
Method Article
Ingénierie des mutations hétérozygotes oncogéniques de gain de fonction dans les cellules souches et progénitrices hématopoïétiques humaines
Dans cet article
Résumé
De nouvelles stratégies pour modéliser fidèlement les mutations somatiques dans les cellules souches et progénitrices hématopoïétiques (HSPC) sont nécessaires pour mieux étudier la biologie des cellules souches hématopoïétiques et les hémopathies malignes. Ici, un protocole pour modéliser les mutations hétérozygotes de gain de fonction dans les HSPC en combinant l’utilisation de CRISPR / Cas9 et la double transduction du donneur rAAV est décrit.
Résumé
Tout au long de leur vie, les cellules souches et progénitrices hématopoïétiques (HSPC) acquièrent des mutations somatiques. Certaines de ces mutations modifient les propriétés fonctionnelles du HSPC telles que la prolifération et la différenciation, favorisant ainsi le développement de tumeurs malignes hématologiques. Une manipulation génétique efficace et précise des HSPC est nécessaire pour modéliser, caractériser et mieux comprendre les conséquences fonctionnelles des mutations somatiques récurrentes. Les mutations peuvent avoir un effet délétère sur un gène et entraîner une perte de fonction (LOF) ou, en revanche, peuvent améliorer la fonction ou même conduire à de nouvelles caractéristiques d’un gène particulier, appelées gain de fonction (GOF). Contrairement aux mutations LOF, les mutations GOF se produisent presque exclusivement de manière hétérozygote. Les protocoles actuels d’édition du génome ne permettent pas le ciblage sélectif des allèles individuels, ce qui entrave la capacité de modéliser les mutations GOF hétérozygotes. Ici, nous fournissons un protocole détaillé sur la façon de concevoir des mutations hétérozygotes du point chaud GOF dans les HSPC humains en combinant la réparation dirigée par homologie médiée par CRISPR / Cas9 et la technologie AAV6 recombinante pour un transfert efficace du modèle de donneur d’ADN. Il est important de noter que cette stratégie utilise un système de rapporteur fluorescent double pour permettre le suivi et la purification des HSPC édités avec succès de manière hétérozygone. Cette stratégie peut être utilisée pour étudier avec précision comment les mutations GOF affectent la fonction HSPC et leur progression vers des hémopathies malignes.
Introduction
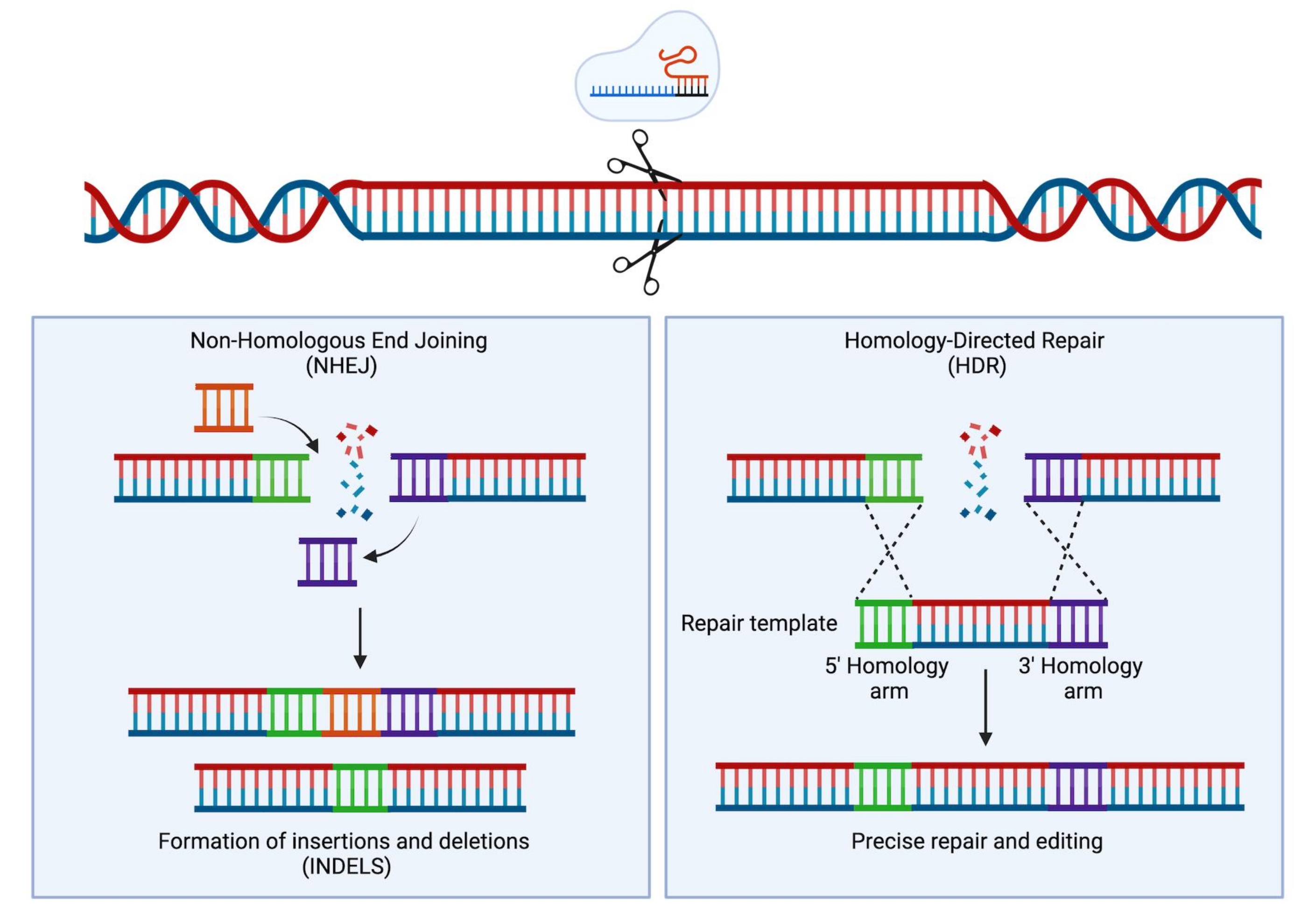
Avec le développement de la technologie CRISPR (Clustered Interspaced Short Palindromic Repeats)/Cas9, un nouvel instrument extrêmement puissant a été ajouté à la boîte à outils des scientifiques. Cette technologie permet l’ingénierie précise du génome et s’est avérée extrêmement utile non seulement à des fins de recherche (examinée dans Hsu et al.1), mais plus récemment a également été traduite avec succès dans le cadre clinique 2,3,4. Les stratégies d’édition CRISPR/Cas9 reposent sur l’activité d’une protéine Cas9 et d’un ARN monoguide (ARNg)5,6,7. Dans la cellule hôte, la protéine Cas9 est guidée vers un site spécifique de l’ADN qui est complémentaire à la séquence d’ARNg et introduira une rupture double brin de l’ADN (DSB). Une fois qu’un DSB est généré, deux mécanismes de réparation principaux et concurrents peuvent se produire : l’assemblage d’extrémité non homologue (NHEJ) et la réparation dirigée par homologie (HDR). NHEJ est un mécanisme de réparation sujet aux erreurs, principalement utilisé conduisant à des insertions et des délétions (indels), tandis que HDR, en utilisant la chromatide sœur comme modèle de réparation, est très précis mais limité à la phase S ou G2 du cycle cellulaire8. En ingénierie génomique, le HDR peut être utilisé pour la modification ciblée de l’ADN en fournissant un modèle de donneur flanqué de bras d’homologie identiques aux deux extrémités de l’ADN du DSB induit par Cas9 (Figure 1). Le type de modèle de donneur utilisé pour le HDR peut avoir un impact considérable sur l’efficacité de l’édition. Pour le génie génétique dans les HSPC humains, le virus adéno-associé de sérotype 6 (AAV6) a récemment été décrit comme un excellent véhicule pour la délivrance de modèles d’ADN simple brin 9,10.
L’ingénierie du génome CRISPR/Cas9 peut être utilisée à des fins thérapeutiques pour corriger les mutations délétères11, mais peut également être utilisée pour introduire des mutations pathogènes dans l’ADN afin de modéliser le développement du cancer12. Le cancer du sang, comme la leucémie, se développe par l’acquisition séquentielle de mutations somatiques dans des HSPC sains13,14. Les événements génétiques précoces entraînent un avantage prolifératif clonal, entraînant une hématopoïèse clonale à potentiel indéterminé (CHIP)15,16. L’acquisition ultérieure de mutations conduira éventuellement à une transformation leucémique et au développement de la maladie. Des mutations somatiques peuvent être trouvées dans les gènes contrôlant l’auto-renouvellement, la survie, la prolifération et la différenciation17.
L’introduction de mutations individuelles via l’ingénierie du génome dans des HSPC sains permet de modéliser précisément ce processus leucémogénique par étapes. Le nombre limité de mutations récurrentes trouvées dans les néoplasmes myéloïdes tels que la leucémie myéloïde aiguë (LAM)18,19 rend cette maladie particulièrement susceptible d’être récapitulée à l’aide d’outils d’ingénierie génomique.
Les mutations somatiques peuvent apparaître sur un seul allèle (mutations monoalléliques/hétérozygotes) ou sur les deux allèles (mutations bialliques/homozygotes) et peuvent avoir des effets profonds sur la fonction du gène, ce qui pourrait entraîner une perte de fonction (LOF) ou un gain de fonction (GOF). Les mutations LOF conduisent à un LOF réduit (si un allèle est affecté) ou complet (si les deux allèles sont affectés) du gène, tandis que les mutations GOF conduisent à une activation accrue ou à une nouvelle fonction du gène. Les mutations GOF sont typiquement hétérozygotes20.
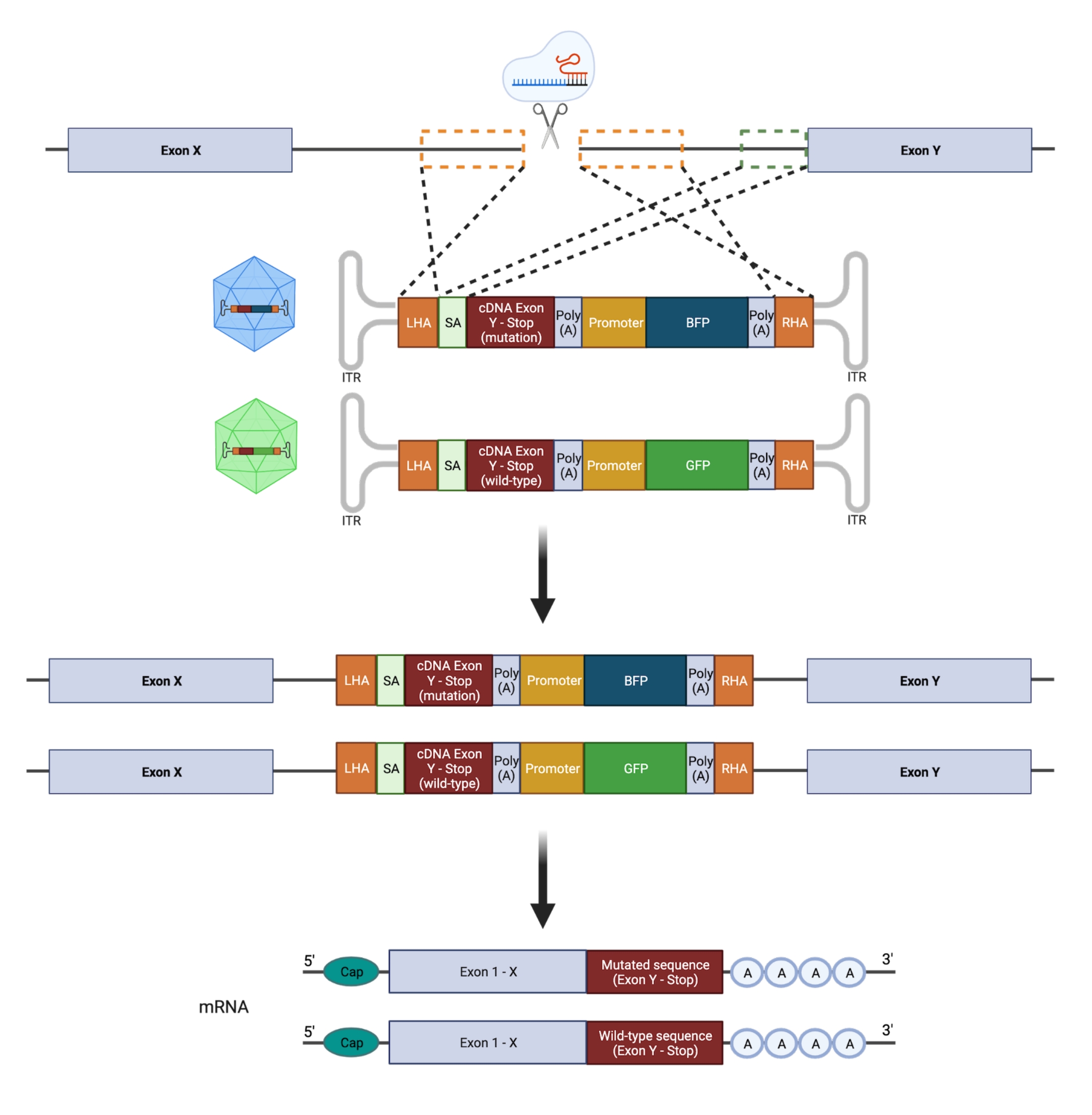
Il est important de noter que la zygotie (hétéro- vs homozygote) a des implications majeures pour la tentative de modéliser fidèlement une mutation; par conséquent, la manipulation ciblée d’un seul allèle d’un gène est nécessaire pour concevoir des mutations GOF hétérozygotes hotspot. Le NHEJ sujet aux erreurs conduit à des indels de différentes longueurs21 qui peuvent entraîner des conséquences biologiques variables et imprévisibles. Cependant, comme le NHEJ est le principal programme de réparation utilisé par les cellules après l’introduction d’un DSB, la plupart des plateformes CRISPR/Cas9 actuellement utilisées pour manipuler les HSPC ne permettent pas de prédire précisément le résultat génétique22,23. En revanche, l’introduction d’une cassure double brin médiée par CRISPR/Cas9 (DSB), combinée à l’utilisation de modèles de donneurs d’ADN à base de vecteurs de virus adéno-associés recombinants (rAAV) pour l’ingénierie du génome via HDR, permet l’insertion de mutations spécifiques aux allèles dans les HSPC humains11,24. L’intégration simultanée d’un mutant et d’une séquence de type sauvage (WT) couplée à des rapporteurs fluorescents distincts sur les allèles individuels peut être réalisée pour sélectionner un génotype hétérozygote (Figure 2). Cette stratégie peut être utilisée comme un outil puissant pour caractériser avec précision les effets des mutations récurrentes, leucémiques et hétérozygotes du point chaud GOF sur la fonction HSPC, l’initiation et la progression de la maladie.
Dans cet article, un protocole détaillé pour l’ingénierie efficace des mutations GOF hétérozygotes récurrentes dans les HSPC humains primaires est fourni. Cette stratégie combine l’utilisation de CRISPR/Cas9 et une double transduction AAV6 pour fournir des modèles de donneurs d’ADN WT et mutants pour la génération prospective de la mutation hétérozygote GOF. À titre d’exemple, l’ingénierie des mutations récurrentes de type 1 (délétion de 52 pb) dans le gène de la calreticuline (CALR) sera montrée25. La mutation GOF hétérozygote dans l’exon 9 de CALR est récurrente dans les troubles myéloprolifératifs tels que la thrombocytémie essentielle (TE) et la myélofibrose primitive (FMP)26. CALR est une protéine résidente du réticulum endoplasmique qui a principalement une fonction de contrôle de la qualité dans le processus de repliement des protéines nouvellement synthétisées. Sa structure peut être divisée en trois domaines principaux: un domaine amino (N)-terminal et un domaine P riche en proline, qui sont impliqués dans la fonction chaperonne de la protéine, et un domaine C, qui est impliqué dans le stockage et la régulationdu calcium 27,28. Les mutations CALR provoquent un décalage de trame de +1, conduisant à la transcription d’une nouvelle extrémité C-terminale étendue et à la perte du signal de rétention du réticulum endoplasmique (ER) (KDEL). Il a été démontré que le CALR mutant se lie au récepteur de la thrombopoïétine (TPO), conduisant ainsi à une signalisation indépendante de la TPO avec une prolifération accrue29.

Figure 1 : Réparation NHEJ et HDR. Représentation schématique simplifiée des mécanismes de réparation NHEJ et HDR suite à l’introduction d’une rupture double brin dans l’ADN. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Vue d’ensemble schématique de la stratégie d’édition HDR biallélique. Représentation schématique montrant l’intégration des modèles de donneurs dans les allèles ciblés suivie de leur traduction en ARNm fonctionnels. Les cases pointillées orange indiquent les régions correspondant au bras d’homologie gauche (LHA) et au bras d’homologie droit (RHA). La taille idéale des AP est de 400 pb chacune. La boîte pointillée verte représente la région correspondant à la séquence SA. La taille de l’AS est de 150 pb. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
Ce protocole exige l’utilisation de CD34+ sains dérivés de donneurs et nécessite l’approbation éthique des comités d’examen des établissements locaux (CISR) et un consentement éclairé signé. Les CSSP CD34+ utilisées dans ce protocole ont été isolées du sang de cordon ombilical (UCB) des accouchements à terme (>34 semaines de gestation). Le consentement éclairé des mères a été obtenu avant l’accouchement et l’approbation éthique pour la collecte d’UCB a été obtenue (approbation IRB: 31-322 ex 18/19) de l’Université de médecine de Graz. Une liste complète des matériaux utilisés dans ce protocole se trouve dans le Tableau des matériaux.
1. Conception de l’ARNg et évaluation de l’efficacité de coupe
- Recherchez l’emplacement de la mutation désirée et le relevé de notes correct dans un outil de base de données en ligne (p. ex., COSMIC, https://cancer.sanger.ac.uk/cosmic).
- Sélectionnez un sgRNA adjacent à la mutation (ciblage exonique) ou dans l’intron précédent (ciblage intronique) à l’aide d’un outil de conception d’ARNg. Dans ce protocole, l’outil en ligne de Benchling est utilisé. Voir le tableau des matériaux pour une liste des outils en ligne possibles pour la conception de sgRNA.
- Sélectionnez l’option + (créer) > Séquence d’ADN > Importer des séquences d’ADN > Importer à partir de bases de données. Entrez le gène souhaité, par exemple, CALR et sélectionnez humain comme espèce > Recherchez > Sélectionnez le bon relevé de notes (par exemple, CALR-201 -ENST00000316448) > Importer. Sélectionnez la région d’intérêt dans laquelle introduire la pause DS (par exemple, intron 7).
- Sélectionnez l’option CRISPR sur le côté droit de l’écran et sélectionnez Concevoir et analyser des guides. Sélectionnez un seul repère, maintenez la longueur du repère à 20 pb et la séquence PAM pour le montage avec SpCas9 (NGG).
- Choisissez un guide avec un score élevé sur la cible (forte chance de modification au locus souhaité) et un score hors cible élevé (faible chance de modification à des loci indésirables). Choisissez au moins trois guides à tester afin de trouver le sgRNA le plus performant. Commandez l’ARNg en tant que sgRNA synthétique chimiquement modifié auprès d’un fournisseur commercial.
REMARQUE: Il est conseillé à ce stade de commander une petite quantité d’ARNg synthétiques pour le criblage initial. Une fois qu’un sgRNA performant a été identifié, procédez avec un ordre plus grand du sgRNA sélectionné. - Décongeler 2 x 10 5-5 x 105 CD34+ HSPC. Transférer les cellules dans 10 mL de RPMI1640 préchauffé complété par des antibiotiques à 1 % (p. ex. pénicilline/streptomycine).
REMARQUE: Les HSPC CD34+ d’une pureté >90% doivent être utilisés pour obtenir les résultats les meilleurs et les plus reproductibles. - Centrifuger à 350 x g à température ambiante (RT) pendant 10 min. Compter les cellules à l’aide d’un hémocytomètre et suspendre les cellules dans le milieu SFEM II supplémenté en pénicilline/streptomycine à 0,2 % (P/S), 100 ng/mL de thrombopoïétine (TPO), 100 ng/mL de facteur de cellules souches (SCF), 100 ng/mL de ligand tyrosine kinase 3 de type FMS (FLT3L), 100 ng/mL d’interleukine-6 (IL-6), 35 nM UM171 et 0,75 μM de StemRegenin1 (SR1) à une concentration de 2,5 x 105 cellules/mL. Incuber à 37 °C/5% CO2 pendant 48 h - 72 h.
REMARQUE : Le support complet sera dorénavant désigné comme support de rétention HSPC. - Recueillir les cellules dans un tube de 15 mL et compter les cellules. Vérifiez la viabilité de la cellule par exclusion du bleu trypan.
- Avant de commencer, allumez le système de transfection et sélectionnez l’option pour les cuvettes. Sélectionnez le programme et la solution de nucléofection appropriés pour les HSPC (voir le tableau des matériaux). Entre 2 x 10 5 et5 x 106 cellules peuvent être nucléofectées dans une cuvette de 100 μL. Mettre de côté un petit nombre de cellules (p. ex., 1 x 10 5-2 x 105) pour les conserver en culture pendant 48 h afin de les utiliser comme témoin WT.
- Préparer le complexe RNP. Dans un tube de 1,5 mL, ajouter 15 μg de Cas9 et 8 μg d’ARNg (rapport molaire 1:2,5) et incuber à 25 °C pendant 10 min dans un bloc chauffant.
- Pendant l’incubation du complexe RNP, centrifuger les cellules à 350 x g à TA pendant 5 min et éliminer le surnageant à l’aide d’une pipette. Suspendre les cellules dans 100 μL de solution de nucléofection.
- Mélanger les cellules avec le complexe RNP et transférer dans la cuvette. Tapotez doucement la cuvette pour éliminer les éventuelles bulles d’air qui auraient pu se former pendant le transfert.
- Insérez la cuvette dans le support du système de transfection et électroporez les cellules avec le programme DZ-100.
- Immédiatement après l’électroporation, ajouter 400 μL de milieu de rétention HSPC préchauffé sans P/S.
- Transférer les cellules à l’aide d’une fine pipette de transfert dans une plaque de culture contenant un milieu de rétention HSPC préchauffé sans P/S. Selon le nombre de cellules, utiliser une plaque de culture appropriée (plaque de 24, 12 ou 6 puits) afin d’atteindre une densité comprise entre 0,25 x 10 6 et 1 x 106 cellules/mL.
- Transférer la plaque dans l’incubateur à 37 °C/5% CO2. Incuber les cellules nucléofectées pendant 6-8 h.
- Après 6-8 h, retirer l’ancien milieu et le remplacer par un milieu de rétention HSPC préchauffé frais complété par du P/S. Suspendre les cellules à une concentration comprise entre 2,5 x 10 5 et 5 x 105 et transférer sur une plaque de culture cellulaire (plaque de 24, 12 ou 6 puits). Incuber les cellules pendant 48 h à 37°C/5% CO2.
- Récolter 2 x 10 5 cellules et centrifuger à 350 x g à TA pendant5 min. Avant de commencer, réglez les blocs chauffants à 65 °C et 98 °C.
- Jeter le surnageant et suspendre les cellules dans 1 mL de 1x DPBS dans un tube de 1,5 mL. Centrifuger à 350 x g à TA pendant 5 min.
- Jeter le surnageant et suspendre les cellules dans 50 μL de solution d’extraction d’ADN. Vortex pendant 15 s.
- Incuber pendant 6 min à 65 °C. Tourbillonner pendant 15 s et incuber pendant 2 min à 98 °C.
- Amplifier la région du DSB par PCR à l’aide d’amorces qui génèrent un amplicon d’environ 400-600 pb avec le DSB au centre. Utilisez 0,5-1 μL d’ADN extrait pour la réaction de PCR.
NOTE: En raison de la composition de la solution d’extraction d’ADN, les concentrations d’ADN ne peuvent pas être quantifiées avec précision par spectrophotométrie. - Faites fonctionner le produit PCR avec une échelle d’ADN sur un gel d’agarose à 1,5 % à 100 V pendant 45-60 min. Placez le gel sur une lumière bleue ou un transilluminateur UV.
- Extraire la bande d’ADN de la bonne taille du gel à l’aide d’une trousse disponible dans le commerce conformément aux instructions du fabricant (voir le tableau des matériaux).
- Séquencer les échantillons par séquençage de Sanger à l’aide de l’amorce directe ou inversée de la PCR. Pour les produits PCR de 400 à 600 pb de longueur, 75 ng d’ADN sont nécessaires pour le séquençage avec une concentration de 5 ng/mL dans un volume total de 15 μL.
- Analyser l’efficacité d’édition des sgRNA en téléchargeant les fichiers de séquençage dans un programme conçu pour calculer l’efficacité d’édition en identifiant l’insertion et les délétions produites par le sgRNA. Sélectionnez le sgRNA le plus performant pour continuer.
REMARQUE : Les outils en ligne dédiés sont répertoriés dans le tableau des matériaux. Cette analyse nécessite les fichiers .ab1 des HSPC transfectés et WT, la séquence sgRNA et la séquence PAM.
2. Construction vectorielle de réparation dirigée par homologie (HDR)
- Conception de modèles HDR
REMARQUE: Deux modèles HDR doivent être conçus: un modèle pour la séquence WT et un modèle pour la séquence mutée.- Importer la séquence génomique et la séquence codante (CDS) du gène désiré dans un logiciel approprié pour le clonage moléculaire (les outils dédiés peuvent être trouvés dans le tableau des matériaux). À partir du fichier de séquence génomique, concevoir le bras d’homologie gauche (HA) en sélectionnant idéalement 400 pb à l’extrémité 5' du DSB. Collez cette séquence dans un nouveau fichier.
- Si un intron est ciblé avec l’ARNg, une séquence d’accepteur d’épissage (SA) doit être incluse. Sélectionnez les 150 derniers pb de l’intron ciblé et collez-le après l’HA gauche. Si l’exon est directement visé, cette séquence n’est pas nécessaire (Figure 3).
- Dans le fichier CDS, sélectionnez l’ADNc qui vous intéresse. Dans le cas où une séquence exonique est ciblée, s’assurer que l’ADNc commence immédiatement en aval du site DSB et comprend tous les exons suivants du gène d’intérêt ainsi que le codon stop. Dans le cas où un intron est ciblé, assurez-vous que l’ADNc commence par le premier codon de l’exon suivant. Codon-optimiser l’ADNc et l’insérer après la séquence SA. Utiliser des outils en ligne dédiés à cette fin (Tableau des matières).
- Insérez un signal de polyadénylation 3' (PolyA) après l’ADNc d’intérêt (p. ex., SV40 ou bGH) (tableau 1). Après la PolyA, insérer une séquence promotrice (p. ex. virus formant le foyer de la rate [SFFV] ou polyubiquitine C [UBC], tableau 1) pour la protéine fluorescente.
- Insérez la séquence de la protéine fluorescente (c.-à-d. GFP, BFP ou mCherry). Insérez un deuxième signal PolyA différent.
REMARQUE: L’insertion d’une séquence PolyA différente évitera des problèmes tels que la recombinaison bactérienne du plasmide ou des problèmes d’alignement de séquence en raison de deux séquences identiques dans le modèle. - À partir du fichier de séquence génomique, concevez le bon HA en sélectionnant idéalement 400 pb à l’extrémité 3' du DSB. Insérez cette séquence après le deuxième PolyA.
- Créez une copie de l’ensemble du modèle et modifiez l’ADNc d’intérêt afin qu’il contienne la séquence de la mutation souhaitée.
- Échangez la protéine fluorescente avec une protéine fluorescente différente. Par exemple, si GFP a été utilisé pour le modèle WT, échangez-le avec BFP ou mCherry pour le modèle mutant. Construisez et clonez les modèles HDR dans le plasmide pAAV-MCS (ou d’autres dorsales appropriées).
REMARQUE: Les fragments requis pour l’assemblage peuvent être produits par PCR ou commandés commercialement et doivent contenir des séquences qui se chevauchent avec leurs fragments voisins. Si la mutation d’intérêt est une mutation ponctuelle, une petite insertion ou une petite délétion, l’ADNc muté peut être produit par PCR en effectuant une mutagénèse dirigée sur le modèle HDR contenant l’ADNc WT. - Transformez Escherichia coli compétent avec le produit assemblé en utilisant la méthode du choc thermique. Avant de commencer, placez les bactéries à décongeler sur la glace pendant 10 min.
- Ajouter 2 μL des produits assemblés à 50 μL de bactéries. Mélanger le contenu en effleurant doucement.
- Déposer les échantillons sur la glace pendant 30 min. Transférer les échantillons dans un thermobloc réglé à 42 °C pendant 30 s.
- Transférer les échantillons sur la glace pendant 5 min. Ajouter 450 μL de milieu SOC à température ambiante et incuber les échantillons à 37 °C pendant 1 h.
- Étaler les échantillons sur des plaques de gélose LB contenant de l’ampicilline. Pour chaque échantillon, étaler 100 μL de trois dilutions différentes de la solution bactérienne (non diluée, 1:5 et 1:10) afin d’obtenir des plaques de gélose LB avec des colonies simples qui peuvent être prélevées. Incuber les plaques pendant la nuit à 37 °C.
- Le lendemain, prélevez trois colonies par échantillon. Transférer les colonies dans des tubes de 15 mL munis de bouchons contenant 4 mL de milieu LB additionné d’ampicilline et incuber pendant une nuit dans un agitateur à 37 °C.
- Aliquote 500 μL de la solution bactérienne de chaque colonie et conservez-la au réfrigérateur pour une utilisation ultérieure. Effectuez une mini-préparation pour extraire l’ADN plasmidique selon les instructions du fabricant.
- Envoyez les échantillons pour le séquençage de Sanger afin de confirmer l’assemblage correct des plasmides. Utilisez suffisamment d’amorces réparties dans tout le plasmide pour assurer une confirmation ininterrompue de la séquence, idéalement en couvrant chaque région deux fois avec des lectures avant et arrière.
- Ajouter 200 μL de la solution bactérienne contenant le plasmide correctement assemblé à 200 mL de milieu LB supplémenté en ampicilline et incuber dans un agitateur pendant une nuit à 37 °C.
- Effectuer une préparation midi ou maxi pour extraire l’ADN plasmidique selon les instructions du fabricant. Conserver l’ADN plasmidique à −20 °C.
- Préparation AAV6 recombinante
REMARQUE: Deux préparations AAV6 distinctes devront être effectuées: une pour le rAAV6 avec le modèle WT HDR et une pour le rAAV6 avec le modèle HDR muté. Cette section décrit les étapes nécessaires à la préparation d’un seul virus.- Décongeler les cellules HEK293T, transférer les cellules dans 10 mL de DMEM préchauffé supplémenté avec 10% FBS, 1% P/S et 25 mM HEPES, et centrifuger à 350 x g à TA pendant 5 min.
- Mettre les cellules en suspension à une concentration de 1 x 105 cellules/mL et les transférer dans une fiole appropriée (p. ex. une fiole de 175 cm2 ). Transvaser la fiole dans un incubateur à 37 °C/5 % de CO2.
REMARQUE: Les cellules HEK293T doivent être décongelées à l’avance afin de permettre aux cellules de récupérer complètement et d’obtenir un nombre suffisant de cellules (11 x 107 cellules sont nécessaires pour la production d’un virus). Il est recommandé d’utiliser les cellules après un minimum de trois passages et de garder les cellules en dessous de 20 passages. Les cellules HEK293T doivent être divisées trois fois par semaine. Tout en maintenant les cellules, celles-ci ne doivent pas dépasser 70% à 80% de confluence. Le DMEM utilisé dans la préparation AAV doit également déjà être complété par de la L-glutamine et du pyruvate de sodium. - Recueillir les cellules dans un tube de 50 mL et centrifuger à 350 x g à TA pendant 5 min.
- Suspendre les cellules dans 20 mL de DMEM complété par 10% FBS, 1% P / S et 25 mM HEPES et compter avec un hémocytomètre. Utilisez le bleu trypan pour l’exclusion des cellules mortes.
- Ensemencer 3 x 106 cellules dans 20 mL de DMEM additionnées de 10 % de FBS, 1 % P/S et 25 mM de HEPES dans une fiole de 175 cm2 . Pour la production d’un virus, préparer au moins quatre fioles de 175 cm2 . Incuber les cellules à 37 °C/5% de CO2 pendant 3 jours.
REMARQUE: Cette étape est mieux effectuée un vendredi car elle permet aux cellules de se développer pendant le week-end. - Récoltez et comptez les cellules HEK293T.
- Pour la préparation de l’un des virus, préparer dix boîtes de 150 mm contenant chacune 11 x 106 HEK293T dans 20 mL de DMEM complétées par 10% FBS, 1% P/S et 25 mM HEPES.
- Placer les plats dans l’incubateur à 37 °C/5% CO2 pendant 24 h. Jeter soigneusement l’ancien milieu et le remplacer par 20 ml de DMEM sans antibiotique complété par 10% FBS, 25 mM HEPES et 1 mM de butyrate de sodium. Avant de continuer, vérifiez que la confluence des cellules n’est pas supérieure à 80%.
- Préparez deux tubes de 15 ml qui contiendront les mélanges de transfection. Au tube 1, ajouter 5 mL de milieu sérique réduit, 60 μg de plasmide HDR muté rAAV6 et 220 μg de pDGM6 (plasmide auxiliaire). Au tube 2, ajouter 5 mL de milieu sérique réduit et 1120 μL de solution de polyéthylènemine à 1 mg/mL (PEI, réactif de transfection).
- Ajouter le contenu du tube 2 au tube 1 et vortex pendant 30 s. Incuber pendant 15 min à TA pour assurer une bonne encapsulation de l’ADN dans les micelles de l’Île-du-Prince-Édouard.
REMARQUE: Ne conservez pas la solution plus de 20 minutes. - Ajouter délicatement goutte à goutte 1,1 mL de solution dans chaque plat et répartir en agitant doucement. Placer les plats dans l’incubateur à 37 °C/5% CO2 pendant 48 h.
- Après 48 h, ajouter 250 μL de 0,5 M d’EDTA à chaque plat et placer les plats dans l’incubateur pendant 10 min. Récoltez les cellules en les lavant de la capsule et transférez-les dans un tube à centrifuger de 500 ml ou plusieurs tubes de 50 ml.
- Centrifuger à 2 000 x g pendant 10 min à 4 °C. Jetez le surnageant. Pour assurer l’élimination complète du surnageant, centrifuger à nouveau à 2 000 x g pendant 1 min à 4 °C.
- Jetez tout surnageant restant. Tout surnageant résiduel pourrait nuire à la purification du virus.
- Desserrez la pastille par vortex et extrayez le virus à l’aide d’une trousse de purification AAV (voir le tableau des matériaux) conformément aux instructions du fabricant. Sinon, effectuer l’extraction par ultracentrifugation à gradient d’iodixanol30,31.
- Aliquote du virus purifié et conserver à −80 °C. Titrer l’AAV fonctionnellement ou quantifier le titre AAV par PCR par gouttelettes numériques (ddPCR)32 afin de déterminer les conditions optimales de transduction.
- Répétez ce processus pour la préparation du plasmide rAAV6 WT HDR.
- Titrage AAV par ddPCR
- Avant de commencer, réglez les blocs chauffants à 65 °C et 98 °C.
- Extraire l’ADN viral en ajoutant 15 μL de solution d’extraction d’ADN à 5 μL du virus. Vortex pendant 15 s.
- Incuber pendant 6 min à 65 °C. Vortex pendant 15 s. Incuber pendant 2 min à 98 °C.
- Utiliser l’ADN extrait (qui est une dilution 1:4 de l’ADN viral) pour préparer des dilutions en série (1:400, 1:40 000, 1:160 000, 1:640 000) avecH2Osans nucléase (nf) pour la ddPCR.
NOTA: Les dilutions peuvent être conservées à −20 °C si la ddPCR n’est pas effectuée immédiatement. - Sélectionnez trois des dilutions pour la ddPCR. Le choix de la dilution à utiliser dépend de la concentration du virus. De préférence, utilisez trois dilutions différentes (par exemple, 1:40 000, 1:160 000 et 1:640 000) pour trouver la meilleure dilution qui se situe dans la limite de détection de la machine.
- Gardez les réactifs sur la glace pendant le travail. Préparez un mélange maître (tous les échantillons seront mesurés en doubles). Pour chaque réaction, préparer ce qui suit : 12,5 μL de ddPCR Supermix pour les sondes, pas de dUTP, 1,25 μL de PrimeTime Std qPCR Assay, AAV-ITR (voir le tableau des matériaux) et 6,25 μL nf-H2O.
- Mélanger 5 μL d’ADN avec 20 μL du mélange maître. Effectuez cette opération pour les dilutions 1:40 000, 1:160 000 et 1:640 000.
- Placez une cartouche dans le porte-cartouche. Ajouter 70 μL d’huile de génération de gouttelettes aux sondes de la rangée appelée huile. Attention à ne pas générer de bulles.
- Ajouter 20 μL du volume de l’échantillon dans la rangée appelée échantillon. Attention à ne pas générer de bulles.
- Scellez la cartouche avec le joint et placez-la dans le générateur de gouttelettes. Générez les gouttelettes en appuyant sur le bouton de démarrage de la machine, retirez le joint et transférez soigneusement, à l’aide d’une pipette multicanal, 40 μL des gouttelettes générées dans une plaque de 96 puits. Travaillez lentement pour éviter la destruction des gouttelettes et des bulles d’air.
- Scellez la plaque à 96 puits avec une feuille de perçage (la bande rouge tournée vers le haut) à l’aide du scellant de plaque PCR. Placez la plaque dans un thermocycleur.
- Réglez le volume à 40 μL, la température du couvercle à 105 °C et les débits de rampe à 2 °C/s. Exécutez le programme PCR comme décrit dans le tableau 2.
- Allumez le lecteur de gouttelettes 30 min avant utilisation. Ouvrez le logiciel sur le bureau.
- Entrez la disposition de la plaque et sélectionnez ABS dans l’onglet expérience et ddPCR Supermix dans l’onglet Supermix. Ensuite, appuyez d’abord sur PRIME, puis cliquez sur FLUSH SYSTEM sur le logiciel pour lancer le système.
- Entrez la plaque dans le lecteur. Démarrez l’exécution et, une fois terminée, exportez les données sous forme de fichier CSV pour une analyse ultérieure sous forme de feuille de calcul.
REMARQUE: Les nombres générés par le logiciel sont des copies du génome (GC) par μL. Ceux-ci doivent être multipliés par les facteurs de dilution : GC/μL x 5 (dilution de l’ADN dans le mélange maître) x dilution initiale (c.-à-d. 40 000 ou 160 000).

Figure 3 : Vue d’ensemble schématique du ciblage intronique et exonique lors du knock-in CRISPR/Cas9 HDR. Comparaison schématique entre les stratégies de ciblage intronique et exonique pour le knock-in CRISPR / Cas9 HDR. (A) Lors du ciblage intronique, une cassure double brin est introduite dans un intron de l’ADN. Le modèle HDR est composé d’un LHA, d’une séquence d’ADNc et d’une ORS. Le ciblage intronique nécessite, en outre, la présence d’un accepteur de tranches contenant le site d’épissure 3', le point de branche et le tractus polypyrimidine. Cela permet un épissage correct. La boîte pointillée verte représente la région correspondant à la séquence SA. La taille de la SA est de 150 pb. (B) Le ciblage exonique repose sur la production d’une cassure double brin directement dans l’exon. Le modèle HDR est composé d’un LHA, d’une séquence d’ADNc et d’une ORS. Les cases pointillées orange indiquent les régions correspondant à la LHA et à l’ORS. La taille idéale des AP est de 400 pb chacune. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Promoteurs | ||||
| Nom | Longueur | Description | ||
| Le | 492 pb | Spleen Focus formant un promoteur de virus. Puissant promoteur de mammifères. Exprimé de manière constitutive | ||
| UBC | 400 pb | Promoteur dérivé du gène humain de l’ubiquitine C. Exprimé de manière constitutive. | ||
| Le | 508 pb | Promoteur dérivé du cytomégalovirus. Il peut contenir une région amplificateur. Exprimé de manière constitutive. Puissant promoteur de mammifères. Peut être réduit au silence. | ||
| EF-1a | 1182 pb | Promoteur alpha du facteur d’élongation 1 de la traduction eucaryote humaine. Exprimé de manière constitutive. Puissant promoteur de mammifères. | ||
| EFS | 200-300 pb | EF-1 alpha forme abrégée sans intron | ||
| CAG | 584 pb | Promoteur hybride de mammifères contenant l’amplificateur précoce du CMV (C), le promoteur de la bêta-actine de poulet (A) et l’accepteur d’épissures pour le gène de la bêta-globine de lapin (G). Exprimé de manière constitutive. | ||
| Signaux de polyadénylation (PolyA) | ||||
| Abréviation | Longueur | Description | ||
| SV40 PolyA | 82-122 pb | Signal de polyadénylation du virus Simian 40 | ||
| bGH PolyA | 224 pb | Signal de polyadénylation de l’hormone de croissance bovine | ||
| rbGlob PolyA | 56 pb | Signal de polyadénylation de la bêta-globine de lapin | ||
Tableau 1 : Promoteurs et signaux de polyadénylation.
| Pas | Température | Heure | Cycles |
| Activation enzymatique | 95°C | 10 minutes | 1x |
| Dénaturation | 94°C | 30 secondes | 42x |
| Recuit | 60°C | 1 minute | |
| Extension | 72°C | 30 secondes | |
| Désactivation enzymatique | 98°C | 10 minutes | 1x |
| Tenir | 4°C | ∞ |
Tableau 2 : Programme de PCR par gouttelettes numériques.
3. Révision des HSPC
- Transfection et transduction des HSPC
- Décongeler les HSPC CD34+ et transférer les cellules dans 10 mL de RPMI préchauffé complété par 1% P/S. Centrifuger à 350 x g à TA pendant 10 min.
- Suspendre les cellules dans le milieu de rétention HSPC à une concentration de 2,5 x 10 5 cellules/mL et incuber à 37 °C/5 % CO2 pendant 48 h - 72 h.
- Récoltez les cellules dans un tube de 15 mL. Comptez les cellules et vérifiez la viabilité cellulaire par exclusion du bleu trypan. Entre 2 x 10 5 et5 x 106 cellules peuvent être nucléofectées dans une seule cuvette. Préparez le nombre approprié de cuvettes en fonction de votre numéro de cellule calculé.
- Préparer le complexe RNP. Dans un tube de 1,5 mL, ajouter 15 μg de Cas9 et 8 μg d’ARNg (rapport molaire 1:2,5) et incuber à 25 °C pendant 10 min dans un bloc chauffant.
- Pendant l’incubation du complexe RNP, centrifuger les cellules à 350 x g pendant 5 min et jeter le surnageant.
- Suspendre les cellules dans 100 μL de solution de nucléofection. Mélanger les cellules avec le complexe RNP et transférer dans la cuvette.
- Tapotez doucement la cuvette pour éliminer les bulles d’air résiduelles qui auraient pu se former pendant le transfert.
- Insérez la cuvette dans le support du système de transfection et électroporez les cellules comme décrit précédemment dans la section sur la conception de l’ARNg.
- Immédiatement après l’électroporation, ajouter 400 μL de milieu de rétention HSPC préchauffé sans P/S et transférer les cellules à l’aide d’une fine pipette de transfert dans une plaque de culture tissulaire contenant 500 μL de milieu de rétention HSPC préchauffé sans P/S. Selon le nombre de cellules, utiliser une plaque de culture appropriée (plaque de 24, 12 ou 6 puits) afin d’atteindre une densité comprise entre 0,25 x 10 6 et 1 x 106 cellules/mL. Transférer la plaque dans l’incubateur à 37 °C/5% CO2.
- Décongeler les flacons contenant les AAV congelés sur de la glace. Transduisez les cellules en pipetant la quantité optimale de chaque rAAV6 sur la suspension cellulaire. Effectuez la transduction dans les 20-30 minutes après l’électroporation pour obtenir des rendements de transduction élevés.
REMARQUE : La concentration optimale de chaque virus (un virus pour le modèle WT HDR et un autre virus distinct pour le modèle HDR muté) doit être déterminée expérimentalement. Habituellement, 5 000 à 10 000 GC/cellule (par exemple, 5 x 109 GC pour 1 x 106 cellules) entraînent une transduction élevée. - Mélangez délicatement la suspension cellulaire avec la pipette. Incuber les cellules transduites pendant 6-8 h à 37 °C/5% CO2. Après 6-8 h, recueillir les cellules dans un tube et centrifuger à 350 x g à TA pendant 5 min.
- Jeter l’ancien milieu et le remplacer par un milieu de rétention HSPC préchauffé frais complété par du P/S.
- Suspendre les cellules à une concentration comprise entre 2,5 x 10 5-5 x 105 cellules/ml et les transférer dans une plaque de culture cellulaire traitée dans les tissus (plaque à 24, 12 ou 6 puits). Incuber les cellules pendant 48 h à 37 °C/5% CO2 avant de procéder au tri.
- Tri en flux des HSPC modifiés porteurs de la mutation GOF hétérozygote
- Récolter les cellules dans un tube de 15 mL et centrifuger à 350 x g à TA pendant 5 min. Retirer le surnageant, suspendre les cellules dans 1 mL de DPBS contenant 0,1% de BSA, et centrifuger à nouveau à 350 x g à TA pendant 5 min.
- Jeter le surnageant et suspendre les cellules dans un volume approprié de DPBS + 0,1% BSA en fonction du nombre de cellules.
NOTE: Il est recommandé de suspendre les cellules à un volume minimum de 200 μL et de ne pas dépasser 1 x 107 cellules/mL afin de réduire le risque de colmatage du trieur. - Transférer les cellules dans un tube FACS stérile muni d’un capuchon. Ajouter du 7-AAD ou un autre colorant de viabilité (selon leur compatibilité avec les protéines fluorescentes) à la suspension cellulaire pour l’exclusion des cellules vivantes / mortes.
- Trier les cellules vivantes qui sont doublement positives pour les protéines rapporteures fluorescentes dans un tube de collecte contenant 200 μL de milieu de rétention HSPC.
REMARQUE : Des contrôles appropriés composés uniquement de cellules transduites avec des AAV doivent être ajoutés pour assurer un contrôle approprié pendant la procédure de tri. - Centrifuger les cellules triées à 350 x g à TA pendant 5 min et suspendre les cellules dans un milieu de rétention HSPC à une concentration de 2,5 x 105 cellules/mL pour une expansion ultérieure en culture ou utiliser les cellules directement pour des essais fonctionnels.
4. Confirmation de la réussite de l’édition de gènes
- Extraction d’ADN génomique
- Avant de commencer, réglez les blocs chauffants à 65 °C et 98 °C. Récolter 2 x 10 5 cellules génétiquement modifiées et purifiées par tri et centrifuger à 350 x g pendant5 min.
- Extrayez l’ADNg comme décrit précédemment. Utilisez la solution d’ADN directement pour la PCR ou stockez-la à -20 °C.
- In-Out PCR
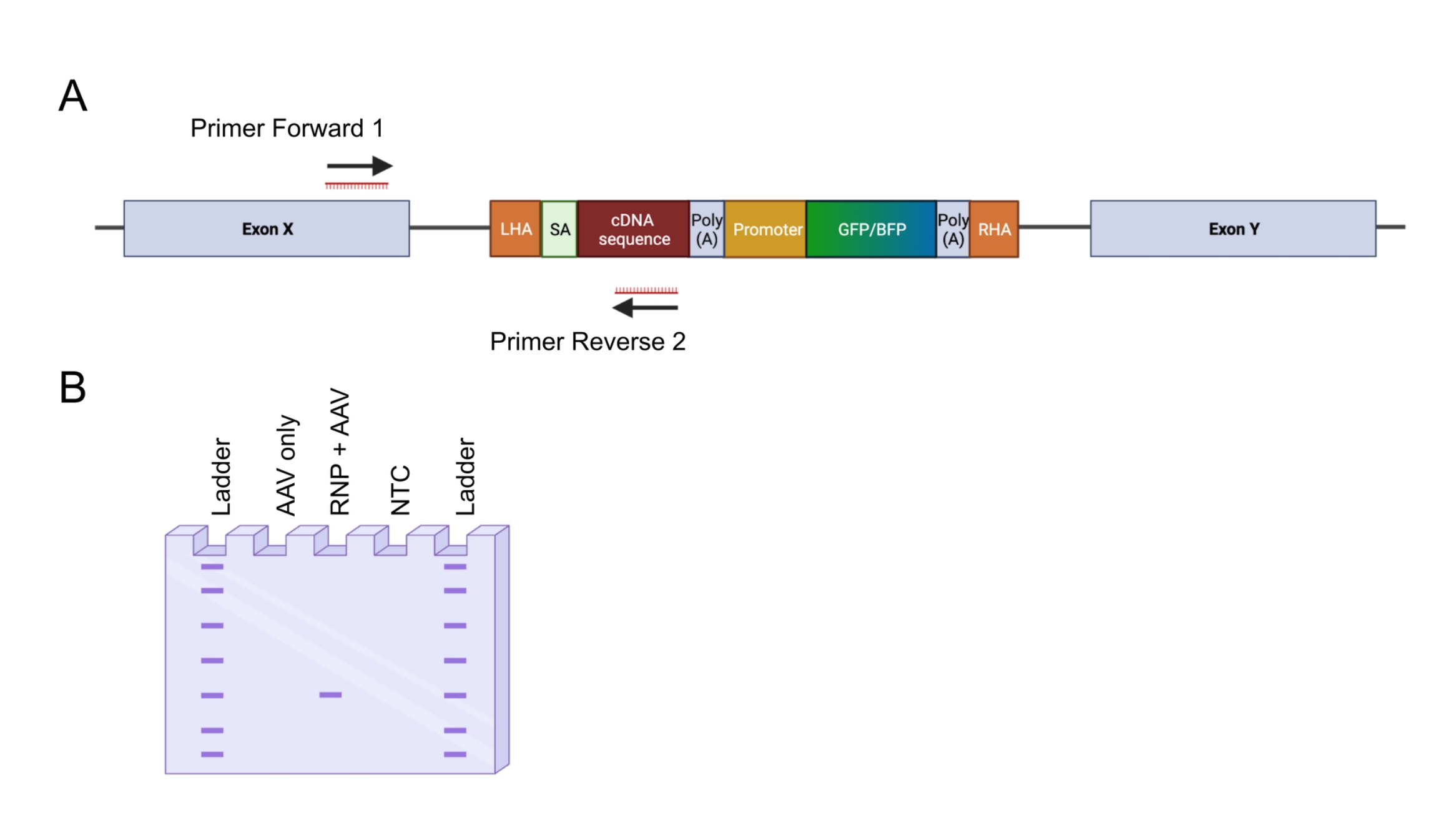
- Concevoir deux amorces pour amplifier le site d’insertion 5' (Figure 4A) : l’amorce avant 1 cible le locus génomique à l’extérieur du bras d’homologie gauche ; L’amorce inverse 2 cible la séquence intégrée.
- Pour la PCR entrée-sortie, préparer le mélange PCR suivant par réaction :
10 μL de PCR Master Mix (2x; voir le tableau des matériaux)
1 μL d’amorce avant 1 (10 μM de stock)
1 μL d’amorce inverse 2 (10 μM de stock)
0,5-1,0 μL d’ADN extrait
H2Osans nucléase jusqu’à un volume final de 20 μL
REMARQUE : Pour plus d’une réaction, il est conseillé de préparer un mélange maître qui contient une réaction supplémentaire pour tenir compte des erreurs de pipetage. Il est important d’inclure un témoin non modèle et un modèle d’ADN provenant d’un échantillon simulé traité. - Exécutez les réactions PCR avec le programme de cyclage thermique approprié (voir les instructions du fabricant).
REMARQUE: Il est conseillé de tester d’abord la paire d’apprêts pour la température de recuit optimale. Cela peut être fait en calculant le Tm, puis en exécutant la PCR avec un thermocycleur qui peut effectuer une température de gradient. - Exécutez les produits de PCR avec une échelle d’ADN sur un gel d’agarose à 1,5% à 100 V pendant 45-60 min (Figure 4B). Placez le gel sur une lumière bleue ou un transilluminateur UV. Extrayez les bandes du gel.
- Extrayez l’ADN des bandes à l’aide d’un kit d’extraction de gel d’ADN. Envoyez les échantillons de PCR extraits avec les amorces appropriées pour le séquençage de Sanger afin de confirmer l’intégration correcte et transparente de l’ADNc souhaité au locus du gène endogène.

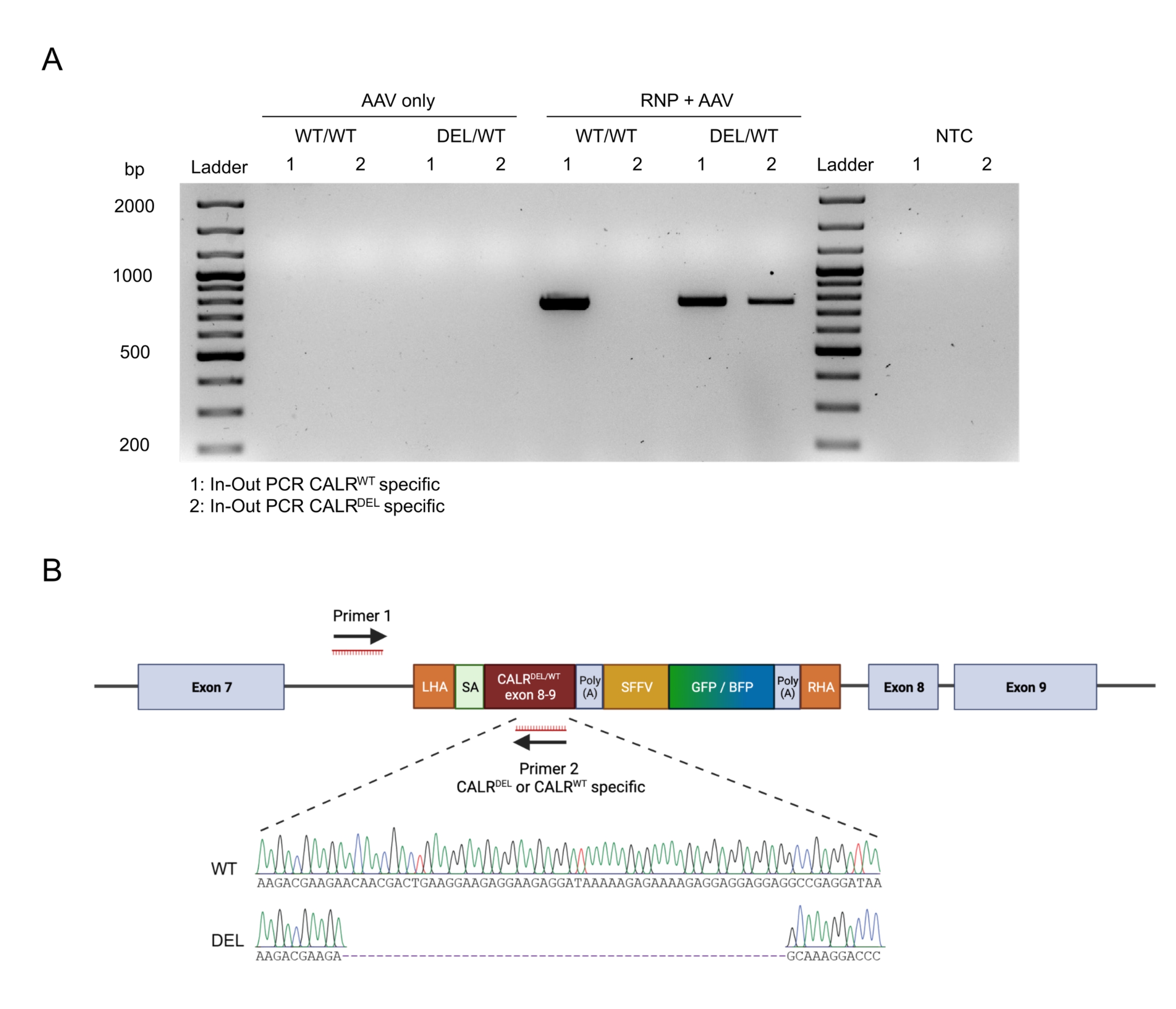
Figure 4 : Validation de l’intégration génomique par PCR in-out. (A) Représentation schématique de la stratégie de PCR in-out. Dans la stratégie représentée, deux amorces ont été conçues. L’amorce avant 1 cible le locus génomique à l’extérieur du LHA, et l’amorce inverse 2 cible la séquence optimisée pour les codons. (B) Représentation schématique d’une électrophorèse sur gel d’agarose. Seules les cellules éditées avec succès (RNP + AAV) généreront un produit PCR pendant la PCR entrée-sortie, tandis que les échantillons non édités (AAV uniquement) ne généreront pas de produit PCR. Abréviation : NTC = contrôle sans modèle. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
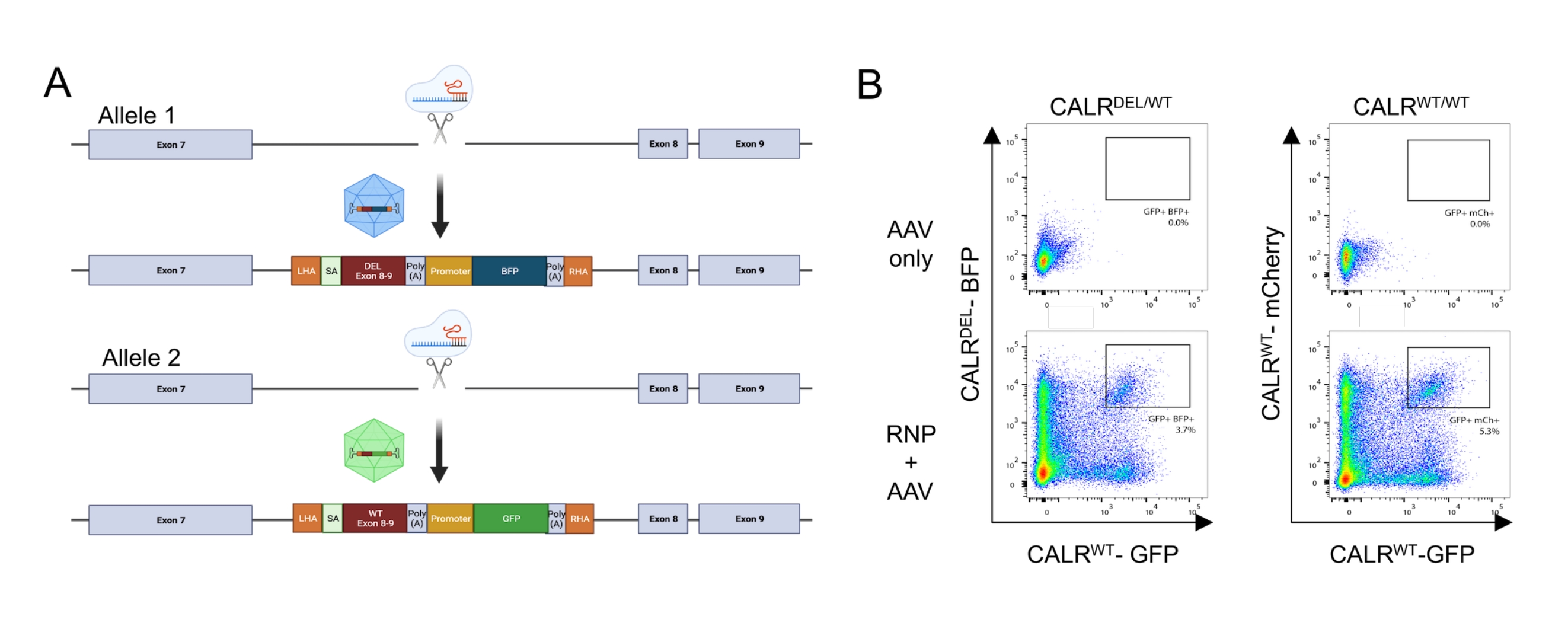
En appliquant le protocole décrit ci-dessus, des mutations hétérozygotes de type 1 CALR ont été introduites de manière reproductible dans les HSPC dérivés du sang de cordon. Cette mutation consiste en une délétion de 52 pb dans l’exon 9 (le dernier exon de CALR), ce qui entraîne un décalage de trame +1, conduisant à la traduction d’un nouveau domaine C-terminal chargé positivement26,33. Pour introduire la mutation CALR au locus du gène endogène, une stratégie de ciblage intronique en amont de l’exon 9 a été adoptée, car cela contournerait tout changement indésirable dans la séquence codante dans les cas où le DSB induit par Cas9 n’était pas réparé via le mécanisme HDR. Dans ce cas précis, un ARNg pour l’intron 7 a été conçu en raison de la disponibilité de séquences élevées sur cible et basses hors cible en combinaison avec des bras d’homologie favorables (absence de répétitions de séquence; Figure 5A).
Deux modèles de donneurs ont ensuite été conçus et regroupés dans des vecteurs AAV6. Afin de permettre un épissage correct des exons endogènes vers l’ADNc intégré, les matrices donneuses contiennent (i) une séquence SA comprenant le site d’épissure 3', le point de branche et le tractus polypyrimidine, (ii) la séquence d’ADNc optimisée pour le codon des exons 8-9, contenant soit le WT (CALRWT), soit la séquence mutée (CALRDEL) ), y compris un codon stop, iii) un signal polyA du virus simien 40 (SV40), iv) une séquence codant pour une protéine fluorescente sous le contrôle du promoteur interne distinct, le promoteur du virus formant le foyer de la rate (SFFV), suivie v) d’un signal polyA de l’hormone de croissance bovine (bGH). Le modèle de donneur contenant l’ADNc CALR WT a été conçu pour contenir une cassette GFP, tandis que le modèle de donneur contenant la séquence d’ADNc CALRDEL a été conçu pour contenir une cassette BFP. L’ensemble de la construction était flanqué d’un HA gauche et droit (figure 5A).
Deux jours après la transfection avec le complexe RNP et la transduction avec les virus rAAV6, les cellules ont été analysées par cytométrie de flux. Quatre populations principales ont pu être détectées : (i) les cellules n’exprimant ni la GFP ni la BFP, représentant des cellules sans édition génomique basée sur HDR, (ii) les cellules positives uniquement pour la GFP, représentant celles qui n’avaient intégré que la construction WT, (iii) les cellules positives uniquement pour la BFP, représentant celles qui n’avaient intégré que la construction mutée, et (iv) les cellules doublement positives GFP et BFP, représentant les cellules qui avaient intégré à la fois les séquences WT et mutées (Figure 5B). Afin d’obtenir des populations pures de HSPC portant la mutation hétérozygote de type 1 CALR , les cellules doublement positives ont été triées par cytométrie de flux. Les HSPC dans lesquels deux séquences WT ont été frappées ont été utilisées comme cellules témoins (GFP+ mCherry+; Figure 5B). Une alternative valable à l’utilisation comme cellules témoins serait les HSPC avec une intégration biallélique des protéines fluorescentes dans un locus Safe Harbor (c.-à-d. AAVS1; non représenté). Le comptage des cellules par exclusion du bleu de trypan effectué sur les HSPC triés a indiqué que plus de 90% des cellules étaient viables.
L’intégration transparente des concepts sur cible a été confirmée par l’application de la stratégie de PCR entrée-sortie (figure 6A). Dans ce cas précis, nous avons effectué deux PCR entrée-sortie distinctes, l’une pour la séquence CALRWT knock-in (voie 1 de l’électrophorèse sur gel de la figure 6A) et l’autre pour la séquence CALR DEL knocked-in (voie 2 de l’électrophorèse sur gel de la figure 6A). Le séquençage de Sanger effectué sur l’ADN extrait des bandes de gel a confirmé l’insertion correcte du WT et des séquences mutées dans les HSPC CALRDEL/WT (Figure 6B).

Figure 5 : Génération de HSPC portant la mutation CALR hétérozygote. (A) Schéma représentatif illustrant la stratégie d’édition pour l’insertion de la mutation CALR hétérozygote. Le complexe RNP cible l’intron entre l’exon 7 et l’exon 8 du gène CALR. Deux AAV, l’un contenant les exons mutés 8-9 et un BFP et l’autre contenant les exons WT 8-9 et un GFP, serviront de modèles de réparation donneur et favoriseront l’intégration de la séquence mutée dans un allèle et l’intégration de la séquence WT dans l’allèle restant. (B) Tracés représentatifs de cytométrie en flux illustrant l’expression de la GFP et du BFP ou de la GFP et de la mCherry 48 h après la transfection et la transduction des HSPC. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6 : Validation de la mutation CALR hétérozygote réussie dans les HSPC. (A) Électrophorèse sur gel à partir des produits de la PCR in-out réalisée sur de l’ADN génomique extrait des témoins AAV, CALR WT/WT et CALRDEL/WT. Une échelle d’ADN de 100 pb a été utilisée. Abréviation : NTC = contrôle sans modèle. (B) Résultats du séquençage de Sanger obtenus à partir des PCR entrées-sorties effectuées sur CALRDEL/WT confirmant l’intégration réussie du WT et des séquences mutées. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
La manipulation génétique efficace et précise des HSPC primaires humaines représente une excellente occasion d’explorer et de comprendre les processus influençant l’hématopoïèse normale et, surtout, la transformation leucémique des cellules hématopoïétiques.
Dans ce protocole, une stratégie efficace pour concevoir des HSPC humains afin qu’ils expriment des mutations hétérozygotes récurrentes du GOF a été décrite. Cette procédure a tiré parti de la technologie CRISPR / Cas9 et des vecteurs rAAV6 en tant que donneurs de modèles d’ADN pour insérer avec précision des séquences d’ADN WT et mutantes dans leurs loci de gènes endogènes. Le couplage des ADNc modifiés (WT et mutant) avec des protéines rapporteures fluorescentes séparées permet l’enrichissement et le suivi des cellules avec un état hétérozygote définitif.
Cette stratégie présente plusieurs avantages par rapport aux méthodes à base de lentiviraux (LV) fréquemment utilisées. L’un des principaux avantages est que le système basé sur CRISPR/Cas9 permet une édition précise dans les loci endogènes, ce qui permet de préserver les promoteurs endogènes et les éléments régulateurs. Cela conduit à une homogénéité dans l’expression du gène édité dans les cellules, un objectif difficilement réalisable lorsqu’une méthode basée sur le VG est utilisée. Le transfert de gènes avec des vecteurs VG conduit à une intégration semi-aléatoire du gène avec une préférence pour les sites transcriptionnellement actifs34. Cela peut se traduire par une surexpression du gène transféré et une hétérogénéité entre les cellules modifiées, ce qui entraîne éventuellement des difficultés à étudier et à analyser le rôle des mutations et des interactions géniques. Un deuxième avantage est que le système décrit, étant un système d’édition spécifique au site, élimine les risques de mutagénèse insertionnelle35.
La stratégie du rapporteur fluorescent double permet l’enrichissement et le suivi précis des cellules qui ont été éditées avec succès sur les deux allèles, un allèle intégrant l’ADNc WT et l’autre allèle intégrant les séquences d’ADNc mutées. Les cellules exprimant un seul rapporteur représentent soit uniquement l’intégration monoallélique, soit l’intégration biallélique de modèles HDR avec le même rapporteur fluorescent. Les deux scénarios ne peuvent être distingués avec précision que si des clones dérivés d’une seule cellule sont produits et analysés individuellement. Cependant, les HSPC n’ont qu’une capacité proliférative limitée in vitro, et lorsqu’ils sont conservés en culture pendant de longues périodes, les HSPC commencent à se différencier en descendants plus matures et perdent leur capacité d’auto-renouvellement et de greffe. Cela rend impossible la sélection et l’expansion de clones unicellulaires hébergeant la mutation hétérozygote souhaitée. L’application de la stratégie de la double protéine fluorescente et l’enrichissement par cytométrie en flux pour les cellules porteuses de la mutation hétérozygote permettent de contourner les problèmes induits par la culture in vitro prolongée.
Dans cet exemple spécifique, il a été démontré avec succès que les HSPC pouvaient être efficacement conçus et triés afin d’obtenir des populations pures de HSPC porteurs de la mutation hétérozygoteCALR DEL/WT.
Cependant, ce système ne se limite pas à l’ingénierie de mutations hétérozygotes par décalage de cadre, mais peut également être facilement adopté pour créer d’autres types de mutations, y compris des mutations faux-sens et non-sens. En appliquant différentes combinaisons d’AAV contenant WT ou des séquences mutées avec différentes protéines rapporteures fluorescentes, ce système peut également être utilisé pour l’introduction de mutations homozygotes (transduction simultanée avec deux AAV porteurs tous deux d’ADNc mutant mais de rapporteurs fluorescents différents) ou même la correction de mutations (transduction simultanée avec deux AAV tous deux porteurs d’ADNc WT mais de rapporteurs fluorescents différents). De plus, il est important de mentionner que cette stratégie ne se limite pas à l’introduction de mutations GOF oncogènes. En fait, le protocole décrit peut être utilisé pour de multiples stratégies alternatives, y compris l’élimination des gènes, le remplacement des gènes 36,37, l’intégration ciblée des transgènes (c.-à-d. les récepteurs antigéniques chimériques)38, et même pour la correction des mutations pathogènes11,39.
La stratégie consistant à combiner CRISPR/Cas9 et AAV6 avec plusieurs rapporteurs fluorescents s’est également avérée applicable à de nombreux autres types de cellules, notamment les lymphocytes T, les cellules plasmacytoïdes dendritiques, les cellules souches pluripotentes induites, les cellules souches neuronales et les cellules souches des voies respiratoires 24,38,40,41,42,43,44 . Cette stratégie peut être mise en œuvre pour la production de lymphocytes T à récepteur antigénique chimérique (CAR) supérieur. Par exemple, il a été récemment publié que l’élimination médiée par CRISPR / Cas9 du gène TGFBR2 dans les cellules CAR T augmente considérablement leur fonction dans le microenvironnement tumoral suppressif riche en TGF-β45. Une telle approche pourrait fournir un protocole en une étape pour à la fois concevoir les cellules T pour exprimer le CAR et pour éliminer le gène TGFBR2 par site insérant spécifiquement le CAR dans les deux allèles du gène TGFBR2. De plus, cette approche pourrait également être utile pour générer des cellules CAR T universelles en intégrant le CAR dans le gène de la constante du récepteur alpha des cellules T (TRAC)46,47.
Pour augmenter la reproductibilité et garantir une édition efficace des cellules, certaines considérations importantes doivent être prises en compte. Les principaux points critiques pour assurer une édition réussie des cellules résident dans (i) la sélection de l’ARNg, (ii) la conception du modèle HDR et (iii) la production de rAAV6.
La sélection d’un sgRNA performant est cruciale car elle déterminera le nombre maximum d’allèles dans lesquels le modèle HDR peut être intégré. Grâce aux nombreux logiciels qui sont maintenant disponibles, la recherche de candidats sgRNA a été simplifiée. En sélectionnant la région d’intérêt, le logiciel peut proposer une série de sgRNA avec un score sur cible et un score hors cible qui indiquent les chances d’édition au locus souhaité et aux loci indésirables, respectivement. Ces scores sont calculés sur la base de modèles de notation48,49 publiés précédemment. Bien qu’il s’agisse d’un bon point de départ pour sélectionner un sgRNA performant, la performance du sgRNA doit être confirmée car sa performance prévue in silico ne correspond pas toujours à un sgRNA efficace in vitro. Par conséquent, il est fortement recommandé de concevoir et de tester au moins trois sgRNA pour augmenter les chances de trouver le meilleur sgRNA. Une fois qu’un véritable sgRNA performant a été identifié, il est suggéré de procéder à la conception du modèle HDR.
Des précautions doivent être prises en considération lors de la conception du modèle HDR. Les bras d’homologie gauche et droite (LHA et RHA, respectivement) devraient s’étendre chacun sur 400 bp en amont et en aval du site de coupe de l’ARNg, respectivement, car des HA plus courts pourraient entraîner une réduction des fréquences HDR. La taille de l’ADNc qui peut être introduit via HDR dépend des capacités d’empaquetage des AAV, qui est d’environ 4,7 kb. En raison des nombreux éléments obligatoires dans le modèle HDR (LHA, RHA, SA, PolyA, promoteur et séquence rapporteur fluorescent), l’espace restant pour l’ADNc muté ou WT est limité. Ceci n’est pas problématique si la mutation souhaitée est située près de l’extrémité 3' d’un gène ou dans des gènes avec un CDS globalement court. Cependant, dans les cas où la mutation est située près du côté de départ transcriptionnel (SCT) des gènes avec un CDS long (dépassant l’espace de tassement restant de l’AAV), cette approche décrite peut ne pas être réalisable. Pour contourner ce problème, une stratégie qui repose sur la division du modèle HDR en deux AAV a été récemment développée par Bak et ses collègues. Cette stratégie repose sur deux intégrations HDR distinctes pour obtenir l’intégration finale transparente d’un grand gène50.
La qualité du virus et son titre sont des facteurs supplémentaires qui peuvent faire ou défaire l’ingénierie génomique réussie des cellules. Pour un rendement optimal, il est important de ne pas laisser le HEK293T atteindre sa pleine confluence tout en étant maintenu en culture. Idéalement, les cellules HEK293T devraient être divisées lorsque la confluence 70%-80% est atteinte. De plus, le HEK293T ne doit pas être cultivé pendant de longues périodes, car cela peut diminuer leur capacité à produire le virus. Les nouvelles cellules HEK293T doivent être décongelées après 20 passages. L’obtention de titres de virus élevés est importante pour augmenter l’efficacité et la reproductibilité des expériences. De faibles titres viraux se traduiront par de grands volumes de solution de rAAV nécessaires à la transduction des HSPC. En règle générale, la solution rAAV ajoutée aux cellules nucléofectées ne doit pas dépasser 20 % du volume total du milieu de rétention HSPC. Des volumes plus élevés de solution AAV peuvent entraîner une augmentation de la mort cellulaire, une prolifération plus faible et une altération de l’efficacité de la transduction. Dans le cas de faibles titres de virus, il est donc recommandé de concentrer davantage le virus.
En résumé, ce protocole offre une approche reproductible pour manipuler les HSPC humains avec précision et efficacité grâce à l’utilisation simultanée de modèles de donneurs CRIPSR/Cas9 et rAAV6 avec des rapporteurs fluorescents doubles supplémentaires. Cette approche s’est avérée être un excellent outil pour étudier la biologie des cellules souches hématopoïétiques normales et les contributions des mutations à la leucémogenèse.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail est soutenu par des subventions du Fonds autrichien pour la science (FWF; numéro P32783 et I5021) à A.R. Un financement supplémentaire à A.R. est également fourni par la Société autrichienne de médecine interne (bourse Joseph Skoda), la Société autrichienne d’hématologie et d’oncologie (OeGHO; Subvention de recherche clinique) et MEFOgraz. T.K. est membre spécial de la Leukemia & Lymphoma Society.
matériels
| Name | Company | Catalog Number | Comments |
| 175 cm2 Cell Culture Flask, Vent Cap, TC-treated | Corning | 431080 | |
| 150 mm x 25 mm dishes | Corning | 430599 | |
| 293T | DSMZ | ACC 635 | https://www.dsmz.de/collection/catalogue/details/culture/ACC-635 |
| 4D Nucleofector Core Unit | Lonza | - | For nucleofection of human HSPCs use the DZ-100 program. |
| 4D Nucleofector X Unit | Lonza | - | |
| 500 ml Centrifuge Tube | Corning | 431123 | |
| 7-AAD | BD Biosciences | 559925 | |
| AAVpro Purification Kit | Takara | 6666 | |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | 1081058 | |
| Avanti JXN-30 Ultracentrifuge | Beckman Coulter | - | |
| Benchling sgRNA design tool | Online tool for sgRNA design: http://www.benchling.com/crispr | ||
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7906-100G | |
| C1000 Touch Thermal Cycler | Bio-Rad | - | |
| Chemically modified synthetic sgRNA | Synthego | Website: https://www.synthego.com/products/crispr-kits/synthetic-sgrna Sequence for the sgRNA targeting intron 7 of CALR: 5’-CGCCTGTAATCCTCGCCCAG-3’ An 80 nucleotide SpCas9 scaffold is added to the 20 nucleotide RNA sequence to complete the sgRNA. Chemical modifications of 2'-O-Methyl are added to the first and last 3 bases and 3' phosphorothioate bonds are added in the first 3 and last 2 bases. *Alternatively chemically modified synthetic sgRNAs can be acquired from IDT (https://eu.idtdna.com/site/order/oligoentry/index/crispr) and Trilink (https://www.trilinkbiotech.com/custom-oligos) | |
| CHOPCHOP sgRNA design tool | Online tool for sgRNA design: http://chopchop.cbu.uib.no | ||
| Costar 24-well Clear TC-treated Multiple Well Plates | Corning | 3526 | |
| CRISPick sgRNA design tool | Online tool for sgRNA design: https://portals.broadinstitute.org/gppx/crispick/public | ||
| CRISPOR sgRNA design tool | Online tool for sgRNA design: http://crispor.tefor.net | ||
| ddPCR 96-Well Plates | Bio-Rad | 12001925 | |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863024 | |
| DG8 Cartridges for QX200/QX100 Droplet Generator | Bio-Rad | 1864008 | |
| DG8 Gaskets for QX200/QX100 Droplet Generator | Bio-Rad | 1863009 | |
| DreamTaq Green PCR Master Mix (2X) | Thermo Scientific | K1081 | |
| Droplet Generation Oil for Probes | Bio-Rad | 1863005 | |
| Dulbecco’s Modified Eagle Medium (DMEM) with high glucose | Sigma-Aldrich | D6429-6X500ML | |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500ML | |
| FACSAria Fusion | BD Biosciences | - | |
| Falcon 5 mL Round Bottom | Corning | 352054 | |
| Fetal Bovine Serum (FBS) Good Forte (heat inactivated), 500 ml | Pan Biotech | P40-47500 | |
| FlowJo 10.8.0 | BD Biosciences | - | |
| GenAgarose L.E. | Inno-train | GX04090 | |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | |
| Gibson Assembly Master Mix | New England Biolabs Inc. (NEB) | E2611L | |
| HEK293T | |||
| HEPES solution | Sigma-Aldrich | H0887-100ML | |
| ICE | Synthego | https://ice.synthego.com | |
| IDT codon optimization tool | IDT | https://www.idtdna.com/pages/tools/codon-optimization-tool | |
| IDT sgRNA design tool | Online tool for sgRNA design: https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM | ||
| LB Broth (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7658-1KG | |
| LB Broth with agar (Lennox) EZMix powder microbial growth medium | Sigma-Aldrich | L7533-1KG | |
| Midori Green Advance | Nippon Genetics | MG04 | |
| Monarch Plasmid Miniprep Kit | NEB | T1010L | |
| Monarch DNA Gel Extraction Kit | NEB | T1020L | |
| NEB 5-alpha Competent E. coli (High Efficiency) | NEB | C2987U | |
| Nuclease-Free Water, 5X100 ml | Ambion | AM9939 | |
| NucleoBond Xtra Midi | Macherey-Nagel | 740410 | |
| Opti-MEM, Reduced Serum Medium, 500 ml | Gibco | 31985070 | |
| P3 Primary Cell 4D-Nucleofetor X Kit L | Lonza | V4XP-3024 | The Lonza Primary P3 solution is supplied as a 2.25 mL P3 Primary Cell Nucleofector Solution and 0.5 mL Supplement 1. To reconstitute, add the Supplement 1 to the P3 Primary Cell Nucleofector Solution and mix. |
| pAAV-MCS2 | Addgene | 46954 | |
| PCR Plate Heat Seal Foil, pierceable | Bio-Rad | 1814040 | |
| pDGM6 | Addgene | 110660 | |
| Penicillin-Streptomycin (P/S) | Gibco | 15140122 | |
| Polyethylenimine (PEI) | Polysciences | 23966 | Add 50 mL of PBS 4.5 pH (made with HCl) to 50 mg of PEI in a tube. Dissolve by placing the tube in a 70°C water bath and vortexing every 10 minutes until the solution is dissolved. After the solution has reached RT, filter sterilze through a 0.22 μm filter, make 1120 μL aliquots, and store at -80°C. |
| Polystyrene Test Tube, with Snap Cap | |||
| Primers | Eurofins | - | Primers were ordered from Eurofins (eurofinsgenomics.eu) as unmodified salt free custom oligos. The primers were designed by using PRIMER-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) Primer 1 Fwd: AAGTGATCCGTTCGCCATGAC; Primer 2 Rev CALR WT specific: ACGTCCTCTTCCTCGTCCTC; Primer 2 Rev CALR DEL specific: CCAACCCTGGAGACACGCTTC |
| PrimeTime qPCR Primer Assay | IDT | - | PrimeTime qPCR Probe Assays (1 probe/2 primers) that can be ordered from IDT (https://eu.idtdna.com/site/order/qpcr/assayentry). Scale: Std - qPCR Assay 500 reactions; Primer 1 Forward (5'-3') : GGAACCCCTAGTGATGGAGTT; Primer 2 Reverse (5'-3'): CGGCCTCAGTGAGCGA; Probe (5'-3'): CACTCCCTCTCTGCGCGCTCG; 5' Dye/3' Quencher: FAM/ZEN/IBFQ; Primer to probe ratio: 3.6 |
| PX1 PCR Plate Sealer | Bio-Rad | 1814000 | |
| QuantaSoft Software | Bio-Rad | ||
| Quick Extract DNA Extraction Solution | Lucigen | QE0905T | |
| QX200 Droplet Generator | Bio-Rad | 1864002 | |
| QX200 Droplet Reader | Bio-Rad | ||
| Recombinant human Flt3-ligand | Peprotech | 300-19 | |
| Recombinant human IL-6 | Peprotech | 200-06 | |
| Recombinant Human SCF | Peprotech | 300-07 | |
| Recombinant Human TPO | Peprotech | 300-18 | |
| RPMI 1640 | Sigma-Aldrich | R8758-6X500ML | |
| SnapGene | Dotmatics | Molecular cloning software https://www.snapgene.com *Alternatively also Benchling (https://www.benchling.com) and Geneious (https://www.geneious.com) can be used. | |
| Soc outgrowth medium | NEB | B9020S | |
| Sodium-butyrate | Sigma-Aldrich | B5887-1G | |
| Stem Regenin 1 (SR1) | Biogems | 1224999 | |
| StemSpan SFEM II | STEMCELL Technologies | 9655 | |
| TAE Buffer (Tris-acetate-EDTA) 50X | Thermo Scientific | B49 | |
| TIDE | http://shinyapps.datacurators.nl/tide/ | ||
| Trypan blue 0.4% | Sigma-Aldrich | T8154-100ML | |
| TrypLE (with phenol red), 500 ml | Thermo Scientific | 16605-028 | |
| UltraPure 0.5: EDTA, pH 8.0, 100 ml | Thermo Scientific | 15575-038 | |
| UM171 | STEMCELL Technologies | 72914 | |
| Vector Builder codon optimization tool | Vector Builder | https://en.vectorbuilder.com/tool/codon-optimization.html |
Références
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Gillmore, J. D., et al. CRISPR-Cas9 in vivo gene editing transthyretin amyloidosis. New England Journal of Medicine. 385 (6), 493-502 (2021).
- Frangoul, H., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine. 384 (3), 252-260 (2021).
- Stadtmauer, E. A., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 367 (6481), (2020).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Veldwijk, M. R., et al. Pseudotyped recombinant adeno-associated viral vectors mediate efficient gene transfer into primary human CD34+ peripheral blood progenitor cells. Cytotherapy. 12 (1), 107-112 (2010).
- Song, L., et al. High-efficiency transduction of primary human hematopoietic stem cells and erythroid lineage-restricted expression by optimized AAV6 serotype vectors in vitro and in a murine xenograft model in vivo. PLoS One. 8 (3), 58757 (2013).
- Dever, D. P., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 539 (7629), 384-389 (2016).
- Drost, J., et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521 (7550), 43-47 (2015).
- Jan, M., et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine. 4 (149), (2012).
- Corces-Zimmerman, M. R., Hong, W. J., Weissman, I. L., Medeiros, B. C., Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America. 111 (7), 2548-2553 (2014).
- Jaiswal, S., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine. 377 (2), 111-121 (2017).
- Genovese, G., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New England Journal of Medicine. 371 (26), 2477-2487 (2014).
- Papaemmanuil, E., et al. Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine. 374 (23), 2209-2221 (2016).
- Cancer Genone Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 368 (22), 2059-2074 (2013).
- Ball, M., List, A. F., Padron, E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 128 (20), 2381-2387 (2016).
- Varmus, H. E. The molecular genetics of cellular oncogenes. Annual Review of Genetics. 18, 553-612 (2003).
- Cox, D. B. T., Platt, R. J., Zhang, F. Therapeutic genome editing: Prospects and challenges. Nature Medicine. 21 (2), 121-131 (2015).
- Tothova, Z., et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell. 21 (4), 547-555 (2017).
- Mandal, P. K., et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 15 (5), 643-652 (2014).
- Bak, R. O., Dever, D. P., Porteus, M. H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols. 13 (2), 358-376 (2018).
- Foßelteder, J., et al. Human gene-engineered calreticulin mutant stem cells recapitulate MPN hallmarks and identify targetable vulnerabilities. Leukemia. , (2023).
- Nangalia, J., et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. New England Journal of Medicine. 369 (25), 2391-2405 (2013).
- Merlinsky, T. R., Levine, R. L., Pronier, E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clinical Cancer Research. 25 (10), 2956-2962 (2019).
- Belčič Mikič, T., Pajič, T., Zver, S., Sever, M. The contemporary approach to CALR-positive myeloproliferative neoplasms. International Journal of Molecular Sciences. 22 (7), 3371 (2021).
- How, J., Hobbs, G. S., Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 134 (25), 2242-2248 (2019).
- Grieger, J. C., Choi, V. W., Samulski, R. J. Production and characterization of adeno-associated viral vectors. Nature Protocols. 1 (3), 1412-1428 (2006).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 6 (6), 973-985 (1999).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2011).
- Klampfl, T., et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New England Journal of Medicine. 369 (25), 2379-2390 (2013).
- Bulcha, J. T., Wang, Y., Ma, H., Tai, P. W. L., Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduction and Targeted Therapy. 6 (1), 1-24 (2021).
- Montini, E., et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. The Journal of Clinical Investigation. 119 (4), 964-975 (2009).
- Vaidyanathan, S., et al. Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Molecular Therapy. 30 (1), 223-237 (2022).
- Cromer, M. K., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nature Medicine. 27 (4), 677-687 (2021).
- Wiebking, V., et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing αβ T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica. 106 (3), 847-858 (2021).
- Wilkinson, A. C., et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nature Communications. 12 (1), 1-9 (2021).
- Dever, D. P., et al. CRISPR/Cas9 genome engineering in engraftable human brain-derived neural stem cells. iScience. 15, 524-535 (2019).
- Laustsen, A., et al. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nature Communications. 9 (1), 1-14 (2018).
- Bak, R. O., et al. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife. 6, 27873 (2017).
- Nakauchi, Y., et al. The cell type-specific 5hmC landscape and dynamics of healthy human hematopoiesis and TET2-mutant preleukemia. Blood Cancer Discovery. 3 (4), 346-367 (2022).
- Vaidyanathan, S., et al. selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 26 (2), 161-171 (2020).
- Tang, N., et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 5 (4), 133977 (2020).
- Georgiadis, C., et al. Long terminal repeat CRISPR-CAR-coupled "universal" T cells mediate potent anti-leukemic effects. Molecular Therapy. 26 (5), 1215-1227 (2018).
- Ren, J., et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical Cancer Research. 23 (9), 2255-2266 (2017).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Doench, J. G., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Bak, R. O., Porteus, M. H. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Reports. 20 (3), 750-756 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.