
Method Article
Analysieren von Tumor-Genexpressionsfaktoren mit dem CorExplorer-Webportal
In diesem Artikel
Zusammenfassung
Wir stellen das CorExplorer-Webportal vor, eine Ressource zur Erforschung von Tumor-RNA-Sequenzierungsfaktoren, die vom Machine Learning-Algorithmus CorEx (Correlation Explanation) gefunden wurden, und zeigen, wie Faktoren im Verhältnis zum Überleben, Datenbankanmerkungen, Protein-Protein-Wechselwirkungen und untereinander, um Einblicke in die Tumorbiologie und therapeutische Interventionen zu gewinnen.
Zusammenfassung
Die Differentialgenexpressionsanalyse ist eine wichtige Technik zum Verständnis von Krankheitszuständen. Der Machine Learning Algorithmus CorEx hat bei der Analyse der differenziellen Expression von Gengruppen in Tumor-RNA-seq in einer Weise nützlich sein, um die Präzisionsonkologie voranzubringen. CorEx produziert jedoch viele Faktoren, die eine Herausforderung darstellen können, um bestehendes Verständnis zu analysieren und mit ihnen in Verbindung zu treten. Um solche Verbindungen zu erleichtern, haben wir eine Website, CorExplorer, erstellt, die es Benutzern ermöglicht, die Daten interaktiv zu untersuchen und häufig gestellte Fragen im Zusammenhang mit ihrer Analyse zu beantworten. Wir haben CorEx auf RNA-seq Genexpressionsdaten für vier Tumortypen trainiert: Eierstock, Lunge, Melanom und kolorektal. Anschließend haben wir entsprechende Überlebens-, Protein-Protein-Wechselwirkungen, Gene Ontology (GO) und Kyoto Encyclopedia of Genes and Genomes (KEGG) Signalanreicherungen und Heatmaps in die Website integriert, um sie mit der Faktorgraphenvisualisierung in Verbindung zu bringen. Hier verwenden wir Beispielprotokolle, um die Verwendung der Datenbank zu veranschaulichen, um die Bedeutung der erlernten Tumorfaktoren im Kontext dieser externen Daten zu verstehen.
Einleitung
Seit seiner Einführung vor etwas mehr als einem Jahrzehnt ist RNA-seq zu einem allgegenwärtigen Werkzeug zur Messung der Genexpression1geworden. Dies liegt daran, dass es eine schnelle und kostengünstige de novo Profilierung des gesamten Transkriptoms einer Probe ermöglicht. Die RNA-seq-Tumordaten spiegeln jedoch eine zugrunde liegende Biologie wider, die an sich komplex und oft unterbeprobt ist, während die Daten selbst hochdimensional und laut sind. Dies stellt eine große Herausforderung für die Extraktion zuverlässiger Signale dar. Der CorEx-Algorithmus nutzt multivariate gegenseitige Informationen, um subtile Muster in solchen Situationen zu finden2,3 . Diese Technik wurde zuvor angepasst, um Eierstocktumor-RNA-seq-Proben aus dem Krebsgenomatlas (TCGA) zu analysieren, und in diesem Zusammenhang schien sie signifikante Vorteile gegenüber häufiger verwendeten Analysemethoden zu haben4.
Obwohl die Verwendung von RNA-Seq in Forschungsanwendungen, auch in der Onkologie, enorm weit verbreitet ist, haben diese Bemühungen nicht zu einer breiten Nutzung für die Zwecke klinischer Interventionen geführt5. Ein Grund dafür ist ein Mangel an benutzerfreundlichen Algorithmen und Software, die auf diese spezifischen Probleme ausgerichtet sind. Um diese Lücke zu schließen, haben wir das CorExplorer-Webportal entwickelt, um Forschern aus einer Vielzahl von Hintergründen die Untersuchung von Genexpressionsfaktoren von Tumor-RNA-seq-Proben zu ermöglichen, wie sie im CorEx Machine Learning-Algorithmus gefunden wurden. Das CorExplorer-Portal unterstützt die interaktive Visualisierung und Abfrage von Faktoren verschiedener Tumortypen wie Lunge, Dickdarm, Melanom und Eierstock6,7,8,9, 10, mit der Absicht, Forschern zu helfen, die Datenkorrelationen zu durchforsten und Kandidatenwege zur Stratifizierung von Patienten zu therapeutischen Zwecken zu identifizieren.
Wir erwarten, dass das CorExplorer-Portal für verschiedene Benutzertypen nützlich sein kann. Das Portal wurde mit Blick auf den Anwender konzipiert, der die breiten Faktoren verstehen möchte, die Unterschiede bei der Tumorgenexpression in öffentlichen Datenbanken antreiben und möglicherweise auch individuelle Genexpressionsprofile in den Kontext von Tumoren mit ähnlichen Merkmale. Zusätzlich zu den hier beschriebenen repräsentativen Protokollen können CorExplorer-Untersuchungen als Ausgangspunkt dienen, um Hypothesen für weitere Tests vorzuschlagen, CorEx-Befunde in Datensätzen außerhalb des CorExplorer zu vergleichen und zu kontrastieren und eine Verbindung herzustellen. pathologische Expressionssignaturen eines oder mehrerer Gene in einem einzelnen Tumor an größere Gruppen, die koordiniert betroffen sein können. Schließlich kann es als benutzerfreundliche Einführung in die Anwendung des maschinellen Lernens auf RNA-seq für diejenigen dienen, die in diesem Bereich beginnen.
Protokoll
1. Untersuchen von Faktoren, die ein Gen von Interesse enthalten
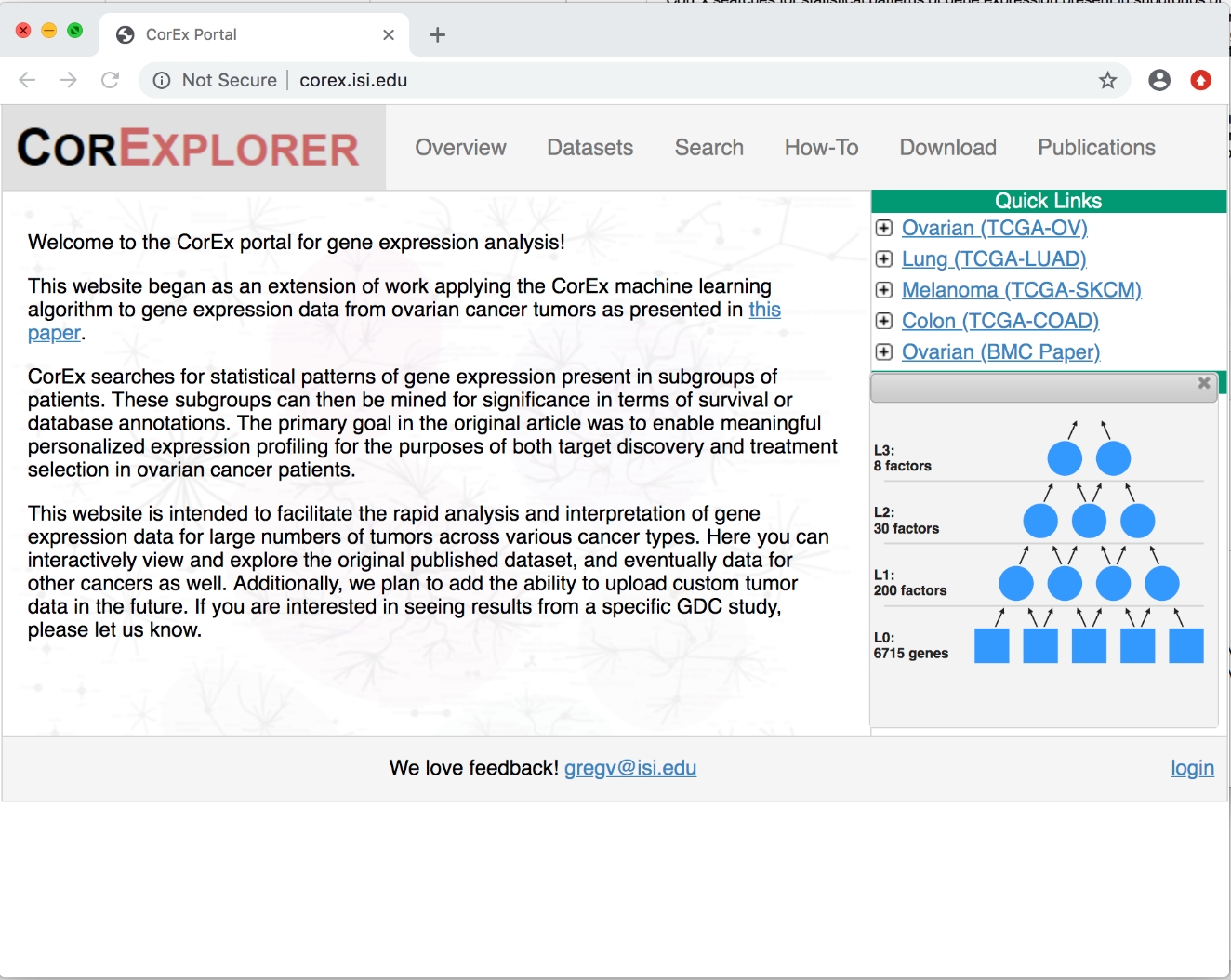
- Öffnen Sie einen Webbrowser und wechseln Sie zu http://corex.isi.edu, der CorExplorer-Startseite.
- Klicken Sie auf der rechten Seite unter Quick Linksauf die Schaltfläche + erweitern neben Ovarian (TCGA-OV), um eine Zusammenfassung des CorEx-Faktordiagramms zu sehen, das auf den TCGA-Eierstockkrebsdaten trainiert wurde (siehe Abbildung 1). Klicken Sie optional auf andere, um sie zu vergleichen.
- Nachdem Sie die Faktordiagramme überprüft haben, klicken Sie aufLunge (TCGA-LUAD), um auf die CorExplorer-Seite für Lungenkrebs RNA-seq zuzugreifen.
- Erkunden Sie das CorEx-Faktordiagramm für ein Gen von Interesse mithilfe des CorExplorer-Fensters "Faktordiagramm".
- Bewegen Sie den Mauszeiger über das Anzeigefenster faktorgraph. Zoomen Sie mit dem Maus-Scrollrad oder Trackpad in das Faktordiagramm, um Details des Diagramms zu sehen, z. B. die wichtigsten Gene in jedem Faktor und die Verbindungen zwischen Knoten in verschiedenen Schichten. Alternativ können Sie auf klicken und ziehen, um den Ansichtsbereich oder einen beliebigen Knoten zu verschieben.
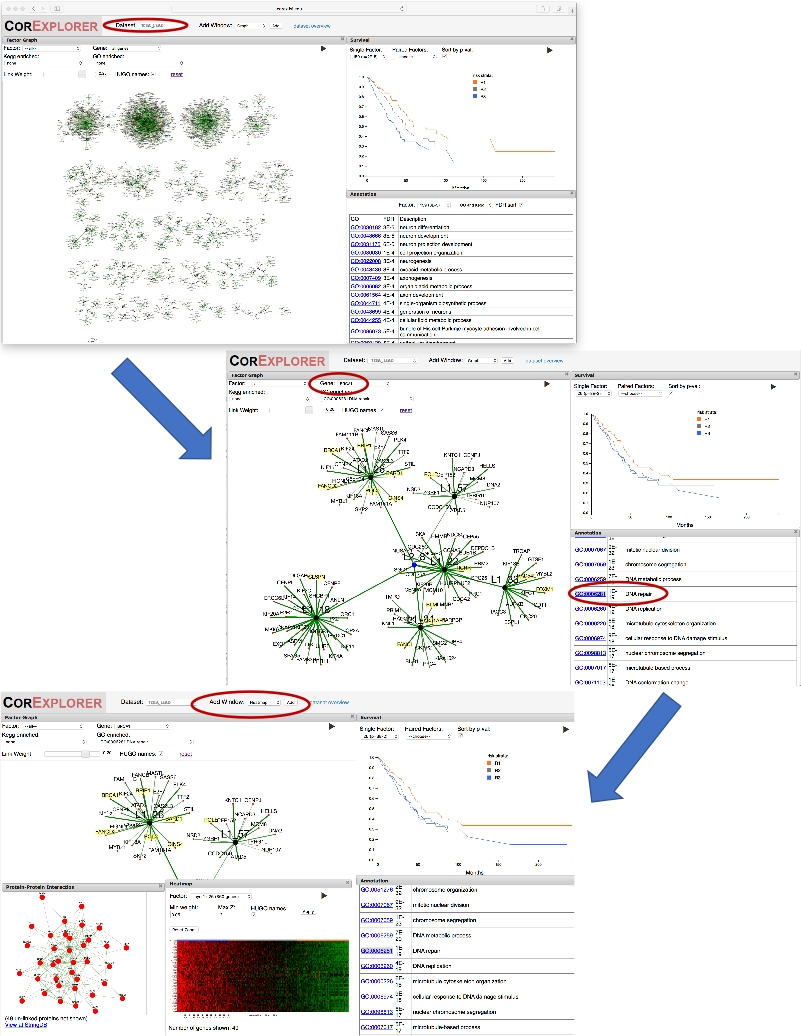
- Um ein Zielgen zu finden (hier verwenden wir BRCA1), klicken Sie auf das Dropdown-Menü Gene oben im Faktordiagrammfenster. Geben Sie 'BRCA1' ein, um es in der Dropdown-Liste auszuwählen, und drücken Sie Return, um den Ansichtszoom auf Faktor 26 zu vergrößern, den Faktor, mit dem BRCA1 am stärksten korreliert ist.
- Positionieren Sie die Maus über die Diagrammanzeige, und scrollen Sie, um den Knoten Ebene 2, L2_8, und die zugehörigen Faktoren, die Nachbarn zu Faktor 26 sind, zu verkleinern. Beachten Sie, dass nur Gene mit einem Gewicht angezeigt werden, das größer als der auf dem Min-Link-Gewichtsregler angegebene Schwellenwert ist.
- Um alle Gene anzuzeigen, die mit dem Faktor verknüpft sind, klicken Sie auf den Knoten L1_26 und wählen Sie zusätzliche Gene im Pop-up-Fenster laden aus. Wenn das Wort "Fertig" angezeigt wird, schließen Sie das Popupfenster.
- Kehren Sie nun zum Kopfabschnitt über dem Faktordiagrammfenster zurück, und greifen Sie den Modifikator Min-Link-Gewichtung an und ziehen Sie ihn. Nun, da der Linkgewichtsregler auf 0,05 nach unten verschoben wird, werden andere Gene im Faktor L1_26, einschließlich BRCA2, in der Gewichtsreihenfolge angezeigt. Optional können Sie Knoten durch Greifen und Ziehen neu positionieren, um das Layout zu verbessern.
- Bestimmen Sie, wie sich die Schichtung der Patienten in Bezug auf den Faktor auf das Überleben auswirkt, indem Sie im Überlebensfenster abfragen.
- Deaktivieren Sie im Überlebensfenster sortieren nach p-val, und wählen Sie dann Faktor 26 im Dropdown-Menü Einzelfaktor aus, um Überlebenskurven für Faktor 26 anzuzeigen.
- Scrollen Sie im Überlebensdiagramm nach unten, um die Anzahl der gefährdeten Patienten entlang der x-Achse anzuzeigen.
- Suchen Sie Assoziationen mit biologischer Funktion, indem Sie im Anmerkungsfenster abfragen.
- Um im Anmerkungsfenster das Dropdown-Menü Faktor nach Faktornummer statt nach False Discovery Rate (FDR) zu sortieren, deaktivieren Sie die FDR-Sortierung.
- Scrollen Sie, und klicken Sie, um Faktor 26 in der Dropdown-Dropdownliste des Anmerkungsfensters auszuwählen, um Annotationen für den Faktor anzuzeigen.
- Scrollen Sie in der Anmerkungsliste nach unten, bis die DNA-Reparatur sichtbar ist, und klicken Sie darauf, um die zugehörigen Gene, die auf der Grafikanzeige gelb hervorgehoben sind, sofort zu sehen. Siehe das mittlere Panel von Abbildung 2.
- Beachten Sie, dass Faktoren verschwinden oder erscheinen, wenn unterschiedliche GO-Begriffe ausgewählt werden, je nachdem, ob sie für Gene mit der ausgewählten Anmerkung angereichert sind, z. B. "intrinsischer apoptotischer Signalweg als Reaktion auf DNA-Schäden".
- Erkunden Sie die Faktoren weiter, indem Sie Fenster mit unterschiedlichen Funktionen hinzufügen.
- Fügen Sie in der oberen Menüleiste ein Protein-Protein-Interaktionsnetzwerk (PPI)-Fenster hinzu, indem Sie PPI aus der Dropdown-Liste Fenster hinzufügen auswählen und dann auf die Schaltfläche Hinzufügen klicken, um dem Anzeigebereich ein PPI-Diagrammfenster hinzuzufügen. Wählen Sie im PPI-Diagrammfenster den Faktor 'Layer1: 26', um die Protein-Protein-Wechselwirkungen anzuzeigen. Beachten Sie die Dichte der Verbindungen.
- Wählen Sie in der oberen Menüleiste anstelle von PPI Heatmap aus der Dropdown-Liste Fenster hinzufügen aus, und klicken Sie dann auf die Schaltfläche Hinzufügen, um dem Anzeigebereich ein Heatmap-Fenster hinzuzufügen. Wählen Sie im Heatmap-Fenster den Faktor 'Layer1: 26', um die Genexpressionsmuster anzuzeigen.
- Schnappen und positionieren Sie das Heatmap-Fenster neu, so dass auch das Überlebensfenster sichtbar ist. Beobachten Sie oben auf der Heatmap, wie der orange/blau/grau gefärbte Balken den Risikoschichten des Patienten im Überlebensdiagramm entspricht. Die Ergebnisse sind unten in Abbildung 2dargestellt.
- Erkunden Sie das CorEx-Faktordiagramm für ein Gen von Interesse mithilfe des CorExplorer-Fensters "Faktordiagramm".
2. Filtern und Interpretieren von CorEx-Faktoren mithilfe von Gengewichts-, Überlebens- und Anmerkungsdaten
- Filtern Sie anhand von Überlebens- und Clusterqualität nach Faktoren von Interesse.
- Wählen Sie im Dropdown-Menü Datensatz oben TCGA_OVCA aus, um zur CorExplorer-Seite für den TCGA Eierstockkrebs RNA-seq zu gelangen.
- Sobald die Seite geladen wurde, beachten Sie aus dem Überlebensfenster, dass der Faktor mit dem größten Überlebensunterschied für verschiedene Schichten 114 ist.
- Wählen Sie oben im Faktordiagrammfenster 'Layer1: 114' aus der Dropdownliste Faktor aus.
- Greifen Sie den Linkgewichtsregler mit der Maus und bewegen Sie ihn bis zu 0,5. Beachten Sie, dass die große Anzahl von Genen im Faktor 114 (1609), ohne dass keines gewicht >0,35 hat, auf eine relativ schwache Clusterbildung hinweist.
- Erweitern Sie als Nächstes die Liste der Faktoren im Überlebensfenster, und wählen Sie den nächstbesten Faktor im Dropdown-Dropdown des Überlebensfensters, Faktor 39, aus, um die zugehörigen Überlebenskurven anzuzeigen.
- Wählen Sie Faktor 39 im Anmerkungsfenster aus, indem Sie darauf klicken. Die signifikanten GO- und KEGG-Anmerkungen werden angezeigt.
- Um ein besseres Verständnis der biologischen Rolle von Genen in Faktor 39 zu erlangen, interpretieren Sie die Faktoren anhand von Nachbarschaftsanmerkungsinformationen wie folgt.
- Wählen Sie oben im Faktordiagrammfenster den Faktor 'Layer1: 39' in der Faktor-Dropdown-Liste aus. Bewegen Sie dann die Maus über das Faktordiagrammfenster und zoomen Sie nach, um den gesamten L2_14-Cluster mit 6 Faktoren anzuzeigen: 14, 32, 39, 42, 52 und 82 (siehe Abbildung 3).
- Um die relative Bedeutung der Faktoren zu verstehen, die mit dem Knoten L2_14 verknüpft sind, betrachten Sie zunächst Überlebensunterschiede für jeden der L2_14-Faktoren. Deaktivieren Sie Sortieren nach p-val im Überlebensfenster, und klicken Sie dann auf jede der Faktorzahlen nacheinander. Beachten Sie dabei, dass nur die Faktoren 14, 32 und 39 eine Überlebensassoziation aufweisen.
- Wählen Sie nun in der oberen Menüleiste PPI aus der Dropdown-Dropdownliste Fenster hinzufügen aus. Drücken Sie Hinzufügen, um dem Anzeigebereich ein PPI-Diagrammfenster hinzuzufügen. Wählen Sie im PPI-Diagrammfenster den Faktor 'Layer1: 52' aus, um die signifikanten Protein-Protein-Wechselwirkungen anzuzeigen. Ein Beispiellayout von Fenstern an dieser Stelle ist in Abbildung 3dargestellt.
- Klicken Sie unten im PPI-Fenster auf den Link Ansicht in StringDB, um eine Verknüpfung mit der StringDB-Onlinedatenbank herzustellen. Klicken Sie auf dem ersten Bildschirm auf Weiter, und wählen Sie dann wie zuvor die Registerkarte Analyse unter dem Netzwerkdiagramm aus, um eine Online-GO-Analyse für die PPI-Netzwerkgene zu erhalten. Die oberste zelluläre Komponente ist der Proteinkomplex "MHC Class II".
- Kehren Sie zum CorExplorer-Registerkarten- und PPI-Fenster zurück, und wählen Sie Faktor 32 aus, diesmal aus der Faktor-Dropdown-Liste. Klicken Sie auf den Link Ansicht in StringDB mit der StringDB-Analyse. Die oberste zelluläre Komponente ist 'MHC Class I Protein complex', im Gegensatz zu Klasse II für Faktor 52 im vorherigen Schritt!
- Gehen Sie schließlich zurück zum PPI-Fenster und wählen Sie "Layer1: 39" aus dem Dropdown-Menü Faktor oben aus. Klicken Sie auf den Link Ansicht in StringDB, um eine Verknüpfung mit der StringDB-Analyse herzustellen.
- Klicken Sie auf dem ersten Bildschirm auf Weiter, und wählen Sie dann die Registerkarte Analyse unter dem Netzwerkdiagramm aus, um eine Online-GO-Analyse für die PPI-Netzwerkgene zu erhalten. Beachten Sie, dass die oberste molekulare Funktion "CXCR3 Chemokin-Rezeptorbindung" ist.
3. Verwenden von Überlebens- und Datenbankanmerkungen zur Suche nach vielversprechenden therapeutischen Kombinationen
- Wechseln Sie zum TCGA Melanom CorExplorer, indem Sie TCGA_SKCM aus dem Dropdown-Menü Dataset auswählen.
- Beachten Sie, dass der Faktor mit dem größten Überlebensunterschied Faktor 171 ist. Untersuchen Sie den Faktor 171 Anmerkungen durch Scrollen und beachten Sie, dass "Immunantwort" und "Zytokin-vermittelter Signalweg" in der Nähe der Spitze sind (wie sie für den obersten Eierstockfaktor waren).
- Um einen komplementären Faktor zu finden, untersuchen Sie die top-überlebensassoziierten Faktoren zusammen mit ihren Top-Anmerkungsbegriffen. Klicken Sie dazu auf den Link Datensatzübersicht in der oberen Menüleiste, um eine separate Registerkarte mit einer Tabelle mit Details zur Datasetverarbeitung sowie einer Zusammenfassung der Top-Faktoren entsprechend dem p-Wert der Überlebensdifferenz zu öffnen. Beachten Sie, dass der erste Nicht-Immunfaktor 88 ist.
- Kehren Sie zur Registerkarte TCGA_SKCM-Browser zurück.
- Wählen Sie Faktor 88 in den Überlebens-, Anmerkungs- und Diagrammfenstern aus. Die obersten GO-Begriffe beziehen sich auf die "rRNA-Verarbeitung" und die "Mitochondrion-Organisation", die sie als von den immunbedingten Faktoren unterscheidend bestätigen.
- Wählen Sie im Überlebensfenster, auf der Dropdown-Liste der gepaarten Faktoren, '88_171', um zu sehen, wie das Überleben für Patienten in der mittleren Schicht für die kombinierten 171 und 88 Expressionsfaktoren verbessert wird. Anmerkungs- und Überlebensvergleiche sind in Abbildung 4dargestellt.
4. Auf der Suchseite Gemeinsamkeiten und Unterschiede der Genexpressionsvariation zwischen Tumortypen finden
- Klicken Sie auf die CorExplorer-Überschrift, um zur Titelseite zurückzukehren.
- Klicken Sie in der oberen Menüleiste auf Suchen, um zu einer Seite zu gelangen, auf der die Suche nach allen Datensätzen auf der CorExplorer-Website möglich ist.
- Geben Sie im Suchfeld Gene 'FLT1' (VEGFR1) ein und drücken Sie Return oder drücken Sie Search. FLT1 ist mit einem relativ hohen Gewicht in den folgenden Faktoren gefunden: OVCA - 76, LUAD - 162, SKCM - 195 und SKCM - 184, sowie COAD - 112 und COAD - 74.
- Alternativ können Sie in allen Datasets nach einem zugehörigen GO-Term suchen. Probieren Sie dies im Feld "GO-Suche" aus, indem Sie 'Angiogenese' eingeben und auf Return drücken oder Searchdrücken. Alle FLT1-Faktoren, mit Ausnahme von SKCM-195, werden als statistisch angereichert für "Angiogenese"-Gene aufgeführt – Faktor 195 hat in der Tat die Anmerkung, aber unterhalb der Standardschwelle 10-8. Die Suchergebnisse hierfür und den vorherigen Schritt sind in Abbildung 5dargestellt.
- Als weitere Beispiele, im GO-Suchfeld, erste Typ "epidermalwachstumsfaktor rezeptor." Nur LUAD ist für diesen Begriff angereichert, ein bekannter Schichtungsfaktor für Lungenkrebs. Geben Sie als Nächstes 'mesenchymal' in das Suchfeld ein. Dieser Begriff ist in Genexpressionsgruppen für OVCA angereichert, wo es ein gut untersuchter Schichtungsfaktor ist.
Ergebnisse
Die Suche nach dem Gen 'BRCA1' im Lungenkrebs-Datensatz zeigt, dass es am stärksten mit dem CorEx-Faktor 26 in Verbindung gebracht wird (Abbildung 2). Go Begriff Anreicherung für diesen Faktor wird als extrem hoch angesehen, mit DNA-Reparatur zeigt eine FDR von nur 1 x 10-19. Die Auswahl lenkt die Aufmerksamkeit auch auf den Cluster der zweiten Ebene L2_8, der sechs eng verwandte Faktoren als untergeordnete Faktoren hat. Die Auswahl von "DNA-Reparatur" in den GO-Term-Anmerkungen oder der GO-angereicherten Dropdown-Liste des Faktordiagramms hebt die zugehörigen Gene in jedem der Faktoren hervor, wobei der Faktor 26 mit Abstand die meisten hat, wie erwartet11. Das Protein-Protein-Interaktionsnetzwerk ist stark vernetzt und unterstützt die eng verknüpfte Funktionalität der Gene in Faktor 26. Das zugehörige Überlebensdiagramm deutet auf einen möglichen Zusammenhang mit dem Überleben der Patienten hin, aber dies müsste in einem größeren Datensatz bestätigt werden.
Beginnend mit dem Überleben kann die Zerlegung von Gründen für ein verbessertes Überleben im Zusammenhang mit bestimmten Genexpressionsgruppen ermöglichen. Als Beispiel wird der Top-Faktor, der das Überleben von Eierstockkrebs beeinflusst, als Nummer 39 angesehen, die stark für Gene angereichert ist, die mit dem Immunsystem assoziiert sind (Abbildung 3). Fünf weitere Faktoren, die mit dem gleichen Level-2-Knoten verbunden sind, sind ebenfalls als immunbezogen angegeben, jedoch scheinen die Überlebensauswirkungen unter ihnen stark variabel zu sein, wobei 39 der höchste und 52 der niedrigste ist. Das Hinzufügen eines Protein-Protein-Interaktionsfensters für einen Faktor zeigt das unmittelbare Interaktionsnetzwerk und ermöglicht eine Verknüpfung mit der StringDB12-Website, um verschiedene Anreicherungen für die PPI-Netzwerkgene abzufragen. Wenn man dies wiederum für jeden der L2_14-Faktoren tut, stellt man fest, dass StringDB-Anreicherungen für die PPI-Netzwerkgene die folgende mögliche Erklärung für die Assoziationen mit dem Überleben nahelegen. Faktor 32 enthält Gene, die den Haupt-Histokompatibilitätskomplex (MHC) der Klasse I bilden, der von zytotoxischen T-Lymphozyten erkannt wird. Faktor 39 entspricht der Zytokinsignalisierung und CXCR3-Rezeptorbindung, bezogen auf CD8+ T-Lymphozyten. Beide Faktoren scheinen Patienten mit relativ hoher Expression der entsprechenden Gene einen signifikanten Überlebensvorteil zu verschaffen. Zytotoxische CD8+ T-Lymphozyten sind in erster Linie für die Anti-Tumor-Immunität verantwortlich. Faktor 52 hingegen besteht aus Genen, die für Proteine im MHC-Komplex der Klasse II kodieren und vor allem von CD4+ T-Helferzellen und nicht direkt durch zytotoxische T-Lymphozyten erkannt werden. Die verbleibenden L2_14-Faktoren spiegeln die allgemeine Aktivierung des Immunsystems wider, die die beiden Arten von Lymphozytenpopulationen nicht unterscheidet. Eine Überlebensassoziation spezifisch für zytotoxische T-Lymphozyten-Erkennung von MCH-Klasse I-Zellantigenen steht im Einklang mit unserem Verständnis der Antitumorimmunität im Allgemeinen und von anderen Krebsarten wie Melanom13,14.
Das Webportal unterstützt die Entdeckung von Faktorenpaaren mit komplementären Funktionen, die auf effektive tumorspezifische Kombinationstherapien hindeuten können. Die Datensatzübersicht kann auf Faktoren gescannt werden, die eine Korrelation mit dem Überleben aufweisen, aber unterschiedliche GO-Anreicherungen aufweisen. Bei Melanomen (TCGA_SKCM; Abbildung 4), es wird gesehen, dass der obere Überlebensfaktor 171 immunbedingt ist, während Faktor 88 unten in der Liste eine Anreicherung für Gene im Zusammenhang mit mit mitochondrion Organisation zeigt. In der Tat wurde dies als Ziel bei Melanom15vorgeschlagen. Das Hinzufügen von Überlebensfenstern zur CorExplorer-Seite ermöglicht den Vergleich der Schichtung mit dem Faktorpaar mit dem jedes Faktors einzeln, was zeigt, dass günstige Genexpressionsmuster aus beiden Gruppen einen besseren Überlebenstrend aufweisen als der für eine der beiden Faktor allein. Die oberste Schicht scheint jedoch nicht verbessert zu werden, was darauf hindeutet, dass eine Immuntherapie nur für einige Patienten die beste Option sein kann.
Gemeinsamkeiten und Unterschiede zwischen Tumoren können durch die Suche in Datensätzen nach Genen oder GO-Begriffen (Abbildung 5) gesehen werden. Als Beispiel ist FLT1 (aka VEGFR1) ein gut studierter pro-angiogener Marker16,17. Wenn es in die Suchleiste gelegt wird, haben alle Tumoren Faktoren, bei denen FLT1 eine wichtige Rolle spielt. Umgekehrt erscheinen bei der Eingabe des GO-Begriffs "Angiogenese" auf der Suchseite 5 von 6 der FLT1-Gruppen mit dieser Bereicherung. Alle FLT1-Faktoren, mit Ausnahme von SKCM-195, werden als statistisch angereichert für "Angiogenese"-Gene aufgeführt. Der sechste Faktor hat in der Tat die Anmerkung, aber unterhalb der Standardschwelle 10-8. Wenn die Gewichtung innerhalb der Faktorliste in einem alternativen Anreicherungsrechner verwendet wird, z.B. Gene Set Enrichment Analysis (GSEA)18, wird festgestellt, dass der sechste Faktor auch für "Angiogenese"-Gene signifikant angereichert ist.
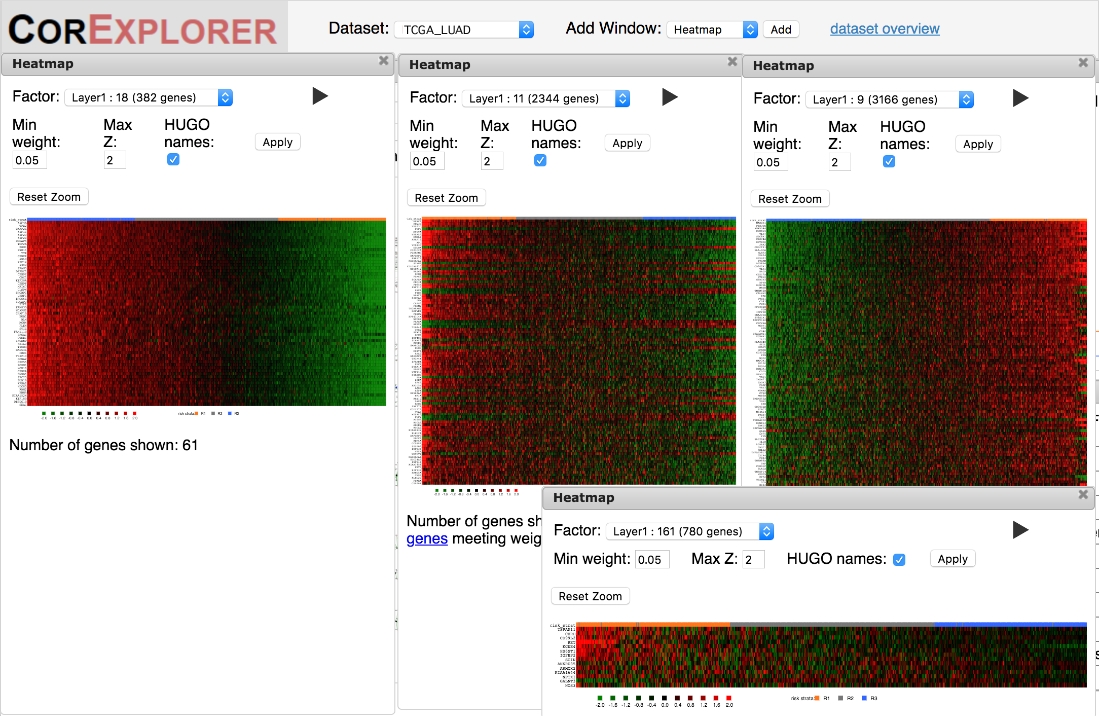
Es ist wichtig, die Heatmaps zu überprüfen, um sicherzustellen, dass das Genexpressionsmuster von ausreichender Qualität ist, um biologische Interpretationen zu unterstützen. Heatmaps, die eine starke klare Variation aufweisen, können entweder eine koordinierte Expression der Faktorgene aufweisen, die von niedrigen bis hohen oder komplexeren Mustern reichen, wobei einige Gene eine niedrige Expression aufweisen, die mit anderen mit hoher Korreliert sind (Abbildung 6). Ein wichtiger Marker einer qualitativ hochwertigen Gruppierung ist das Vorhandensein mehrerer Gene mit einer glatten Variation der Expression als Funktion der Faktorpunktzahl. Die Faktor-Heatmaps zeigen Nach Faktor-Score geordnete Stichproben an, daher sollte ein glatter Farbverlauf von links nach rechts bewegt werden. Dies kann jedoch auf mindestens zwei verschiedene Arten fehlschlagen. Am häufigsten können die Korrelationen extrem laut sein (Abbildung 5C), was die Robustheit und nützlichheit von Rückschlüssen auf das Überleben und/oder die biologische Funktion in Frage stellt. Außerdem entsprechen Muster, die nur in einer kleinen Minderheit von Stichproben vorkommen, möglicherweise nicht dem Modell von drei Ausdruckszuständen, die vom CorEx-Algorithmus angenommen werden, was zu einer irreführenden Klassifizierung der Stichproben führt (rechte Seite von Abbildung 5D).

Abbildung 1: CorExplorer-Titelseite. Nachdem Sie unter Quick Linksauf + neben Ovarian Cancer geklickt haben, werden die Faktordiagrammdetails angezeigt. Das hierarchische CorEx-Modell besteht aus Eingabevariablen (in diesem Fall Genexpression) auf der unteren Ebene und abgeleiteten latenten Faktoren in den höheren Schichten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Verwendung eines Gennamens als Leitfaden für die Exploration. Die Abbildung zeigt eine Reihe von Screenshots, die die Erforschung von CorEx-Lungenkrebsfaktoren veranschaulichen, die stark mit BRCA1 zusammenhängen. Wenn Sie zunächst im Dropdown-Feld "Gene" für das Faktordiagramm "BRCA1" auswählen, vergrößert die Diagrammansicht den Faktor, für den BRCA1 das größte Gewicht hat. Das Verkleinern eines Bits umrahmt den Layer zwei Knoten L2_8, der diesen Faktor mit anderen verwandten verbindet. Überleben und Anmerkungen können verglichen werden: Ein Klick auf den GO-Begriff DNA-Reparatur hebt kommentierte Gene hervor. Ein PPI-Fenster wird hinzugefügt, um die Netzwerkinteraktionen für Gene im Faktor anzuzeigen. Die Verwendung der Schaltfläche Fenster hinzufügen, um eine Heatmap hinzuzufügen, zeigt die Assoziation von Expressionsmustern mit dem Überleben, was darauf hindeutet, dass eine erhöhte Expression von DNA-Reparaturgenen mit einem verminderten Überleben verbunden sein kann. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Verwendung klinischer Daten (Überleben) als Richtschnur für die Exploration. Die Erforschung des top-überlebensassoziierten Faktors (39) für Eierstockkrebs zeigt interessante Zusammenhänge zwischen benachbarten Faktoren. Nach der Auswahl von Faktor 39 im Faktordiagramm und dem Verkleinern eines Bits wird der Layer-Zwei-Faktor, der mit Faktor 39 verknüpft ist, fünf weitere zugeordnete Faktoren haben. Ein zusätzliches Überlebensfenster ermöglicht einen direkten Vergleich der damit verbundenen Überlebensunterschiede. Die Faktoren 39 und 32 weisen beide eine positive Überlebenskorrelation auf, im Gegensatz zu Faktor 52, was nicht der Fall ist. Die Protein-Protein-Interaktionsnetzwerke sind alle gut definiert. Die Verknüpfung mit StringDB ermöglicht den Vergleich der GO-Anmerkungen (nicht dargestellt): Faktor 39 ist mit einem Zytokin-Signalnetzwerk im Zusammenhang mit zytotoxischer CD8+ T-Lymphozytenaktivierung verbunden und Faktor 32 wird von MHC-Antigen der Klasse I dominiert, das Proteine darstellt, die Proteine präsentieren, die Proteine präsentieren, die Triggererkennung durch solche Lymphozyten; Die benachbarten Faktoren werden jedoch von anderen Komponenten des Immunsystems wie CD4+ Helfer-T-Zellen dominiert und zeigen keine Überlebenskorrelation. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Die Untersuchung der oberen Überlebensfaktoren deutet auf mögliche therapeutische Kombinationen hin. Der Link "Datensätze" in der Menüleiste der Startseite führt zu einer knappen Tabelle mit Überlebensfaktoren, die nach p-Wert sortiert sind, zusammen mit der obersten GO-Anmerkung (nicht angezeigt). Unter Verwendung dieser Informationen für Melanome erscheint die Kombination von Faktor 171 für die Immunfunktion mit Faktor 88 für die Mitochondrion-Organisation komplementär. Die Abbildung zeigt Beschriftungsfenster für jeden der Faktoren nebeneinander, um sie zu kontrastieren. Überlebenskurven für Patienten, die einzeln oder zusammen durch die beiden Faktoren geschichtet werden, deuten darauf hin, dass die Kombination das Überlebensgefälle im Vergleich zu einem faktor nur erhöht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Die Suchseite erleichtert die Pan-Krebs-Analyse. Gene oder GO biologische Prozessbegriffe können über den Suchlink von der Homepage über alle Datensätze hinweg gesucht werden. Die Abbildung zeigt Suchergebnisse für das Gen FLT1 und den GO-Begriff "Angiogenese". Die Ergebnisse zeigen das Vorhandensein von FLT1 in Faktoren, die mit dem Begriff "Angiogenese" bei Krebserkrankungen mit Anmerkungen beschriftet sind. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Heatmaps können verwendet werden, um Korrelationen zwischen Genen und Proben qualitativ nach Faktor-Score zu bewerten. Qualitativ hochwertige Genexpressionsbeziehungen werden durch eine glatte Abstufung angezeigt, wenn Patienten in den Heatmaps nach Faktor-Score sortiert werden. Die linke Heatmap für Faktor 18 ist ein Beispiel. Die Muster können auch komplexe Signaturen des Auf- und Ab-Ausdrucks wie in der mittleren großen Heatmap für Faktor 11 umfassen. Niedrigere Qualitätsmuster zeigen manchmal abrupte Veränderungen im Ausdruck für eine Untergruppe von Patienten wie in der Faktor 9 Heatmap auf der rechten seite oder einfache sehr laute Korrelationen wie in der Faktor 161 Heatmap unten rechts. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Wir haben die CorExplorer-Website vorgestellt, einen öffentlich zugänglichen Webserver zur interaktiven Erforschung von maximal korrelierten Genexpressionsfaktoren, die vom CorEx-Algorithmus aus Tumor-RNA-seq gelernt wurden. Wir haben gezeigt, wie die Website verwendet werden kann, um Patienten entsprechend der Tumorgenexpression zu schichten, und wie eine solche Schichtung der biologischen Funktion und dem Überleben entspricht.
Andere Webserver für die RNA-Seq-Analyse wurden gebaut. Differential- und Co-Expressionsanalyse für Tumoren kann untersucht und mit anderen Datentypen in cbioPortal19,20integriert werden. Die Server GenePattern21, Mev22und Morpheus23enthalten etablierte Clustering-Techniken wie PcA (Principal Component Analysis), kmeans oder self-organizing maps (SOMs). Zu den innovativeren Bemühungen gehören CamurWeb24, basierend auf einem automatisierten regelerzeugenden Klassifikatoren, und TACCO25, das zufällige Waldklassifikatoren und Lassos implementiert. Der hier verwendete CorEx-Algorithmus optimiert multivariate Informationen, um eine Hierarchie von Faktoren zu finden, die Muster in Daten erklären. Das nichtlineare und hierarchische Faktorlernen scheint eine verbesserte Interpretationsfähigkeit im Vergleich zu den linearen globalen Faktoren zu erzielen, die über PCA4gefunden werden. Darüber hinaus ermöglicht die feinkörnige Analyse von Probensignalen präzise Tumorvergleiche gegenüber häufiger verwendeten breiten Subtypen. Diese Kombination aus überlappender und hierarchischer Faktoranalyse unterscheidet den CorExplorer von den meisten anderen Ansätzen und erfordert neue Werkzeuge für die Visualisierung und Zusammenfassung.
Ein kritischer Teil der CorExplorer-Faktoranalyse ist die Fähigkeit, nicht nur mehrere, sondern auch über 100 Faktoren mit informativen Genmustern zu untersuchen, die innerhalb einer sich überlappenden Hierarchie platziert sind. Der CorExplorer erleichtert den Abbau dieser unzähligen Faktoren für biologische und klinische Assoziationen und ermöglicht eine außergewöhnlich detaillierte Charakterisierung einzelner Tumoren. Das unbeaufsichtigte Erlernen einer so großen Anzahl von Faktoren bedeutet, dass nicht alle für die Krankheitsbiologie relevant sein werden. In einem solchen Fall ist es wichtig, entweder Anmerkungen oder bekannte Gene zu verwenden, um Interessensfaktoren herauszuziehen oder nach Faktoren zu suchen, die mit klinischen Daten wie dem Überleben in Verbindung stehen. Somit ermöglicht der CorExplorer Benutzern, diesen sehr wichtigen Filterschritt zu implementieren. Das Vorhandensein von Faktor-Gen-Mustern in einem Tumor kann sogar einen Ansatz für eine personalisierte onkologische Behandlung nahelegen. Darüber hinaus die Vielzahl der Faktor-Scores für jeden Tumor, die für die Entdeckung von potenziell nützlichen therapeutischen Kombinationen ermöglicht.
Es ist manchmal der Fall, dass keine signifikanten GO-Anmerkungen für Faktoren erscheinen, die stark mit dem Überleben korrelieren. Während dies aufgrund von lauten oder unter Stichproben datenreichen Daten auftreten kann, gibt es andere mögliche Ursachen, wie z. B. eine Clustergröße, die zu klein ist, um signifikante Anreicherungswerte zu registrieren, oder die Gruppe, die ein "Korb" einzelner Gene aus verschiedenen Pfaden ohne kohärente biologische Verband. Darüber hinaus kann eine Kategorie von Anmerkungen, die sich vom biologischen Prozess KEGG und GO unterscheiden, z. B. Zellkompartimt, angemessen sein. Auf diese kann zugegriffen werden, indem sie mit StringDB verknüpft werden, wie im Protokoll gezeigt. Die Gene Ontology Anreicherungsanalyse auf der CorExplorer-Website berücksichtigt derzeit nicht die Gengewichtung in einem Faktor, obwohl dies wahrscheinlich in naher Zukunft behoben werden wird. Beachten Sie, dass unter "Fenster hinzufügen" eine Genlistenoption verfügbar ist, die den Download der vollständigen Faktor-Gen-Liste für die weitere Analyse mit externen Tools ermöglicht.
Für die Zwecke der Website wurde CorEx für jedes der Datasets fünfmal ausgeführt, und die Ausführung, die zu der größten Gesamtkorrelation führte, wurde beibehalten. Eine statistische Darstellung der Ergebnisse mehrerer Durchläufe kann informativer sein und ist ein Ziel für die zukünftige Arbeit. Darüber hinaus ist der Satz von Tumortypen, die auf dem Server verfügbar sind, eher klein, aber wir erwarten, dass sich dies im Laufe der Zeit entsprechend dem Benutzerinteresse ausdehnt.
Wie oben beschrieben, visualisiert der CorExplorer CorEx RNA-seq-Faktor-Beziehungen zusammen mit klinischen und Datenbankinformationen und ermöglicht so eine Vielzahl verschiedener Verhörmodi. Wir hoffen, dass dieses Tool zu weiteren Arbeiten führen wird, um die Leistungsfähigkeit der RNA-Seq-Analyse für die Entdeckung und klinische Anwendung in der Onkologie zu nutzen.
Offenlegungen
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Danksagungen
GV wurde durch den DARPA Award W911NF-16-0575 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Public server for CorExplorer website | USC | http://corex.isi.edu | Intel Xeon E5-2690 4-core 2.6 GHz, 8GB RAM. Backend architecture is LAMP: Linux, Apache, MySQL, PHP. |
| Web browser | Google/Apple | Chrome/Safari | Verified web browsers. |
Referenzen
- Petryszak, R., et al. The RNASeq-er API-a gateway to systematically updated analysis of public RNA-seq data. Bioinformatics. 33, 2218-2220 (2017).
- Steeg, G. V., Galstyan, A. Maximally Informative Hierarchical Representations of High-Dimensional Data. Proceedings of the Eighteenth International Conference on Artificial Intelligence and Statistics (AISTATS). , (2015).
- Ver Steeg, G., Galstyan, A. Discovering structure in high-dimensional data through correlation explanation. Advances in Neural Information Processing Systems. , (2014).
- Pepke, S., Ver Steeg, G. Comprehensive discovery of subsample gene expression components by information explanation: therapeutic implications in cancer. BMC medical Genomics. 10, 12 (2017).
- Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D., Craig, D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature Reviews Genetics. 17, 257 (2016).
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511, 543 (2014).
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330 (2012).
- Akbani, R., et al. Genomic classification of cutaneous melanoma. Cell. 161, 1681-1696 (2015).
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609 (2011).
- Grossman, R. L., et al. Toward a shared vision for cancer genomic data. New England Journal of Medicine. 375, 1109-1112 (2016).
- Moynahan, M. E., Chiu, J. W., Koller, B. H., Jasin, M. Brca1 controls homology-directed DNA repair. Molecular Cell. 4, 511-518 (1999).
- Szklarczyk, D., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 47, 607-613 (2018).
- Durgeau, A., Virk, Y., Corgnac, S., Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Frontiers in Immunology. 9, 14 (2018).
- Sato, E., et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 102, 18538-18543 (2005).
- De Moura, M. B., et al. Mitochondrial respiration-an important therapeutic target in melanoma. PLoS One. 7, 40690 (2012).
- Folkman, J., Merler, E., Abernathy, C., Williams, G. Isolation of a tumor factor responsible for angiogenesis. Journal of Experimental Medicine. 133, 275-288 (1971).
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biological and Pharmaceutical Bulletin. 34, 1785-1788 (2011).
- Subramanian, A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 102, 15545-15550 (2005).
- Cerami, E., et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2, 401-404 (2012).
- Gao, J., et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signalling. 6, 1 (2013).
- Reich, M., et al. GenePattern 2.0. Nature Genetics. 38, 500 (2006).
- Wang, Y. E., Kutnetsov, L., Partensky, A., Farid, J., Quackenbush, J. WebMeV: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. Cancer Research. 77, 11-14 (2017).
- . Morpheus Available from: https://software.broadinstitute.org/morpheus (2019)
- Weitschek, E., Lauro, S. D., Cappelli, E., Bertolazzi, P., Felici, G. CamurWeb: a classification software and a large knowledge base for gene expression data of cancer. BMC Bioinformatics. 19, 354 (2018).
- Chou, P. -. H., et al. tACCo, a Database Connecting transcriptome Alterations, pathway Alterations and Clinical outcomes in Cancers. Scientific Reports. 9, 3877 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten