
Method Article
Analisando fatores de expressão gênica do tumor com o portal da Web CorExplorer
Neste Artigo
Resumo
Apresentamos o portal Web CorExplorer, um recurso para a exploração de fatores de sequenciamento de RNA tumoral encontrados pelo algoritmo de aprendizado de máquina CorEx (explicação de correlação), e mostrar como os fatores podem ser analisados em relação à sobrevivência, anotações de banco de dados, interações proteína-proteína, e um outro para ganhar a introspecção na biologia do tumor e em intervenções terapêuticas.
Resumo
A análise diferencial da expressão gênica é uma técnica importante para a compreensão dos Estados da doença. O algoritmo de aprendizado de máquina CorEx mostrou utilidade na análise da expressão diferencial de grupos de genes no tumor RNA-Seq de uma forma que pode ser útil para o avanço da Oncologia de precisão. No entanto, o CorEx produz muitos fatores que podem ser desafiadores para analisar e conectar-se à compreensão existente. Para facilitar tais conexões, construímos um site, CorExplorer, que permite aos usuários explorar interativamente os dados e responder a perguntas comuns relacionadas à sua análise. Nós treinamos CorEx em dados da expressão de gene do RNA-Seq para quatro tipos do tumor: ovariano, pulmão, melanoma, e colorectal. Nós incorporamos então a sobrevivência correspondente, interações da proteína-proteína, Ontology do gene (GO) e a enciclopédia de Kyoto de genes e genomas (KEGG) enriquments da via, e Heatmaps no Web site para a associação com a visualização do gráfico do fator. Aqui nós empregamos protocolos do exemplo para ilustrar o uso da base de dados para compreender o significado dos fatores de tumor aprendidos no contexto destes dados externos.
Introdução
Desde sua introdução pouco mais de uma década atrás, RNA-Seq tornou-se uma ferramenta onipresente para medir a expressão gênica1. Isto é porque permite o perfilamento rápido e barato de de novo do transcriptoma inteiro de uma amostra. Entretanto, os dados do tumor de RNA-Seq refletem uma biologia subjacente que seja intrinsecamente complexa e frequentemente sob-amostrado, quando os dados próprios forem elevado-dimensionais e ruidosos. Isso representa um desafio significativo para a extração de sinais confiáveis. O algoritmo Corex aproveita as informações mútuas multivariadas para encontrar padrões sutis em tais situações2,3. Esta técnica foi previamente adaptada para analisar amostras de RNA-Seq do tumor ovariano do Atlas do genoma do cancro (TCGA) e neste contexto pareceu ter vantagens significativas sobre métodos de análise mais comumente usados4.
Embora o uso de RNA-Seq seja enormemente generalizado em aplicações de pesquisa, inclusive em Oncologia, esses esforços não levaram a uma ampla utilização para fins de intervenções clínicas5. Parte da razão para isso é a falta de algoritmos de fácil utilização e software direcionados a esses problemas específicos. Para ajudar a preencher essa lacuna, projetamos o portal da Web do CorExplorer para permitir que pesquisadores de uma variedade de origens estudem fatores de expressão gênica de amostras de RNA-Seq tumorais, conforme encontrado pelo algoritmo de aprendizado de máquina CorEx. O portal corexplorer suporta visualização interativa e consulta de fatores de vários tipos de tumores diferentes, incluindo pulmão, cólon, melanoma e ovariano6,7,8,9, 10, com a intenção de ajudar os pesquisadores a peneirar as correlações de dados e identificar as vias candidatas para estratificar os pacientes para fins terapêuticos.
Esperamos que o portal CorExplorer pode ser útil para vários tipos de usuários. O portal foi projetado com o usuário em mente que deseja compreender os fatores amplos que impulsionam as diferenças de expressão gênica tumoral em bases de dados públicas e possivelmente também colocam perfis individuais de expressão gênica no contexto de tumores com semelhante Características. Além dos protocolos representativos aqui descritos, as investigações do CorExplorer podem servir como ponto de partida para sugerir hipóteses para testes posteriores, comparar e contrastar os achados do CorEx em conjuntos de dados fora do CorExplorer e conectar assinaturas da expressão patológica de um ou de alguns genes em um tumor individual aos grupos maiores que podem ser afetados coordenadamente. Finalmente, pode servir como uma introdução fácil de usar para a aplicação do aprendizado de máquina ao RNA-Seq para aqueles que começ começados no campo.
Protocolo
1. explorando fatores que contenham um gene de interesse
- Abra um navegador da Web e vá para http://corex.isi.edu, a página inicial do CorExplorer.
- No lado direito em links rápidos, clique no botão + Expand ao lado de ovariano (tcga-OV) para ver um resumo do gráfico do fator Corex que foi treinado nos dados do câncer de ovário tcga (mostrado na Figura 1). Opcionalmente, clique em outros para comparar.
- Uma vez terminado inspecionando os gráficos de fatores, clique emPulmão (TCGA-LUAD)para acessar a página CorExplorer para RNA-Seq de câncer de pulmão.
- Explore o gráfico do fator CorEx para um gene de interesse usando a janela CorExplorer ' factor Graph '.
- Mova o cursor do mouse sobre a janela de exibição do gráfico de fatores. Aumente o zoom no gráfico de fatores usando a roda de rolagem do mouse ou o trackpad para ver detalhes do gráfico, como os genes mais importantes em cada fator e as conexões entre nós em diferentes camadas. Como alternativa, clique e arraste para mover a área de exibição ou qualquer nó.
- Para encontrar um gene alvo (aqui vamos usar BRCA1), clique no menu drop-down gene na parte superior da janela do gráfico do fator. Digite ' BRCA1 ' para selecioná-lo na lista suspensa e pressione Return para fazer o zoom da vista para o fator 26, o fator com o qual BRCA1 está mais fortemente correlacionado.
- Reposicione o mouse sobre a exibição do gráfico e role para reduzir para ver o nó de nível 2, L2_8 e seus fatores associados que são vizinhos ao fator 26. Observe que somente genes com peso maior que o limiar indicado no controle deslizante de peso do link mínimo são mostrados.
- Para ver todos os genes associados ao fator, clique no nó L1_26 e selecione carregar genes adicionais na janela pop-up. Quando a palavra ' done ' for exibida, feche a janela pop-up.
- Agora volte para a seção de cabeçalho acima da janela do gráfico do fator e agarre e arraste o modificador de peso de link min . Agora, como o controle deslizante de peso do link é movido para baixo para 0, 5, outros genes no fator L1_26, incluindo BRCA2, aparecerá na ordem de peso. Opcionalmente, reposicione os nós agarrando e arrastando para melhorar o layout.
- Determinar como a estratificação dos pacientes em relação ao fator afeta a sobrevida por meio da consulta na janela de sobrevida.
- Na janela de sobrevivência, desmarque ordenar por p-Val, em seguida, selecione o fator 26 no menu suspenso de fator único para mostrar as curvas de sobrevivência para o fator 26.
- Role para baixo o gráfico de sobrevivência para mostrar o número de pacientes em risco ao longo do eixo x.
- Encontre associações com função biológica consultando dentro da janela anotação.
- Na janela de anotação, para classificar o menu suspenso de fator por número de fator em vez de taxa de descoberta falsa (FDR), desmarque a classificação FDR.
- Role e clique para selecionar o fator 26 no menu suspenso da janela de anotação para mostrar anotações de enriquecimento para o fator.
- Role para baixo a lista de anotações até que o reparo do DNA esteja visível e clique nele para ver imediatamente os genes associados destacados em amarelo na exibição do gráfico. Consulte o painel do meio da Figura 2.
- Observe que os fatores desaparecem ou aparecem como diferentes termos de GO são selecionados, de acordo com se eles são ou não enriquecidos para genes com a anotação selecionada, por exemplo, "via de sinalização apoptótica intrínseca em resposta a danos de DNA."
- Explore os fatores ainda mais adicionando janelas com funcionalidade diferente.
- Na barra de menu superior, adicione uma janela de rede de interação proteína-proteína (PPI) selecionando PPI na lista suspensa Adicionar janela e, em seguida, clique no botão Adicionar para adicionar uma janela de gráfico PPI à área de exibição. Na janela do gráfico PPI, escolha o fator ' Layer1:26 ' para mostrar as interações proteína-proteína. Observe a densidade de conexões.
- Na barra de menus superior, em vez de PPI, selecione heatmap no menu suspenso Adicionar janela e, em seguida, clique no botão Adicionar para adicionar uma janela heatmap à área de exibição. Na janela heatmap, escolha o fator ' Layer1:26 ' para mostrar os padrões de expressão gênica.
- Agarre e reposicione a janela do heatmap de modo que a janela da sobrevivência esteja igualmente visível. Ao longo da parte superior do heatmap, observe como a barra colorida laranja/azul/cinza corresponde aos estratos de risco do paciente no gráfico de sobrevivência. Os resultados são mostrados na parte inferior da Figura 2.
- Explore o gráfico do fator CorEx para um gene de interesse usando a janela CorExplorer ' factor Graph '.
2. filtrando e interpretando fatores CorEx usando dados de peso, sobrevivência e anotação do gene
- Filtre por fatores de interesse usando a sobrevivência e a qualidade do cluster.
- No menu suspenso conjunto de dados na parte superior, selecione TCGA_OVCA para ir para a página corexplorer para o RNA-Seq de câncer de ovário tcga.
- Uma vez que a página foi carregada, note a partir da janela de sobrevivência que o fator com o maior diferencial de sobrevivência para diferentes estratos é 114.
- Na parte superior da janela do gráfico de fatores, selecione ' Layer1:114 ' na lista suspensa fator .
- Agarre o slider do peso da ligação com o rato e mova-o até 0,5. Observe que o grande número de genes no fator 114 (1609), com nenhum peso > 0,35, indica um agrupamento relativamente fraco.
- Em seguida, expanda a lista de fatores na janela de sobrevivência e selecione o próximo melhor fator no menu suspenso da janela de sobrevivência, fator 39, para mostrar suas curvas de sobrevivência associadas.
- Selecione o fator 39 na janela de anotação clicando nele. As anotações significativas GO e KEGG são mostradas.
- Para obter uma melhor compreensão do papel biológico dos genes no fator 39, interpretar os fatores usando informações de anotação de vizinhança da seguinte maneira.
- Na parte superior da janela do gráfico de fatores, selecione o fator ' Layer1:39 ' na lista suspensa de fatores. Em seguida, mova o mouse sobre a janela do gráfico do fator e aumente o zoom para revelar todo o cluster L2_14 com 6 fatores: 14, 32, 39, 42, 52 e 82 (mostrado na Figura 3).
- Para entender a significância relativa dos fatores vinculados ao nó L2_14, comece exibindo diferenciais de sobrevida para cada um dos fatores L2_14. Desmarque ordenar por p-Val na janela de sobrevivência e, em seguida, clique em cada um dos números de fator em sucessão. Fazendo isso, observe que apenas os fatores 14, 32 e 39 exibem uma associação de sobrevivência.
- Agora a partir da barra de menu superior, selecione PPI da janela Adicionar DropDown mais uma vez. Pressione Adicionar para adicionar uma janela de gráfico PPI à área de exibição. Na janela do gráfico PPI, selecione o fator ' Layer1:52 ' para mostrar as interações proteína-proteína que são significativas. Um layout de exemplo do Windows neste ponto é mostrado na Figura 3.
- Clique no link Exibir no StringDB na parte inferior da janela PPI para vincular o banco de dados online do stringdb. Clique em continuar a partir da primeira tela e, em seguida, selecione a guia análise abaixo do gráfico de rede como antes para obter uma análise go on-line para os genes da rede PPI. O principal componente celular é o complexo proteico da classe II de MHC.
- Retorne à aba de CorExplorer e à janela de PPI e selecione o fator 32, esta vez da lista suspensa do fator. Clique no link View em stringdb out para a análise stringdb. O principal componente celular é ' MHC classe I complexo proteico, ' em contraste com a classe II para o fator 52 no passo anterior!
- Finalmente, volte para a janela PPI e selecione ' Layer1:39 ' no menu suspenso do fator na parte superior. Clique na exibição de link em stringdb para vincular a análise de stringdb.
- Clique em continuar a partir da primeira tela e, em seguida, selecione a guia análise abaixo do gráfico de rede para obter uma análise go on-line para os genes da rede PPI. Observe que a função molecular superior é ' CXCR3 ligação do receptor de quimiocina '.
3. usando as anotações de sobrevivência e banco de dados para procurar combinações terapêuticas promissoras
- Alterne para o melanoma TCGA CorExplorer selecionando TCGA_SKCM no menu suspenso conjunto de dados .
- Observe que o fator com o maior diferencial de sobrevida é o fator 171. Examine as anotações do fator 171 rolando e observe que a "resposta imune" e a "via de sinalização mediada por citocinas" estão perto do topo (como eram para o fator ovariano superior).
- Para encontrar um fator complementar, examine os principais fatores associados à sobrevivência, juntamente com seus termos de anotação superior. Para fazer isso, clique no link visão geral do conjunto de dados na barra de menu superior para abrir uma guia separada contendo uma tabela com detalhes de processamento de conjunto de dados, bem como um resumo dos principais fatores de acordo com o valor de p do diferencial de sobrevivência. Note que o primeiro fator não imune é 88.
- Retorne à aba do navegador TCGA_SKCM .
- Selecione o fator 88 nas janelas de sobrevivência, anotação e gráfico. Os vários termos GO estão relacionados ao ' processamento de rRNA ' e ' organização das mitocrodrias ', confirmando-o como distinto dos fatores imunes.
- Na janela de sobrevivência, na lista suspensa de fatores emparelhados, selecione ' 88_171 ' para ver como a sobrevida é melhorada para pacientes no estrato médio para os fatores de expressão combinados 171 e 88. As comparações de anotação e sobrevivência são ilustradas na Figura 4.
4. encontrando semelhanças e diferenças de variação da expressão gênica entre tipos de tumores usando a página de pesquisa
- Clique no cabeçalho Corexplorer para regressar à página inicial.
- Clique em Pesquisar na barra de menu superior para ir para uma página que permite pesquisar sobre todos os conjuntos de dados no site do corexplorer.
- Na caixa de pesquisa do gene , digite ' FLT1 ' (VEGFR1) e aperte Return ou pressione Search. FLT1 é encontrado com um peso relativamente alto nos seguintes fatores: OVCA-76, LUAD-162, SKCM-195 e SKCM-184, bem como COAD-112 e COAD-74.
- Como alternativa, procure um termo GO relacionado em todos os conjuntos de dados. Tente isso na caixa ' GO Search ' digitando ' angiogênese ' e batendo Return ou pressionando Search. Todos os fatores FLT1, com exceção do SKCM-195, são listados como estatisticamente enriquecidos para genes de ' angiogênese ' – o fator 195, na verdade, tem a anotação, mas abaixo do limiar 10-8 padrão. Os resultados da pesquisa para este e o passo anterior são mostrados na Figura 5.
- Como outros exemplos, na caixa de pesquisa GO, primeiro digite ' receptor de fator de crescimento epidérmico. ' Somente o LUAD é enriquecido para este termo, um fator de estratificação bem conhecido para o câncer de pulmão. Em seguida, digite ' mesenchymal ' na caixa de pesquisa. Este termo é enriquecido em grupos de expressão gênica para OVCA, onde é um fator de estratificação bem estudado.
Resultados
A busca pelo gene ' BRCA1 ' no conjunto de dados do câncer de pulmão revela-se mais fortemente associada ao fator CorEx 26 (Figura 2). O enriquecimento do termo GO para este fator é visto para ser extremamente elevado, com reparo do ADN que exibe um FDR de somente 1 x 10-19. A seleção também chama a atenção para o cluster de segundo nível L2_8 que tem seis fatores intimamente relacionados como crianças. Selecionando ' reparo de DNA ' em ambas as anotações de termo GO ou o gráfico de fator GO enriquecido DropDown destaca genes associados em cada um dos fatores, com o fator 26 tendo de longe o máximo, como esperado11. A rede da interação proteína-proteína é conectada fortemente, suportando mais a funcionalidade firmemente lig dos genes no fator 26. O gráfico de sobrevida associado sugere uma possível associação com a sobrevida do paciente, mas isso teria que ser confirmado em um conjunto de dados maior.
Começar com a sobrevivência pode permitir a dissecção das razões para a sobrevivência melhorada associada com os grupos particulares da expressão de Gene. Como exemplo, o fator de topo que influencia a sobrevida do câncer de ovário é visto como o número 39, que é fortemente enriquecido para genes associados ao sistema imunológico (Figura 3). Outros cinco fatores associados ao mesmo nó de nível 2 também são indicados para serem imunes, porém o impacto da sobrevida parece ser fortemente variável entre eles, sendo 39 o mais alto e 52 sendo o mais baixo. Adicionar uma janela de interação proteína-proteína para um fator mostra a rede de interação imediata e permite o link para o site StringDB12 para consultar vários enriquments para os genes da rede PPI. Fazendo isto para cada um dos fatores L2_14 por sua vez, se encontra que os enriquments de StringDB para os genes da rede do PPI sugerem a seguinte explanação possível para as associações com sobrevivência. O fator 32 contem os genes que compõem o complexo principal da proteína da classe I do complexo da histocompatibilidade (MHC), que é reconhecido por linfócitos T citotóxica. O fator 39 corresponde à sinalização de citocinas e à ligação do receptor CXCR3, relacionada aos linfócitos T CD8 +. Ambos os fatores parecem conferir uma vantagem significativa de sobrevida para os pacientes que exibem expressão relativamente alta dos genes correspondentes. Os linfócitos T citotóxicos CD8 + são os responsáveis principalmente pela imunidade antitumoral. O fator 52, de um lado, é compreendido dos genes que codificam para proteínas no complexo da classe II de MHC que são reconhecidos primeiramente por pilhas do ajudante de CD4 + t um pouco do que diretamente por linfócitos t citotóxica. Os fatores L2_14 restantes refletem a ativação generalizada do sistema imunitário que não diferencia os dois tipos de populações do linfócito. Uma associação da sobrevivência específica ao reconhecimento citotóxica do linfócito de T de antígenos celulares da classe mim de MCH é consistente com nossa compreensão da imunidade antitumoral geralmente e de outros cancros tais como a melanoma13,14.
O portal da Web suporta a descoberta de pares de fatores com funções complementares que podem sugerir terapias de combinação eficazes de tumores específicos. A visão geral do conjunto de dados pode ser verificada para fatores que mostram uma correlação com a sobrevivência, mas têm enriquments GO distintos. Para melanoma (TCGA_SKCM; Figura 4), vê-se que o fator de sobrevivência superior 171 é imune relacionado, enquanto o fator 88 abaixo da lista mostra o enriquecimento para genes relacionados à organização das mitocódrias. Na verdade, isso tem sido sugerido como um alvo no melanoma15. A adição de janelas de sobrevivência à página CorExplorer permite a comparação da estratificação usando o par de fatores para cada fator individualmente, mostrando que os padrões favoráveis de expressão gênica de ambos os grupos exibem uma tendência de sobrevida melhor do que a de qualquer fator sozinho. O estrato superior não parece ser melhorado entretanto, sugerindo a imunoterapia somente pode ser a melhor opção para alguns pacientes.
As semelhanças e diferenças entre os tumores podem ser vistas pesquisando em conjuntos de dados para genes ou termos GO (Figura 5). Como exemplo, FLT1 (aka VEGFR1) é um marcador pró-angiogênico bem estudado16,17. Quando ele é colocado na barra de pesquisa, todos os tumores têm fatores em que FLT1 desempenha um papel importante. Inversamente, quando o termo GO ' angiogênese ' é entrada na página de pesquisa, 5 de 6 dos grupos FLT1 aparecem com esse enriquecimento. Todos os fatores FLT1, com exceção do SKCM-195, são listados como estatisticamente enriquecidos para genes de ' angiogênese '. O sexto fator, na verdade, tem a anotação, mas abaixo do limite padrão de 10-8. Quando a ponderação dentro da lista de fatores é utilizada em uma calculadora de enriquecimento alternativa, por exemplo, Gene Set enriquecimento análise (GSEA)18, o sexto fator é encontrado para ser significativamente enriquecido para ' angiogênese ' genes também.
É importante verificar os Heatmaps para garantir que o padrão de expressão gênica seja de qualidade adequada para suportar interpretações biológicas. Os Heatmaps que mostram uma forte variação clara podem apresentar expressão coordenada dos genes de fator variando de padrões de baixo a alto ou mais complexos, com alguns genes com baixa expressão correlacionados com outros com alta (Figura 6). Um marcador chave de um agrupamento de alta qualidade é a presença de vários genes com uma variação suave na expressão em função do escore fatorial. Os Heatmaps do fator mostram as amostras ordenadas de acordo com a contagem do fator, assim deve haver um inclinação liso que move-se da esquerda para a direita. No entanto, isso pode deixar de acontecer em pelo menos duas maneiras diferentes. Mais comumente, as correlações podem ser extremamente barulhentas (Figura 5C), questionando a robustez e utilidade de quaisquer inferências quanto à sobrevida e/ou função biológica. Além disso, os padrões que acontecem apenas em uma pequena minoria de amostras podem não estar em conformidade com o modelo de três Estados de expressão assumidos pelo algoritmo CorEx, resultando em uma classificação enganosa das amostras (lado direito da Figura 5D).

Figura 1: Página inicial do CorExplorer. Depois de clicar em + Next para câncer de ovário em links rápidos, detalhes do gráfico de fatores são mostrados. O modelo hierárquico CorEx é composto de variáveis de entrada (expressão gênica neste caso) na camada inferior e inferidos fatores latentes nas camadas superiores. Por favor clique aqui para ver uma versão maior desta figura.
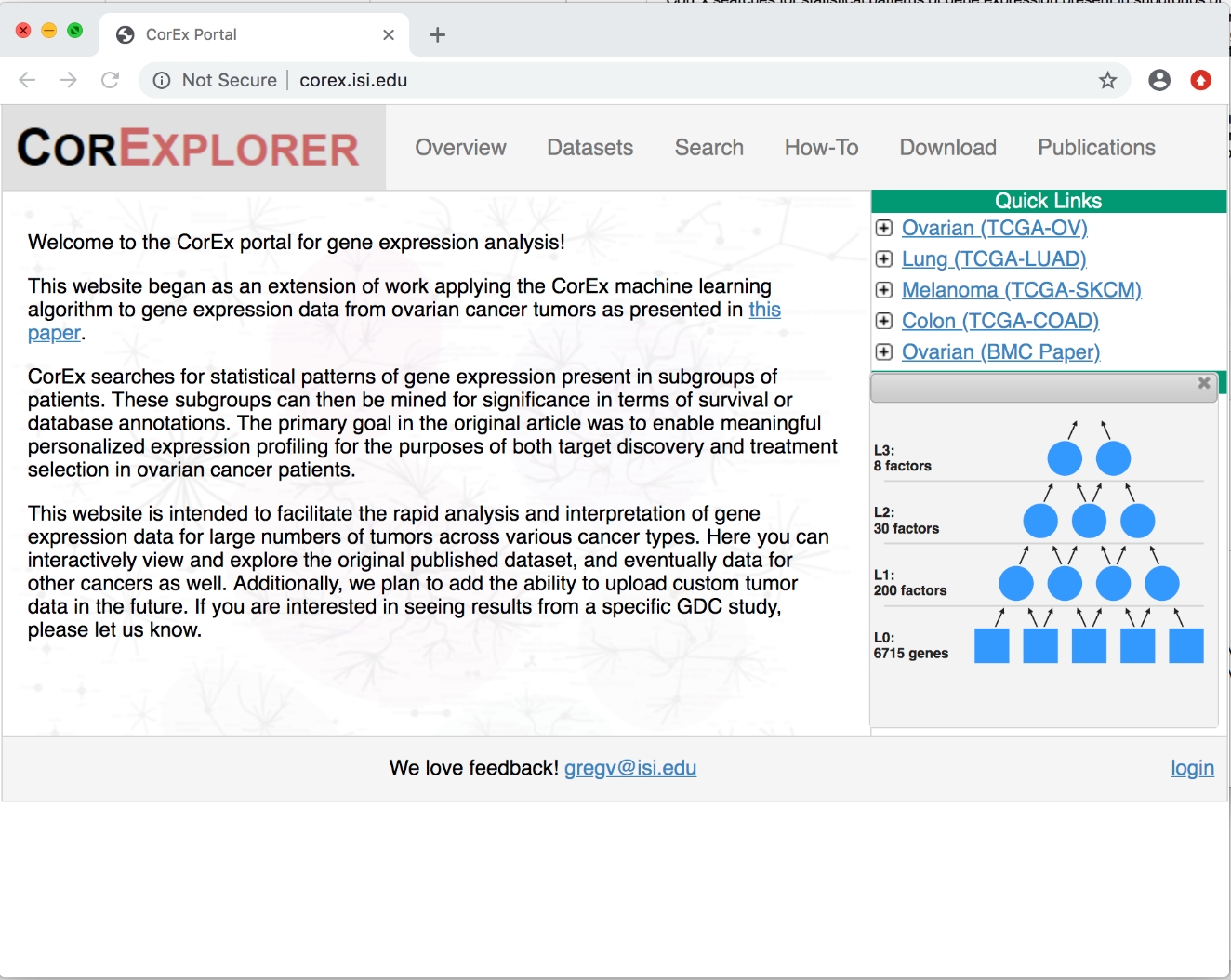{kind=link}

Figura 2: usando um nome de gene para orientar a exploração. A figura mostra uma série de capturas de tela ilustrando a exploração de fatores de câncer de pulmão CorEx fortemente relacionados com BRCA1. Primeiro, selecionando ' BRCA1 ' na caixa suspensa gene para o gráfico de fator faz com que a exibição de gráfico para ampliar o fator para o qual BRCA1 tem maior peso. Ampliar um pouco quadros a camada dois nó L2_8 conectando esse fator para outros relacionados. A sobrevivência e as anotações podem ser comparadas: clicar no termo GO Repair DNA destaca genes anotados. Uma janela PPI é adicionada para mostrar as interações de rede para genes no fator. Usando o botão Adicionar janela para adicionar um mapa de calor mostra Associação de padrões de expressão com a sobrevivência, sugerindo aumento da expressão de genes de reparo de DNA pode estar associada com a diminuição da sobrevida. Por favor clique aqui para ver uma versão maior desta figura.
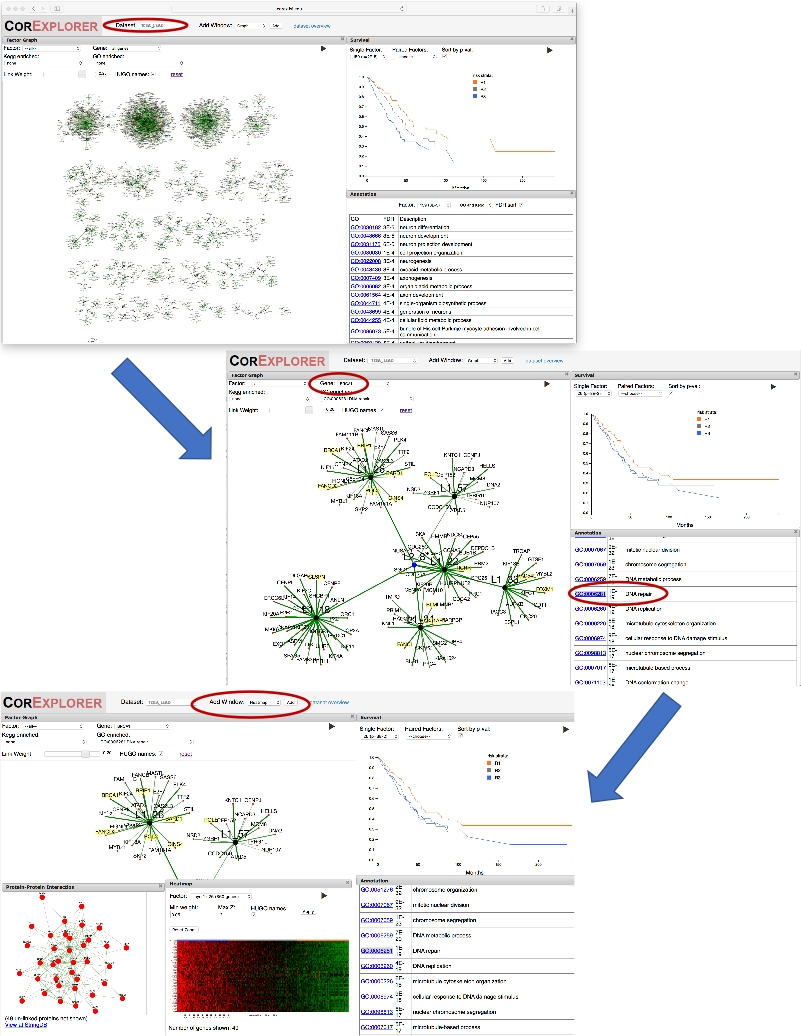{kind=link}

Figura 3: utilização de dados clínicos (sobrevivência) para orientar a exploração. Explorando o fator de sobrevivência-associado superior (39) para o cancro ovariano revela relacionamentos interessantes entre fatores vizinhos. Depois de selecionar o fator 39 no gráfico de fatores e ampliar um pouco, o fator de camada dois vinculado ao fator 39 é visto para ter cinco outros fatores associados. Uma janela de sobrevivência adicional permite a comparação direta dos diferenciais de sobrevida associados. Os fatores 39 e 32 ambos mostram uma correlação positiva de sobrevida, em contraste com o fator 52, o que não. As redes de interação proteína-proteína estão bem definidas. Vincular a StringDB permite a comparação das anotações GO (não mostradas): o fator 39 está associado a uma rede de sinalização de citocinas relacionada à ativação citotóxica de linfócitos T CD8 + e o fator 32 é dominado pelo antígeno de classe I de MHC que apresenta proteínas que desencadear o reconhecimento por tais linfócitos; os fatores vizinhos, entretanto, são dominados por outros componentes do sistema imunitário tais como pilhas de T do ajudante de CD4 + e não mostram nenhuma correlação da sobrevivência. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: explorar os principais fatores de sobrevida sugere possíveis combinações terapêuticas. O link ' conjuntos de dados ' na barra de menu da página inicial leva a uma tabela concisa de fatores de sobrevivência ordenados por valor de p, juntamente com a anotação GO superior (não mostrada). Usando esta informação para o melanoma, a combinação de fator 171 para a função imune com fator 88 para a organização do mitodrion parece complementar. A figura mostra janelas de anotação para cada um dos fatores lado a lado para contrastá-los. Curvas de sobrevida para pacientes estratificados pelos dois fatores individualmente ou em conjunto indicam que a combinação aumenta o diferencial de sobrevida em comparação com qualquer fator isoladamente. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: a página de pesquisa facilita a análise de Pan-cancer. Genes ou termos de processo biológico GO podem ser pesquisados em todos os conjuntos de dados usando o link de pesquisa da página inicial. A figura mostra os resultados da pesquisa para o gene FLT1 e o termo GO ' angiogênese '. Os resultados mostram a presença de FLT1 em fatores anotados com o termo ' angiogênese ' através de cânceres. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: os Heatmaps podem ser utilizados para avaliar qualitativamente as correlações entre genes e amostras de acordo com o escore fatorial. As relações da expressão de gene da alta qualidade são mostradas pela gradação Lisa quando os pacientes são requisitados pela contagem do fator nos Heatmaps. O mapa de calor mais à esquerda para o fator 18 é um exemplo. Os padrões também podem abranger assinaturas complexas de cima e para baixo expressão como no grande heatmap médio para o fator 11. Padrões de qualidade inferiores às vezes mostram mudanças abrupta na expressão para um subgrupo de pacientes como no mapa de calor do fator 9 à direita ou simples correlações muito barulhentas como no fator 161 heatmap no canto inferior direito. Por favor clique aqui para ver uma versão maior desta figura.
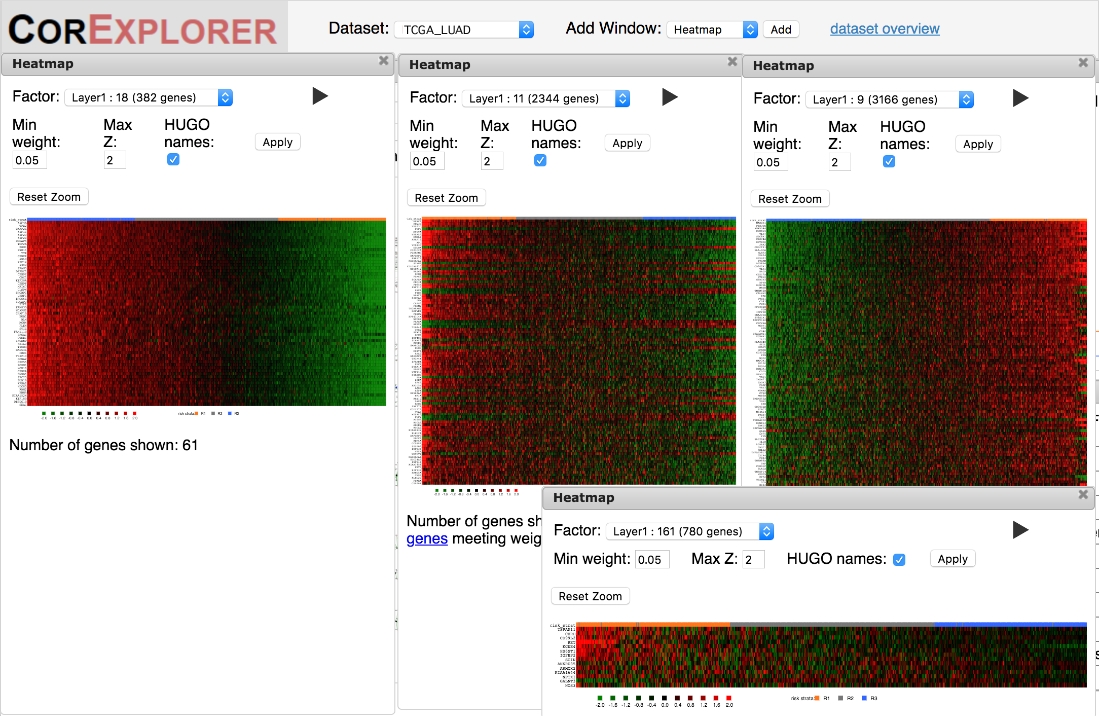{kind=link}
Discussão
Apresentamos o site do CorExplorer, um servidor Web acessível publicamente para exploração interativa de fatores de expressão gênica maximamente correlacionados, aprendidos a partir de RNA-Seq tumoral pelo algoritmo CorEx. Mostramos como o site pode ser usado para estratificar pacientes de acordo com a expressão gênica tumoral, e como tal estratificação corresponde à função biológica e à sobrevida.
Outros servidores Web para análise de RNA-Seq foram construídos. A análise diferencial e de coexpressão para tumores pode ser examinada e integrada com outros tipos de dados no cbioportal19,20. Os servidores GenePattern21, MEV22e Morpheus23, incorporam técnicas de clustering estabelecidas, como análise de componentes principais (PCA), KMeans ou mapas autoorganizantes (Soms). Esforços mais inovadores incluem CamurWeb24, com base em um classificador automatizado de geração de regras, e tacco25, que implementa classificadores de floresta aleatórios e lassos. O algoritmo CorEx usado aqui otimiza as informações multivariadas para encontrar uma hierarquia de fatores que explicam padrões em dados. A aprendizagem fatorial não linear e hierárquica parece produzir uma melhor interpretabilidade em relação aos fatores lineares globais encontrados via PCA4. Adicionalmente, a análise fine-grained da técnica de sinais da amostra permite comparações precisas do tumor vis-à-vis subtypes largos mais geralmente usados. Essa combinação de sobreposição e análise fatorial hierárquica distingue o CorExplorer da maioria das outras abordagens e necessita de novas ferramentas para visualização e compactação.
Uma parte crítica da análise fatorial CorExplorer é a capacidade de explorar não apenas vários, mas mais de 100 fatores com padrões de genes informativos que são colocados dentro de uma hierarquia sobreposta. O CorExplorer facilita a mineração destes fatores miríade para associações biológicas e clínicas e permite a caracterização excepcionalmente detalhada de tumores individuais. A aprendizagem não supervisionada de um número tão grande de factores significa que nem todos serão relevantes para a biologia da doença. Nesse caso, é essencial usar anotações ou genes conhecidos para retirar fatores de interesse ou procurar fatores associados a dados clínicos, como a sobrevida. Assim, o CorExplorer permite que os usuários implementem essa etapa de filtragem muito importante. A presença de testes padrões do gene do fator em um tumor pode mesmo sugerir uma aproximação ao tratamento personalizado da oncologia. Além disso, a multiplicidade de escores fatoriais para cada tumor que permite a descoberta de combinações terapêuticas potencialmente úteis.
Às vezes, é o caso de nenhuma anotação GO significativa aparecer para fatores altamente correlacionados com a sobrevivência. Embora isso possa ocorrer devido a ruídos ou em dados amostrados, há outras causas possíveis, como um tamanho de cluster que é muito pequeno para registrar pontuações de enriquecimento significativas ou o grupo sendo um "cesto" de genes únicos de diferentes vias sem coerência biológica Associação. Além disso, uma categoria de anotação diferente do processo biológico KEGG e GO, por exemplo, compartimento celular, pode ser apropriada. Estes podem ser acessados ligando para StringDB como demonstrado no protocolo. A análise de enriquecimento de ontologia genética no site do CorExplorer atualmente não conta para a ponderação do gene em um fator, embora isso provavelmente será remediado em um futuro próximo. Nota uma opção de lista de genes está disponível em ' Adicionar janela ' que permite o download da lista de genes do fator completo para análise posterior com ferramentas externas.
Para os propósitos do site, o CorEx foi executado em cada um dos conjuntos de dados cinco vezes e a execução que resultou na maior correlação total global foi mantida. Ter uma representação estatística dos resultados de várias execuções pode ser mais informativo e é um objetivo para o trabalho futuro. Além disso, o conjunto de tipos de tumores disponíveis no servidor é bastante pequeno, mas esperamos que isso se expanda ao longo do tempo de acordo com o interesse do usuário.
Conforme descrito acima, o CorExplorer visualiza as relações de fator CorEx RNA-Seq juntamente com informações clínicas e de banco de dados, permitindo assim uma variedade de diferentes modos de interrogação. Nós estamos esperançosos que esta ferramenta conduzirá a um trabalho mais adicional para utilizar o poder da análise do RNA-Seq para a descoberta e a aplicação clínica na oncologia.
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
GV foi apoiado pelo prêmio DARPA W911NF-16-0575.
Materiais
| Name | Company | Catalog Number | Comments |
| Public server for CorExplorer website | USC | http://corex.isi.edu | Intel Xeon E5-2690 4-core 2.6 GHz, 8GB RAM. Backend architecture is LAMP: Linux, Apache, MySQL, PHP. |
| Web browser | Google/Apple | Chrome/Safari | Verified web browsers. |
Referências
- Petryszak, R., et al. The RNASeq-er API-a gateway to systematically updated analysis of public RNA-seq data. Bioinformatics. 33, 2218-2220 (2017).
- Steeg, G. V., Galstyan, A. Maximally Informative Hierarchical Representations of High-Dimensional Data. Proceedings of the Eighteenth International Conference on Artificial Intelligence and Statistics (AISTATS). , (2015).
- Ver Steeg, G., Galstyan, A. Discovering structure in high-dimensional data through correlation explanation. Advances in Neural Information Processing Systems. , (2014).
- Pepke, S., Ver Steeg, G. Comprehensive discovery of subsample gene expression components by information explanation: therapeutic implications in cancer. BMC medical Genomics. 10, 12 (2017).
- Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D., Craig, D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature Reviews Genetics. 17, 257 (2016).
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511, 543 (2014).
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330 (2012).
- Akbani, R., et al. Genomic classification of cutaneous melanoma. Cell. 161, 1681-1696 (2015).
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609 (2011).
- Grossman, R. L., et al. Toward a shared vision for cancer genomic data. New England Journal of Medicine. 375, 1109-1112 (2016).
- Moynahan, M. E., Chiu, J. W., Koller, B. H., Jasin, M. Brca1 controls homology-directed DNA repair. Molecular Cell. 4, 511-518 (1999).
- Szklarczyk, D., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 47, 607-613 (2018).
- Durgeau, A., Virk, Y., Corgnac, S., Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Frontiers in Immunology. 9, 14 (2018).
- Sato, E., et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 102, 18538-18543 (2005).
- De Moura, M. B., et al. Mitochondrial respiration-an important therapeutic target in melanoma. PLoS One. 7, 40690 (2012).
- Folkman, J., Merler, E., Abernathy, C., Williams, G. Isolation of a tumor factor responsible for angiogenesis. Journal of Experimental Medicine. 133, 275-288 (1971).
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biological and Pharmaceutical Bulletin. 34, 1785-1788 (2011).
- Subramanian, A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 102, 15545-15550 (2005).
- Cerami, E., et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2, 401-404 (2012).
- Gao, J., et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signalling. 6, 1 (2013).
- Reich, M., et al. GenePattern 2.0. Nature Genetics. 38, 500 (2006).
- Wang, Y. E., Kutnetsov, L., Partensky, A., Farid, J., Quackenbush, J. WebMeV: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. Cancer Research. 77, 11-14 (2017).
- . Morpheus Available from: https://software.broadinstitute.org/morpheus (2019)
- Weitschek, E., Lauro, S. D., Cappelli, E., Bertolazzi, P., Felici, G. CamurWeb: a classification software and a large knowledge base for gene expression data of cancer. BMC Bioinformatics. 19, 354 (2018).
- Chou, P. -. H., et al. tACCo, a Database Connecting transcriptome Alterations, pathway Alterations and Clinical outcomes in Cancers. Scientific Reports. 9, 3877 (2019).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados