Sinal de poliadenilação do hormônio do crescimento bovino | ||
| rbGlob PolyA | 56 pb | Sinal de poliadenilação da globina beta do coelho | ||
Tabela 1: Promotores e sinais de poliadenilação.
| Passo | Temperatura | Hora | Ciclos |
| Ativação enzimática | 95°C | 10 minutos | 1x |
| Desnaturação | 94°C | 30 segundos | 42x |
| Recozimento | 60°C | 1 minuto | |
| Extensão | 72°C | 30 segundos | |
| Desativação enzimática | 98°C | 10 minutos | 1x |
| Segurar | 4°C | ∞ |
Tabela 2: Programa de PCR de gotículas digitais.
3. Edição de HSPCs
- Transfecção e transdução de HSPCs
- Descongelar HSPCs CD34+ e transferir as células para 10 mL de RPMI pré-aquecido suplementado com 1% de P/S. Centrífuga a 350 x g em RT por 10 min.
- Suspender as células em meio de retenção HSPC até uma concentração de 2,5 x 10 5 células/mL e incubar a 37 °C/5 % de CO2 por 48 h - 72 h.
- Colha as células em um tubo de 15 mL. Conte as células e verifique a viabilidade celular por exclusão azul de tripano. Entre 2 x 10 5 e5 x 106 células podem ser nucleofectadas em uma única cuvete. Prepare o número apropriado de cuvetes, dependendo do seu número de célula calculado.
- Preparar o complexo RNP. Num tubo de 1,5 ml, adicionar 15 μg de Cas9 e 8 μg de sgRNA (razão molar 1:2,5) e incubar a 25 °C durante 10 min num bloco de aquecimento.
- Enquanto o complexo RNP está incubando, centrifugar as células a 350 x g por 5 min e descartar o sobrenadante.
- Suspender as células em 100 μL de solução de nucleofeção. Misture as células com o complexo RNP e transfira para a cubeta.
- Bata suavemente na cubeta para remover quaisquer bolhas de ar residuais que possam ter se formado durante a transferência.
- Insira a cubeta no suporte do sistema de transfecção e eletroporate as células conforme descrito anteriormente na seção de projeto de sgRNA.
- Imediatamente após a eletroporação, adicionar 400 μL de meio de retenção de HSPC pré-aquecido sem P/S e transferir as células com uma pipeta de transferência fina para uma placa de cultura de tecidos contendo 500 μL de meio de retenção de HSPC pré-aquecido sem P/S. Dependendo do número celular, use uma placa de cultura apropriada (placa de 24, 12 ou 6 poços) para atingir uma densidade entre 0,25 x 10 6 e 1 x 106 células/mL. Transferir a placa para a incubadora a 37 °C/5% de CO2.
- Descongele os frascos para injetáveis que contenham os rAAVs congelados no gelo. Transduzir as células pipetando a quantidade ideal de cada rAAV6 para a suspensão celular. Realize a transdução dentro de 20-30 minutos após a eletroporação para obter altas eficiências de transdução.
NOTA: A concentração ideal de cada vírus (um vírus para o modelo WT HDR e outro vírus separado para o modelo HDR mutado) deve ser determinada experimentalmente. Normalmente, 5.000-10.000 GC/célula (por exemplo, 5 x 109 GC para 1 x 106 células) resultam em alta transdução. - Misture suavemente a suspensão celular com a pipeta. Incubar as células transduzidas durante 6-8 h a 37 °C/5% de CO2. Após 6-8 h, coletar as células em um tubo e centrífuga a 350 x g em RT por 5 min.
- Descarte o meio antigo e substitua por meio de retenção HSPC pré-aquecido fresco suplementado com P/S.
- Suspender as células a uma concentração entre 2,5 x 105-5 x 105 células/ml e transferir para uma placa de cultura celular tratada com tecido (placa de 24, 12 ou 6 poços). Incubar as células durante 48 h a 37 °C/5% de CO2 antes de proceder à triagem.
- Classificação de fluxo dos HSPCs projetados com a mutação GOF heterozigótica
- Colher as células em um tubo de 15 mL e centrifugar a 350 x g em RT por 5 min. Remova o sobrenadante, suspenda as células em 1 mL de DPBS contendo 0,1% de BSA e centrifugar novamente a 350 x g no RT por 5 min.
- Rejeitar o sobrenadante e suspender as células num volume adequado de DPBS + BSA a 0,1%, dependendo do número de células.
NOTA: Recomenda-se suspender as células a um volume mínimo de 200 μL e não exceder 1 x 107 células/mL, a fim de reduzir o risco de entupimento do classificador. - Transfira as células para um tubo FACS estéril equipado com uma tampa. Adicione 7-AAD ou outro corante de viabilidade (dependendo da sua compatibilidade com as proteínas fluorescentes) à suspensão celular para exclusão de células vivas/mortas.
- Classifique as células vivas que são duplamente positivas para as proteínas repórteres fluorescentes em um tubo de coleta contendo 200 μL de meio de retenção HSPC.
NOTA: Controles apropriados que consistem em células transduzidas apenas com AAVs devem ser adicionados para garantir o controle adequado durante o procedimento de classificação. - Centrifugar as células classificadas a 350 x g no RT por 5 min e suspender as células em meio de retenção HSPC a uma concentração de 2,5 x 105 células/mL para maior expansão em cultura ou usar as células diretamente para ensaios funcionais.
4. Confirmação da edição de genes bem-sucedida
- Extração de DNA genômico
- Antes de começar, ajuste os blocos de aquecimento a 65 °C e 98 °C. Colha 2 x 10 5 células editadas e purificadas pelo genoma e classifique a 350 x g por5 min.
- Extraia o gDNA conforme descrito anteriormente. Utilize a solução de ADN diretamente para PCR ou guarde a -20 °C.
- In-Out PCR
- Projetar dois primers para amplificar o local de inserção de 5' (Figura 4A): o primer forward 1 tem como alvo o locus genômico fora do braço de homologia esquerdo; primer reverso 2 tem como alvo a sequência integrada.
- Para a PCR de entrada-saída, prepare a seguinte mistura de PCR por reação:
10 μL de PCR Master Mix (2x; ver Tabela de Materiais)
1 μL de primer para a frente 1 (estoque de 10 μM)
1 μL de primer reverso 2 (estoque de 10 μM)
0,5-1,0 μL de DNA extraído
H2O livre de nuclease até um volume final de 20 μL
NOTA: Para mais de uma reação, é aconselhável preparar uma mistura mestre que contenha uma reação extra para explicar os erros de pipetagem. É importante incluir um controle não moldado e um DNA molde de uma amostra tratada simuladamente. - Execute as reações de PCR com o programa de ciclo térmico apropriado (consulte as instruções do fabricante).
NOTA: É aconselhável primeiro testar o par de primer para a temperatura de recozimento ideal. Isso pode ser feito calculando o Tm e, em seguida, executando a PCR com um termociclador que pode executar uma temperatura gradiente. - Execute os produtos de PCR com uma escada de DNA em um gel de agarose a 1,5% a 100 V por 45-60 min (Figura 4B). Coloque o gel em um transiluminador de luz azul ou UV. Extirpe as bandas do gel.
- Extraia o DNA das bandas usando um kit de extração de gel de DNA. Envie as amostras de PCR extraídas com primers apropriados para sequenciamento de Sanger para confirmar a integração correta e perfeita do cDNA desejado no locus genético endógeno.

Figura 4: Validação da integração genômica por PCR in-out. (A) Representação esquemática da estratégia de PCR in-out. Na estratégia retratada, foram desenhados dois primers. O primer forward 1 tem como alvo o locus genômico fora do LHA, e o primer reverse 2 tem como alvo a sequência otimizada para códons. (B) Representação esquemática de uma eletroforese em gel de agarose. Somente células editadas com êxito (RNP + AAV) gerarão um produto de PCR durante a PCR de entrada-saída, enquanto as amostras não editadas (somente AAV) não gerarão um produto de PCR. Abreviação: NTC = controle não-modelo. Por favor, clique aqui para ver uma versão maior desta figura.
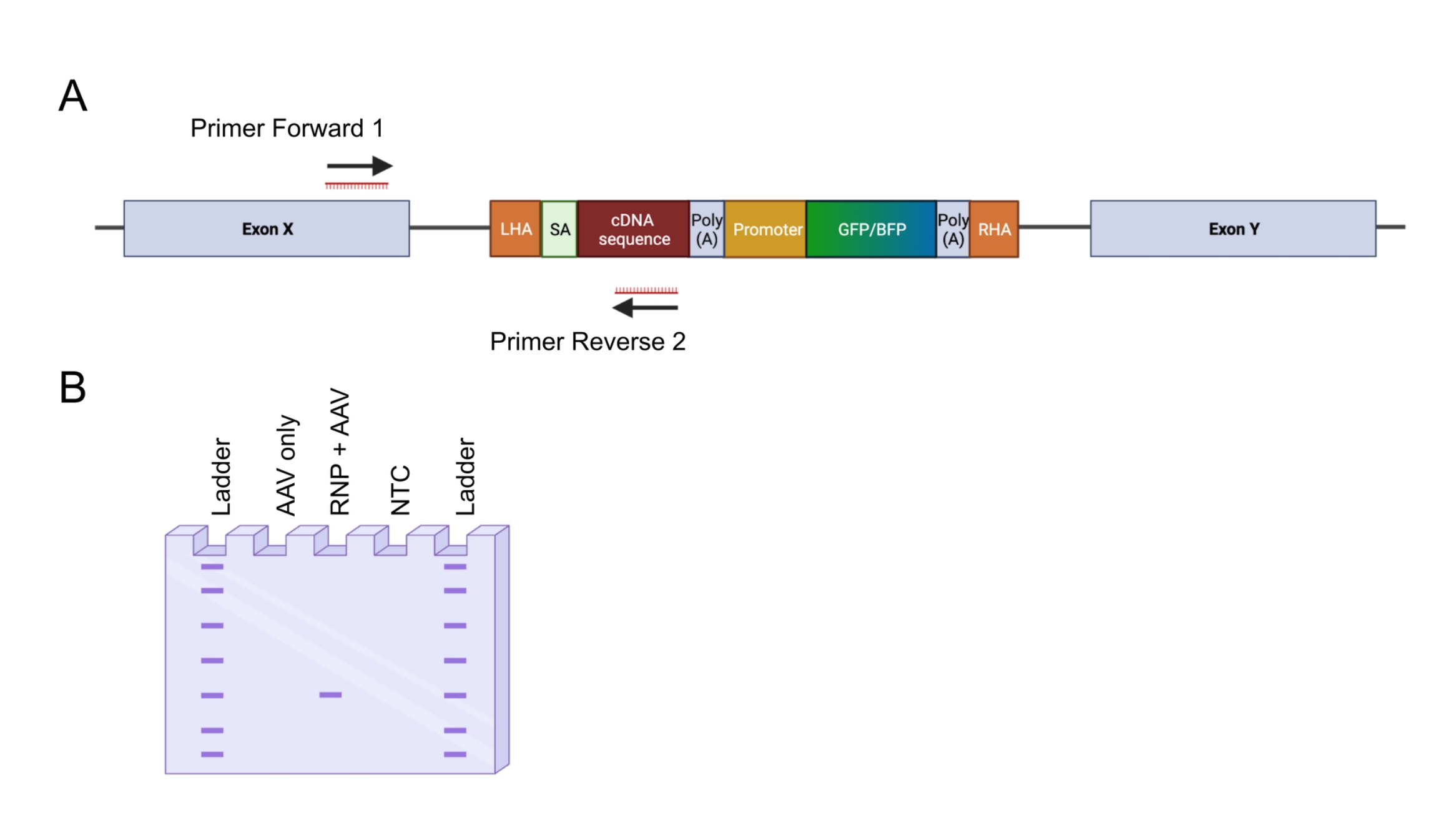{kind=link}
Resultados
Ao aplicar o protocolo acima descrito, mutações CALR heterozigotas tipo 1 foram introduzidas de forma reprodutível em HSPCs derivados do sangue do cordão umbilical. Essa mutação consiste em uma deleção de 52 pb no éxon 9 (o último éxon da CALR), o que resulta em um deslocamento de quadro +1, levando à tradução de um novo domínio C-terminal carregado positivamente26,33. Para introduzir a mutação CALR no locus genético endógeno, uma estratégia de direcionamento intrônico a montante do éxon 9 foi adotada, uma vez que isso contornaria quaisquer alterações indesejadas na sequência de codificação nos casos em que o DSB induzido por Cas9 não fosse reparado através do mecanismo HDR. Neste caso específico, um sgRNA para o íntron 7 foi projetado devido à disponibilidade de sequências altas no alvo e baixas fora do alvo em combinação com braços de homologia favoráveis (falta de repetições de sequência; Figura 5A).
Dois modelos de doadores foram então projetados e embalados em vetores AAV6. A fim de permitir o splicing correto dos éxons endógenos para o cDNA integrado, os modelos doadores contêm (i) uma sequência SA incluindo o sítio de splice de 3', o ponto de ramificação e o trato de polipirimidina, (ii) a sequência de cDNA otimizada para códons dos éxons 8-9, contendo a WT (CALR WT) ou a sequência mutada (CALRDEL ) incluindo um códon de paragem, (iii) um sinal poliA do vírus símio 40 (SV40), (iv) uma sequência codificadora de uma proteína fluorescente sob o controlo do promotor interno separado, o promotor do vírus formador de foco do baço (SFFV), seguido de (v) um sinal poliA da hormona do crescimento bovino (bGH). O molde doador contendo o cDNA CALR WT foi projetado para conter um GFP, enquanto o molde doador contendo a sequência CALRDEL cDNA foi projetado para conter um BFP. Todo o construto foi ladeado por um AH esquerdo e direito (Figura 5A).
Dois dias após a transfecção com o complexo RNP e a transdução com os vírus rAAV6, as células foram analisadas por citometria de fluxo. Quatro populações principais puderam ser detectadas: (i) células que não expressam nem a GFP nem o PBF, representando células sem edição genômica baseada em HDR, (ii) células positivas apenas para GFP, representando aquelas que integraram apenas o construto WT, (iii) células positivas apenas para PBF, representando aquelas que integraram apenas a construção mutada, e (iv) células duplamente positivas para GFP e PBF, representando as células que integraram as sequências WT e mutada (Figura 5B). Para a obtenção de populações puras de HSPCs portadoras da mutação CALR heterozigótica tipo 1, as células duplamente positivas foram classificadas por citometria de fluxo. HSPCs nos quais duas sequências de WT foram batidas foram utilizadas como células controle (GFP+ mCherry+; Figura 5B). Uma alternativa válida para uso como células de controle seriam HSPCs com uma integração bialélica das proteínas fluorescentes em um locus de porto seguro (ou seja, AAVS1; não mostrado). A contagem celular por exclusão azul de tripano realizada nos HSPCs classificados indicou que mais de 90% das células eram viáveis.
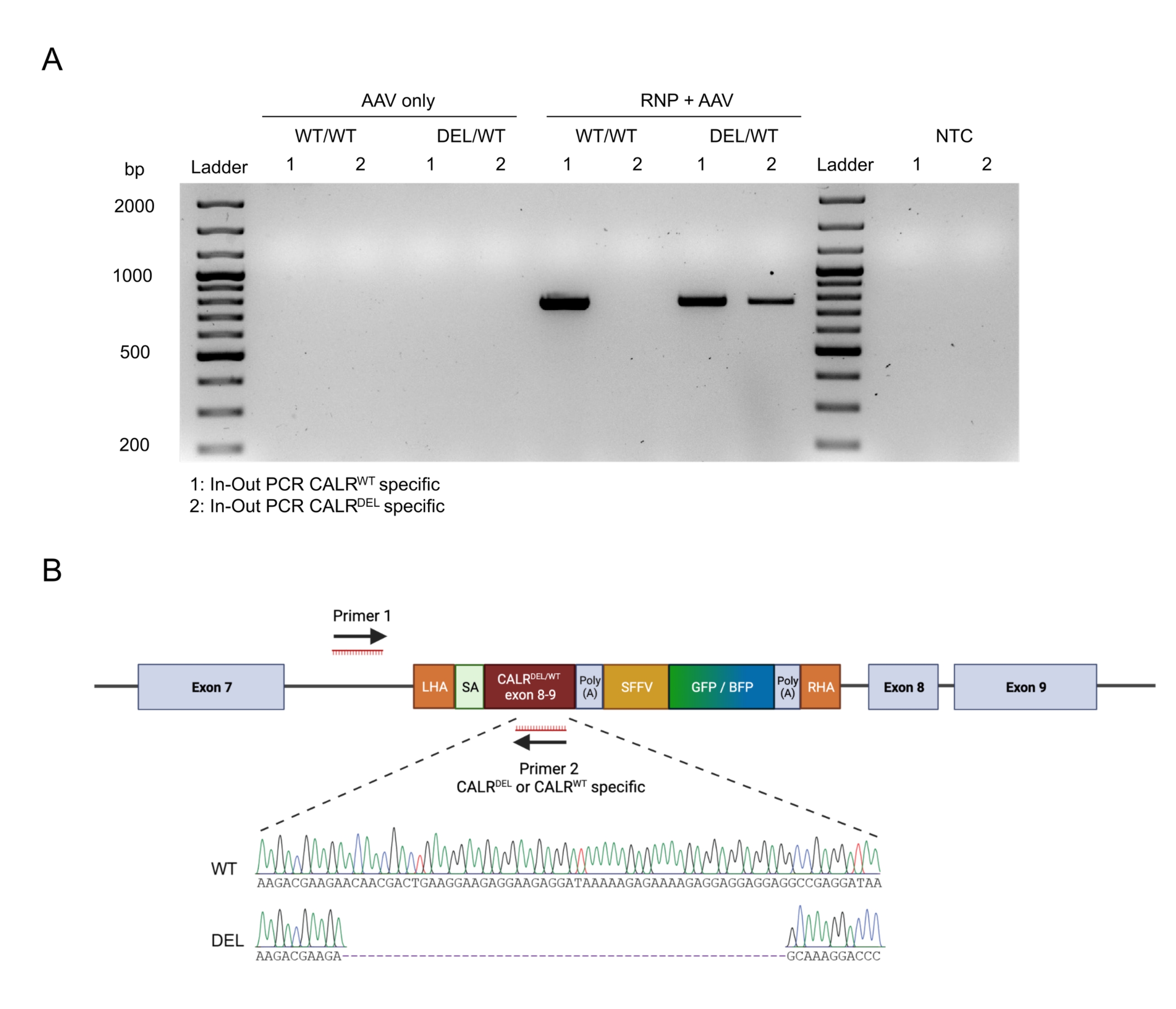
A integração perfeita no alvo dos construtos foi confirmada pela aplicação da estratégia de PCR in-out (Figura 6A). Neste caso específico, foram realizadas duas PCRs in-out separadas, uma para a sequência CALR WT knocked-in (faixa 1 da eletroforese em gel na Figura 6A) e outra para a sequência CALRDEL knocked-in (faixa 2 da eletroforese em gel da Figura 6A). O sequenciamento de Sanger realizado no DNA extraído das bandas de gel confirmou a inserção correta das sequências WT e mutada nos HSPCs CALRDEL/WT (Figura 6B).

Figura 5: Geração de HSPCs portadores da mutação CALR heterozigótica. (A) Esquema representativo que descreve a estratégia de edição para a inserção da mutação CALR heterozigótica. O complexo RNP tem como alvo o íntron entre o éxon 7 e o éxon 8 do gene CARR. Dois AAVs, um contendo os éxons mutantes 8-9 e um PBF e o outro contendo os éxons WT 8-9 e uma GFP, servirão como modelos de reparo do doador e promoverão a integração da sequência mutada em um alelo e a integração da sequência WT no alelo restante. (B) Gráficos representativos de citometria de fluxo que descrevem a expressão de GFP e PBF ou PFG e mCherry 48 h após a transfecção e transdução de HSPCs. Clique aqui para ver uma versão maior desta figura.
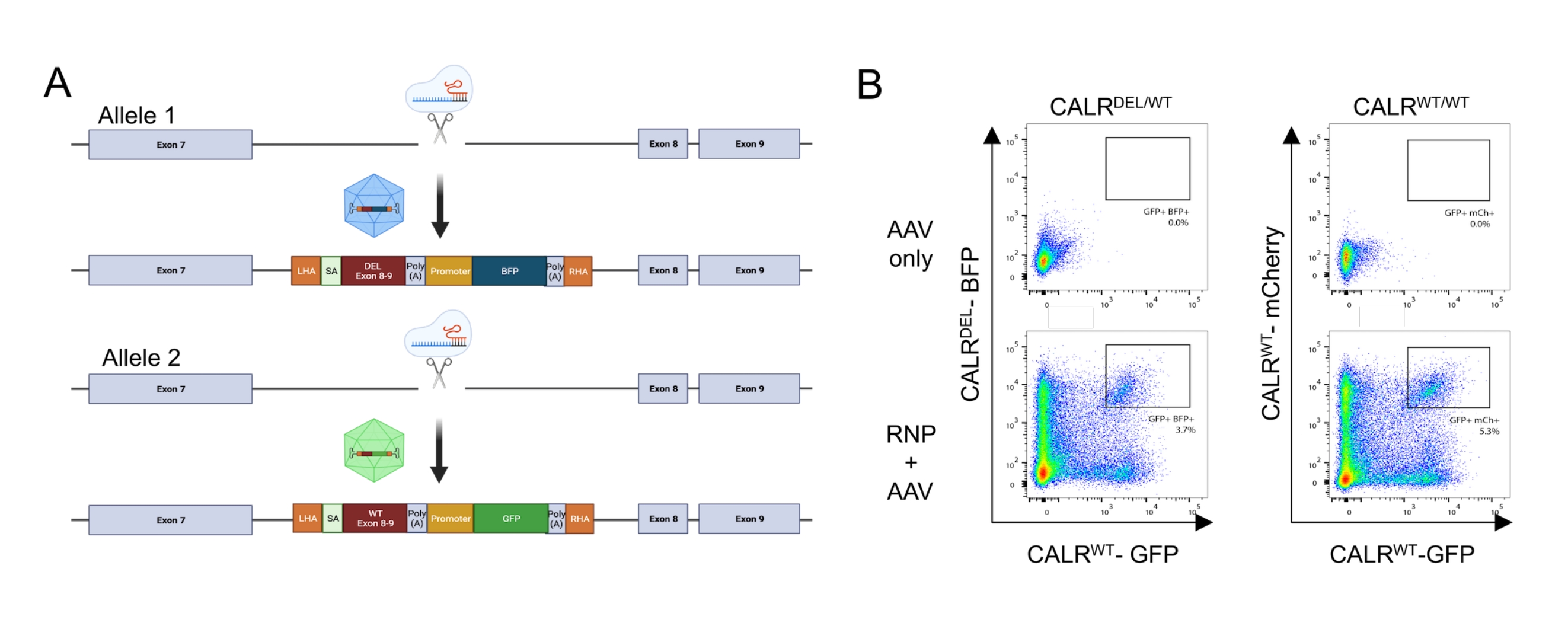{kind=link}

Figura 6: Validação da mutação CALR heterozigótica bem-sucedida em HSPCs. (A) Eletroforese em gel a partir dos produtos da PCR in-out realizada no DNA genômico extraído de controles AAV, CALR WT/WT e CALRDEL/WT. Utilizou-se uma escada de DNA de 100 pb. Abreviação: NTC = controle não-modelo. (B) Resultados de sequenciamento de Sanger obtidos a partir das PCRs in-out realizadas em CALR DEL/WT confirmando a integração bem-sucedida das sequênciasWT e mutadas. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
A manipulação genética eficiente e precisa de HSPCs primários humanos representa uma grande oportunidade para explorar e entender os processos que influenciam a hematopoiese normal e, mais importante, a transformação leucêmica das células hematopoiéticas.
Neste protocolo, uma estratégia eficiente para projetar HSPCs humanos para expressar mutações recorrentes heterozigotas GOF foi descrita. Este procedimento aproveitou a tecnologia CRISPR/Cas9 e os vetores rAAV6 como doadores de modelos de DNA para inserir com precisão WT e sequências de DNA mutante em seus loci genéticos endógenos. O acoplamento dos cDNAs modificados (WT e mutantes) com proteínas repórteres fluorescentes separadas permite o enriquecimento e o rastreamento de células com um estado heterozigoto definitivo.
Esta estratégia apresenta várias vantagens em comparação com os métodos baseados em lentivirais (VE) frequentemente utilizados. Uma das principais vantagens é que o sistema baseado em CRISPR/Cas9 permite uma edição precisa nos locos endógenos, resultando na preservação dos promotores endógenos e elementos regulatórios. Isso leva à homogeneidade na expressão do gene editado nas células, um objetivo dificilmente alcançável quando um método baseado em VE é usado. A transferência gênica com vetores do VE leva à integração semi-aleatória do gene com preferência por sítios transcricionalmente ativos34. Isso pode se traduzir em superexpressão do gene transferido e heterogeneidade entre as células editadas, resultando em dificuldades para investigar e analisar o papel das mutações e interações gênicas. Uma segunda vantagem é que o sistema descrito, sendo um sistema de edição site-specific, elimina os riscos de mutagênese insercional35.
A estratégia de repórter fluorescente duplo permite o enriquecimento preciso e o rastreamento de células que foram editadas com sucesso em ambos os alelos, com um alelo integrando o cDNA WT e o outro alelo integrando as sequências de cDNA mutadas. As células que expressam apenas um único repórter representam apenas a integração monoalélica ou a integração bialélica de modelos HDR com o mesmo repórter fluorescente. Ambos os cenários só podem ser distinguidos com precisão se clones derivados de uma única célula forem produzidos e analisados individualmente. No entanto, os HSPCs têm apenas capacidade proliferativa limitada in vitro e, quando mantidos em cultura por longos períodos de tempo, os HSPCs começam a se diferenciar em progênie mais madura e perdem sua capacidade de auto-renovação e enxerto. Isso inviabiliza a seleção e a expansão de clones unicelulares que abrigam a mutação heterozigótica desejada. A aplicação da estratégia de proteína fluorescente dupla e o enriquecimento por citometria de fluxo para células portadoras da mutação heterozigótica permitem contornar os problemas induzidos pela cultura in vitro prolongada.
Neste exemplo específico, foi demonstrado com sucesso que os HSPCs poderiam ser eficientemente projetados e classificados para obter populações puras de HSPCs portadores da mutação heterozigótica CALRDEL/WT.
No entanto, este sistema não se limita à engenharia de mutações heterozigotas de mudança de quadro, mas também pode ser facilmente adotado para criar outros tipos de mutação, incluindo mutações missense e nonsense. Ao aplicar diferentes combinações de AAVs contendo WT ou sequências mutadas com diferentes proteínas repórteres fluorescentes, este sistema também pode ser utilizado para a introdução de mutações homozigotas (transdução simultânea com dois rAAVs, ambos portadores de cDNA mutante, mas diferentes repórteres fluorescentes) ou mesmo a correção de mutações (transdução simultânea com dois AAVs, ambos portadores de cDNA WT, mas diferentes repórteres fluorescentes). Além disso, é importante mencionar que essa estratégia não se limita à introdução de mutações GOF oncogênicas. De fato, o protocolo descrito pode ser utilizado para múltiplas estratégias alternativas, incluindo knock-out de genes, substituição gênica36,37, knock-in direcionado de transgenes (ou seja, receptores de antígenos quiméricos)38 e até mesmo para a correção de mutações causadoras de doenças11,39.
A estratégia de combinar CRISPR/Cas9 e AAV6 com múltiplos repórteres fluorescentes também demonstrou ser aplicável em muitos outros tipos de células, incluindo células T, células dendríticas plasmocitóides, células-tronco pluripotentes induzidas, células-tronco neuronais e células-tronco das vias aéreas 24,38,40,41,42,43,44 . Esta estratégia pode ser implementada para a produção de células T do receptor de antígeno quimérico superior (CAR). Por exemplo, foi publicado recentemente que o knock-out mediado por CRISPR/Cas9 do gene TGFBR2 em células T CAR aumenta muito sua função no microambiente tumoral rico em TGF-β supressor45. Tal abordagem poderia fornecer um protocolo de uma etapa para projetar as células T para expressar o CAR e para nocautear o gene TGFBR2 por local, inserindo especificamente o CAR em ambos os alelos do gene TGFBR2. Além disso, essa abordagem também pode ser útil para gerar células T CAR universais, integrando o CAR no gene da constante do receptor de células T alfa (TRAC)46,47.
Para aumentar a reprodutibilidade e garantir uma edição eficiente das células, algumas considerações importantes precisam ser cuidadas. Os principais pontos críticos para garantir o sucesso da edição das células residem em (i) a seleção do sgRNA, (ii) o design do modelo HDR e (iii) a produção de rAAV6.
A seleção de um sgRNA de bom desempenho é crucial, pois determinará o número máximo de alelos nos quais o modelo HDR pode ser integrado. Devido aos inúmeros softwares que agora estão disponíveis, a busca por sgRNAs candidatos foi simplificada. Ao selecionar a região de interesse, o software pode propor uma série de sgRNAs com uma pontuação on-target e uma pontuação off-target que indicam as chances de edição no locus desejado e loci indesejados, respectivamente. Esses escores são calculados com base em modelos de pontuação publicados anteriormente48,49. Embora este seja um bom ponto de partida para selecionar um sgRNA de bom desempenho, o desempenho do sgRNA precisa ser confirmado, pois seu desempenho previsto in silico nem sempre corresponde a um sgRNA in vitro eficiente. Portanto, é altamente recomendável projetar e testar pelo menos três sgRNAs para aumentar as chances de encontrar o melhor sgRNA. Uma vez que um verdadeiro sgRNA de bom desempenho tenha sido identificado, sugere-se prosseguir com o design do modelo HDR.
Precauções devem ser levadas em consideração ao projetar o modelo HDR. Os braços de homologia esquerdo e direito (LHA e RHA, respectivamente) devem abranger 400 pb a montante e a jusante do local de corte do sgRNA, respectivamente, pois os AHs mais curtos podem resultar em frequências HDR reduzidas. O tamanho do cDNA que pode ser introduzido via HDR depende dos recursos de embalagem dos AAVs, que é de aproximadamente 4,7 kb. Devido aos numerosos elementos obrigatórios dentro do modelo HDR (LHA, RHA, SA, PolyA, promotor e sequência de repórter fluorescente), o espaço restante para o cDNA mutado ou WT é limitado. Isso não é problemático se a mutação desejada estiver localizada perto da extremidade 3' de um gene ou em genes com um CDS curto geral. No entanto, nos casos em que a mutação está localizada perto do lado inicial transcricional (TSS) dos genes com um CDS longo (excedendo o espaço de empacotamento restante do AAV), essa abordagem descrita pode não ser viável. Para contornar esse problema, uma estratégia que se baseia na divisão do modelo HDR em dois AAVs foi recentemente desenvolvida por Bak e colegas. Essa estratégia depende de duas integrações separadas mediadas por HDR para obter a integração final perfeita de um grande gene50.
A qualidade do vírus e seu título são fatores adicionais que podem fazer ou quebrar a engenharia do genoma bem-sucedida das células. Para um rendimento ideal, é importante não deixar que o HEK293T atinja a confluência total enquanto é mantido em cultura. Idealmente, as células HEK293T devem ser divididas quando a confluência de 70% a 80% for atingida. Além disso, o HEK293T não deve ser cultivado por longos períodos de tempo, pois isso pode diminuir sua capacidade de produzir vírus. Novas células HEK293T precisam ser descongeladas após 20 passagens. A obtenção de altos títulos de vírus é importante para aumentar a eficiência e a reprodutibilidade dos experimentos. Baixos títulos virais se traduzirão em grandes volumes de solução de rAAV necessários para a transdução dos HSPCs. Como regra geral, a solução de rAAV adicionada às células nucleofectadas não deve exceder 20% do volume total do meio de retenção do HSPC. Volumes mais altos de solução de AAV podem levar ao aumento da morte celular, menor proliferação e eficiências de transdução prejudicadas. No caso de baixos títulos de vírus, recomenda-se, portanto, concentrar ainda mais o vírus.
Em resumo, este protocolo oferece uma abordagem reprodutível para manipular HSPCs humanos de forma precisa e eficiente através do uso simultâneo de modelos de doadores CRIPSR/Cas9 e rAAV6 com repórteres fluorescentes duplos adicionais. Esta abordagem provou ser uma ótima ferramenta no estudo da biologia normal das células-tronco hematopoiéticas e as contribuições que as mutações fazem para a leucemogênese.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho é apoiado por subvenções do Austrian Science Fund (FWF; número P32783 e I5021) para a A.R. O financiamento adicional à A.R. também é fornecido pela Sociedade Austríaca de Medicina Interna (Joseph Skoda Fellowship), a Sociedade Austríaca de Hematologia e Oncologia (OeGHO; Bolsa de Investigação Clínica) e MEFOgraz. T.K. é membro especial da Sociedade de Leucemia e Linfoma.
Materiais
| Name | Company | Catalog Number | Comments |
| 175 cm2 Cell Culture Flask, Vent Cap, TC-treated | Corning | 431080 | |
| 150 mm x 25 mm dishes | Corning | 430599 | |
| 293T | DSMZ | ACC 635 | https://www.dsmz.de/collection/catalogue/details/culture/ACC-635 |
| 4D Nucleofector Core Unit | Lonza | - | For nucleofection of human HSPCs use the DZ-100 program. |
| 4D Nucleofector X Unit | Lonza | - | |
| 500 ml Centrifuge Tube | Corning | 431123 | |
| 7-AAD | BD Biosciences | 559925 | |
| AAVpro Purification Kit | Takara | 6666 | |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | 1081058 | |
| Avanti JXN-30 Ultracentrifuge | Beckman Coulter | - | |
| Benchling sgRNA design tool | Online tool for sgRNA design: http://www.benchling.com/crispr | ||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7906-100G | |
| C1000 Touch Thermal Cycler | Bio-Rad | - | |
| Chemically modified synthetic sgRNA | Synthego | Website: https://www.synthego.com/products/crispr-kits/synthetic-sgrna Sequence for the sgRNA targeting intron 7 of CALR: 5’-CGCCTGTAATCCTCGCCCAG-3’ An 80 nucleotide SpCas9 scaffold is added to the 20 nucleotide RNA sequence to complete the sgRNA. Chemical modifications of 2'-O-Methyl are added to the first and last 3 bases and 3' phosphorothioate bonds are added in the first 3 and last 2 bases. *Alternatively chemically modified synthetic sgRNAs can be acquired from IDT (https://eu.idtdna.com/site/order/oligoentry/index/crispr) and Trilink (https://www.trilinkbiotech.com/custom-oligos) | |
| CHOPCHOP sgRNA design tool | Online tool for sgRNA design: http://chopchop.cbu.uib.no | ||
| Costar 24-well Clear TC-treated Multiple Well Plates | Corning | 3526 | |
| CRISPick sgRNA design tool | Online tool for sgRNA design: https://portals.broadinstitute.org/gppx/crispick/public | ||
| CRISPOR sgRNA design tool | Online tool for sgRNA design: http://crispor.tefor.net | ||
| ddPCR 96-Well Plates | Bio-Rad | 12001925 | |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863024 | |
| DG8 Cartridges for QX200/QX100 Droplet Generator | Bio-Rad | 1864008 | |
| DG8 Gaskets for QX200/QX100 Droplet Generator | Bio-Rad | 1863009 | |
| DreamTaq Green PCR Master Mix (2X) | Thermo Scientific | K1081 | |
| Droplet Generation Oil for Probes | Bio-Rad | 1863005 | |
| Dulbecco’s Modified Eagle Medium (DMEM) with high glucose | Sigma-Aldrich | D6429-6X500ML | |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500ML | |
| FACSAria Fusion | BD Biosciences | - | |
| Falcon 5 mL Round Bottom | Corning | 352054 | |
| Fetal Bovine Serum (FBS) Good Forte (heat inactivated), 500 ml | Pan Biotech | P40-47500 | |
| FlowJo 10.8.0 | BD Biosciences | - | |
| GenAgarose L.E. | Inno-train | GX04090 | |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | |
| Gibson Assembly Master Mix | New England Biolabs Inc. (NEB) | E2611L | |
| HEK293T | |||
| HEPES solution | Sigma-Aldrich | H0887-100ML | |
| ICE | Synthego | https://ice.synthego.com | |
| IDT codon optimization tool | IDT | https://www.idtdna.com/pages/tools/codon-optimization-tool | |
| IDT sgRNA design tool | Online tool for sgRNA design: https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM | ||
| LB Broth (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7658-1KG | |
| LB Broth with agar (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7533-1KG | |
| Midori Green Advance | Nippon Genetics | MG04 | |
| Monarch Plasmid Miniprep Kit | NEB | T1010L | |
| Monarch DNA Gel Extraction Kit | NEB | T1020L | |
| NEB 5-alpha Competent E. coli (High Efficiency) | NEB | C2987U | |
| Nuclease-Free Water, 5X100 ml | Ambion | AM9939 | |
| NucleoBond Xtra Midi | Macherey-Nagel | 740410 | |
| Opti-MEM, Reduced Serum Medium, 500 ml | Gibco | 31985070 | |
| P3 Primary Cell 4D-Nucleofetor X Kit L | Lonza | V4XP-3024 | The Lonza Primary P3 solution is supplied as a 2.25 mL P3 Primary Cell Nucleofector Solution and 0.5 mL Supplement 1. To reconstitute, add the Supplement 1 to the P3 Primary Cell Nucleofector Solution and mix. |
| pAAV-MCS2 | Addgene | 46954 | |
| PCR Plate Heat Seal Foil, pierceable | Bio-Rad | 1814040 | |
| pDGM6 | Addgene | 110660 | |
| Penicillin-Streptomycin (P/S) | Gibco | 15140122 | |
| Polyethylenimine (PEI) | Polysciences | 23966 | Add 50 mL of PBS 4.5 pH (made with HCl) to 50 mg of PEI in a tube. Dissolve by placing the tube in a 70°C water bath and vortexing every 10 minutes until the solution is dissolved. After the solution has reached RT, filter sterilze through a 0.22 μm filter, make 1120 μL aliquots, and store at -80°C. |
| Polystyrene Test Tube, with Snap Cap | |||
| Primers | Eurofins | - | Primers were ordered from Eurofins (eurofinsgenomics.eu) as unmodified salt free custom oligos. The primers were designed by using PRIMER-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) Primer 1 Fwd: AAGTGATCCGTTCGCCATGAC; Primer 2 Rev CALR WT specific: ACGTCCTCTTCCTCGTCCTC; Primer 2 Rev CALR DEL specific: CCAACCCTGGAGACACGCTTC |
| PrimeTime qPCR Primer Assay | IDT | - | PrimeTime qPCR Probe Assays (1 probe/2 primers) that can be ordered from IDT (https://eu.idtdna.com/site/order/qpcr/assayentry). Scale: Std - qPCR Assay 500 reactions; Primer 1 Forward (5'-3') : GGAACCCCTAGTGATGGAGTT; Primer 2 Reverse (5'-3'): CGGCCTCAGTGAGCGA; Probe (5'-3'): CACTCCCTCTCTGCGCGCTCG; 5' Dye/3' Quencher: FAM/ZEN/IBFQ; Primer to probe ratio: 3.6 |
| PX1 PCR Plate Sealer | Bio-Rad | 1814000 | |
| QuantaSoft Software | Bio-Rad | ||
| Quick Extract DNA Extraction Solution | Lucigen | QE0905T | |
| QX200 Droplet Generator | Bio-Rad | 1864002 | |
| QX200 Droplet Reader | Bio-Rad | ||
| Recombinant human Flt3-ligand | Peprotech | 300-19 | |
| Recombinant human IL-6 | Peprotech | 200-06 | |
| Recombinant Human SCF | Peprotech | 300-07 | |
| Recombinant Human TPO | Peprotech | 300-18 | |
| RPMI 1640 | Sigma-Aldrich | R8758-6X500ML | |
| SnapGene | Dotmatics | Molecular cloning software https://www.snapgene.com *Alternatively also Benchling (https://www.benchling.com) and Geneious (https://www.geneious.com) can be used. | |
| Soc outgrowth medium | NEB | B9020S | |
| Sodium-butyrate | Sigma-Aldrich | B5887-1G | |
| Stem Regenin 1 (SR1) | Biogems | 1224999 | |
| StemSpan SFEM II | STEMCELL Technologies | 9655 | |
| TAE Buffer (Tris-acetate-EDTA) 50X | Thermo Scientific | B49 | |
| TIDE | http://shinyapps.datacurators.nl/tide/ | ||
| Trypan blue 0.4% | Sigma-Aldrich | T8154-100ML | |
| TrypLE (with phenol red), 500 ml | Thermo Scientific | 16605-028 | |
| UltraPure 0.5: EDTA, pH 8.0, 100 ml | Thermo Scientific | 15575-038 | |
| UM171 | STEMCELL Technologies | 72914 | |
| Vector Builder codon optimization tool | Vector Builder | https://en.vectorbuilder.com/tool/codon-optimization.html |
Referências
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Gillmore, J. D., et al. CRISPR-Cas9 in vivo gene editing transthyretin amyloidosis. New England Journal of Medicine. 385 (6), 493-502 (2021).
- Frangoul, H., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine. 384 (3), 252-260 (2021).
- Stadtmauer, E. A., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 367 (6481), (2020).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Veldwijk, M. R., et al. Pseudotyped recombinant adeno-associated viral vectors mediate efficient gene transfer into primary human CD34+ peripheral blood progenitor cells. Cytotherapy. 12 (1), 107-112 (2010).
- Song, L., et al. High-efficiency transduction of primary human hematopoietic stem cells and erythroid lineage-restricted expression by optimized AAV6 serotype vectors in vitro and in a murine xenograft model in vivo. PLoS One. 8 (3), 58757 (2013).
- Dever, D. P., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 539 (7629), 384-389 (2016).
- Drost, J., et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521 (7550), 43-47 (2015).
- Jan, M., et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine. 4 (149), (2012).
- Corces-Zimmerman, M. R., Hong, W. J., Weissman, I. L., Medeiros, B. C., Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America. 111 (7), 2548-2553 (2014).
- Jaiswal, S., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine. 377 (2), 111-121 (2017).
- Genovese, G., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New England Journal of Medicine. 371 (26), 2477-2487 (2014).
- Papaemmanuil, E., et al. Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine. 374 (23), 2209-2221 (2016).
- Cancer Genone Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 368 (22), 2059-2074 (2013).
- Ball, M., List, A. F., Padron, E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 128 (20), 2381-2387 (2016).
- Varmus, H. E. The molecular genetics of cellular oncogenes. Annual Review of Genetics. 18, 553-612 (2003).
- Cox, D. B. T., Platt, R. J., Zhang, F. Therapeutic genome editing: Prospects and challenges. Nature Medicine. 21 (2), 121-131 (2015).
- Tothova, Z., et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell. 21 (4), 547-555 (2017).
- Mandal, P. K., et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 15 (5), 643-652 (2014).
- Bak, R. O., Dever, D. P., Porteus, M. H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols. 13 (2), 358-376 (2018).
- Foßelteder, J., et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia. , (2023).
- Nangalia, J., et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New England Journal of Medicine. 369 (25), 2391-2405 (2013).
- Merlinsky, T. R., Levine, R. L., Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clinical Cancer Research. 25 (10), 2956-2962 (2019).
- Belčič Mikič, T., Pajič, T., Zver, S., Sever, M. The contemporary approach to CALR-positive myeloproliferative neoplasms. International Journal of Molecular Sciences. 22 (7), 3371 (2021).
- How, J., Hobbs, G. S., Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 134 (25), 2242-2248 (2019).
- Grieger, J. C., Choi, V. W., Samulski, R. J. Production and characterization of adeno-associated viral vectors. Nature Protocols. 1 (3), 1412-1428 (2006).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 6 (6), 973-985 (1999).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2011).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Bulcha, J. T., Wang, Y., Ma, H., Tai, P. W. L., Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduction and Targeted Therapy. 6 (1), 1-24 (2021).
- Montini, E., et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. The Journal of Clinical Investigation. 119 (4), 964-975 (2009).
- Vaidyanathan, S., et al. Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Molecular Therapy. 30 (1), 223-237 (2022).
- Cromer, M. K., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nature Medicine. 27 (4), 677-687 (2021).
- Wiebking, V., et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica. 106 (3), 847-858 (2021).
- Wilkinson, A. C., et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nature Communications. 12 (1), 1-9 (2021).
- Dever, D. P., et al. CRISPR/Cas9 genome engineering in engraftable human brain-derived neural stem cells. iScience. 15, 524-535 (2019).
- Laustsen, A., et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nature Communications. 9 (1), 1-14 (2018).
- Bak, R. O., et al. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife. 6, 27873 (2017).
- Nakauchi, Y., et al. The cell type-specific 5hmC landscape and dynamics of healthy human hematopoiesis and TET2-mutant preleukemia. Blood Cancer Discovery. 3 (4), 346-367 (2022).
- Vaidyanathan, S., et al. selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 26 (2), 161-171 (2020).
- Tang, N., et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 5 (4), 133977 (2020).
- Georgiadis, C., et al. Long terminal repeat CRISPR-CAR-coupled "universal" T cells mediate potent anti-leukemic effects. Molecular Therapy. 26 (5), 1215-1227 (2018).
- Ren, J., et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical Cancer Research. 23 (9), 2255-2266 (2017).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Doench, J. G., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Bak, R. O., Porteus, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Reports. 20 (3), 750-756 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados