
Method Article
Analisi dei fattori di espressione del gene del tumore con il portale Web CorExplorer
In questo articolo
Riepilogo
Introduciamo il portale web CorExplorer, una risorsa per l'esplorazione dei fattori di sequenziamento dell'RNA tumore trovati dall'algoritmo di apprendimento automatico CorEx (Spiegazione di correlazione), e mostriamo come i fattori possono essere analizzati rispetto alla sopravvivenza, alle annotazioni del database, proteine-proteine, e l'un l'altro per ottenere informazioni sulla biologia del tumore e gli interventi terapeutici.
Abstract
L'analisi dell'espressione genica differenziale è una tecnica importante per comprendere gli stati della malattia. L'algoritmo di apprendimento automatico CorEx ha dimostrato utilità nell'analisi dell'espressione differenziale di gruppi di geni nel tumore RNA-seq in un modo che può essere utile per far progredire l'oncologia di precisione. Tuttavia, CorEx produce molti fattori che possono essere difficili da analizzare e connettersi alla comprensione esistente. Per facilitare tali connessioni, abbiamo costruito un sito web, CorExplorer, che consente agli utenti di esplorare in modo interattivo i dati e rispondere a domande comuni relative alla sua analisi. Abbiamo addestrato CorEx sui dati dell'espressione genica RNA-seq per quattro tipi di tumore: ovarico, polmone, melanoma e colorettale. Abbiamo quindi incorporato la sopravvivenza corrispondente, le interazioni proteina-proteina, l'ontologia genica (GO) e l'Enciclopedia Kyoto dei geni e dei genomi (KEGG) e le mappe di calore nel sito web per l'associazione con la visualizzazione del grafico dei fattori. Qui utilizziamo protocolli di esempio per illustrare l'uso del database per comprendere il significato dei fattori tumorali appresi nel contesto di questi dati esterni.
Introduzione
Dalla sua introduzione poco più di un decennio fa, RNA-seq è diventato uno strumento onnipresente per misurare l'espressione genica1. Questo perché consente una profilazione de novo rapida ed economica dell'intero trascrittoma di un campione. Tuttavia, i dati del tumore dell'RNA-seq riflettono una biologia sottostante intrinsecamente complessa e spesso sottocampionata, mentre i dati stessi sono ad alta dimensione e rumorosi. Questo rappresenta una sfida significativa per l'estrazione di segnali affidabili. L'algoritmo CorEx sfrutta le informazioni reciproche multivariate per trovare modelli sottili in tali situazioni2,3 . Questa tecnica è stata precedentemente adattata per analizzare campioni di RNA-seq del tumore ovarico dal The Cancer Genome Atlas (TCGA) e in questo contesto sembrava avere vantaggi significativi rispetto ai metodi di analisi più comunemente utilizzati4.
Anche se l'uso di RNA-seq è enormemente diffuso nelle applicazioni di ricerca, anche in oncologia, tali sforzi non hanno portato ad un ampio utilizzo ai fini degli interventi clinici5. Parte della ragione di questo è la mancanza di algoritmi user-friendly e software mirati a questi problemi specifici. Per contribuire a colmare questa lacuna, abbiamo progettato il portale web CorExplorer per consentire ai ricercatori provenienti da una varietà di background di studiare i fattori di espressione genica dei campioni tumorali di RNA-seq, come trovato dall'algoritmo di apprendimento automatico CorEx. Il portale CorExplorer supporta la visualizzazione interattiva e l'interrogazione di fattori provenienti da diversi tipi di tumore, tra cui polmone, colon, melanoma e ovarico6,7,8,9, 10, con l'intento di aiutare i ricercatori a vagliare le correlazioni dei dati e identificare i percorsi candidati per stratificare i pazienti a fini terapeutici.
Ci aspettiamo che il portale CorExplorer possa essere utile a diversi tipi di utenti. Il portale è stato progettato con l'utente in mente che desidera comprendere i grandi fattori che guidano le differenze di espressione genica tumorale nei database pubblici e possibilmente anche inserire profili di espressione genica individuali nel contesto di tumori con simili Caratteristiche. Oltre ai protocolli rappresentativi qui descritti, le indagini di CorExplorer possono servire come punto di partenza per suggerire ipotesi per ulteriori test, per confrontare e confrontare i risultati CorEx su set di dati al di fuori del CorExplorer e per collegare firme di espressione patologica di uno o pochi geni in un singolo tumore a gruppi più grandi che possono essere influenzati coordinamente. Infine, può servire come introduzione user-friendly all'applicazione dell'apprendimento automatico all'RNA-seq per coloro che hanno iniziato nel campo.
Protocollo
1. Esplorare i fattori contenenti un gene di interesse
- Aprire un browser Web e passare alla http://corex.isi.edu, la home page di CorExplorer.
- Sul lato destro sotto Collegamenti rapidi, fare clic sul pulsante di espansione s accanto a Ovarian (TCGA-OV) per visualizzare un riepilogo del grafico del fattore CorEx che è stato addestrato sui dati del cancro ovarico TCGA (mostrato figura 1). Facoltativamente, fare clic su altri utenti per confrontare.
- Una volta terminata l'ispezione dei grafici dei fattori, fare clic suPolmone (TCGA-LUAD)per accedere alla pagina CorExplorer per il cancro del polmone RNA-seq.
- Esplora il grafico dei fattori CorEx per un gene di interesse utilizzando la finestra CorExplorer 'Factor Graph'.
- Spostare il cursore del mouse sulla finestra di visualizzazione del grafico dei fattori. Eseguire lo zoom nel grafico del fattore utilizzando la rotellina di scorrimento del mouse o il trackpad per visualizzare i dettagli del grafico, ad esempio i geni più importanti in ogni fattore e le connessioni tra i nodi a livelli diversi. In alternativa, fare clic e trascinare per spostare l'area della vista o qualsiasi nodo.
- Per trovare un gene bersaglio (qui useremo BRCA1), fare clic sul menu a discesa Gene nella parte superiore della finestra del grafico dei fattori. Digitare 'BRCA1' per selezionarlo nell'elenco a discesa e premere Invio per fare in modo che la vista si esegua il fattore 26, il fattore con cui BRCA1 è più fortemente correlato.
- Riposizionare il mouse sulla visualizzazione del grafico e scorrere per ridurre lo zoom per visualizzare il nodo livello 2, L2_8, e i fattori associati che sono vicini al fattore 26. Si noti che vengono visualizzati solo i geni con peso superiore alla soglia indicata sul cursore Peso collegamento Min.
- Per visualizzare tutti i geni associati al fattore, fare clic sul nodo L1_26 e selezionare Carica geni aggiuntivi nella finestra pop-up. Quando viene visualizzata la parola 'Fatto', chiudere la finestra pop-up.
- Ora torna alla sezione di intestazione sopra la finestra del grafico dei fattori e prendi e trascina il modificatore Peso del collegamento Min. Ora, mentre il cursore del peso del collegamento viene spostato verso il basso a 0,05, altri geni nel fattore L1_26, incluso BRCA2, appariranno in ordine di peso. Facoltativamente, riposizionare i nodi afferrando e trascinando per migliorare il layout.
- Determinare in che modo la stratificazione dei pazienti rispetto al fattore influisce sulla sopravvivenza eseguendo query nella finestra di sopravvivenza.
- Nella finestra di sopravvivenza, deselezionare Ordina per p-val, quindi selezionare il fattore 26 nel menu a discesa Fattore singolo per mostrare le curve di sopravvivenza per il fattore 26.
- Scorrere verso il basso il grafico di sopravvivenza per mostrare il numero di pazienti a rischio lungo l'asse x.
- Trovare associazioni con la funzione biologica eseguendo query all'interno della finestra Annotazione.
- Nella finestra di annotazione, per ordinare il menu a discesa Fattore in base al numero di fattori anziché al falso tasso di individuazione (FDR), deselezionare Ordinamento FDR.
- Scorrere e fare clic per selezionare il fattore 26 nell'elenco a discesa della finestra di annotazione per visualizzare le annotazioni di arricchimento per il fattore.
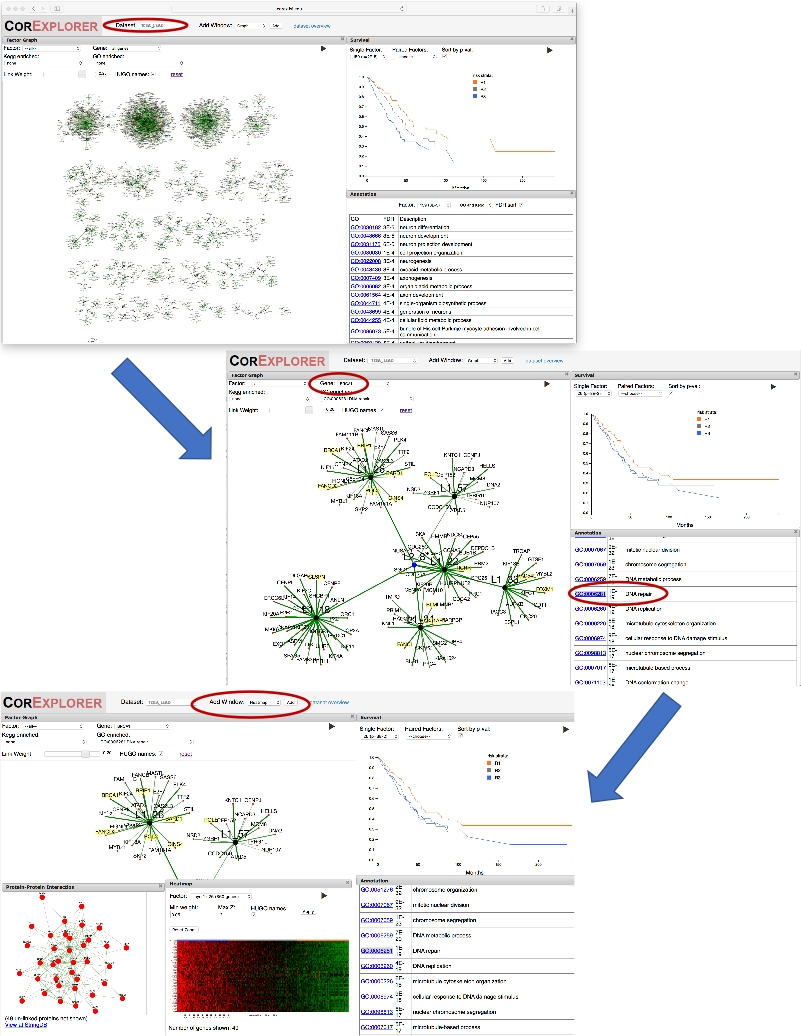
- Scorrere verso il basso l'elenco delle annotazioni fino a visualizzare la riparazione del DNA e fare clic su di esso per visualizzare immediatamente i geni associati evidenziati in giallo sul display del grafico. Vedere il pannello centrale della figura 2.
- Si noti che i fattori scompaiono o appaiono come diversi termini GO vengono selezionati, a seconda che siano arricchiti o meno per i geni con l'annotazione selezionata, ad esempio "percorso di segnalazione apoptotico intrinseco in risposta al danno al DNA".
- Esplora ulteriormente i fattori aggiungendo finestre con funzionalità diverse.
- Dalla barra dei menu in alto, aggiungi una finestra di interazione proteina-proteina (PPI) selezionando PPI dal menu a discesa Aggiungi finestra, quindi fai clic sul pulsante Aggiungi per aggiungere una finestra del grafico PPI all'area di visualizzazione. Nella finestra del grafico PPI, scegliere il fattore 'Layer1: 26' per mostrare le interazioni proteina-proteina. Si noti la densità delle connessioni.
- Dalla barra dei menu superiore, anziché PPI,selezionare Heatmap dal menu a discesa Aggiungi finestra, quindi fare clic sul pulsante Aggiungi per aggiungere una finestra heatmap all'area di visualizzazione. Nella finestra della mappa termica, scegliere il fattore 'Layer1: 26' per mostrare i modelli di espressione genica.
- Afferrare e riposizionare la finestra della mappa termica in modo che sia visibile anche la finestra di sopravvivenza. Lungo la parte superiore della mappa termica, osservare come la barra di colore arancione/blu/grigio corrisponde agli strati di rischio del paziente sul grafico di sopravvivenza. I risultati sono riportati nella parte inferiore della figura 2.
- Esplora il grafico dei fattori CorEx per un gene di interesse utilizzando la finestra CorExplorer 'Factor Graph'.
2. Filtraggio e interpretazione dei fattori CorEx utilizzando i dati relativi al peso genico, alla sopravvivenza e all'annotazione
- Filtrare per i fattori di interesse utilizzando la sopravvivenza e la qualità del cluster.
- Dal menu a discesa Set di dati nella parte superiore, selezionare TCGA_OVCA per passare alla pagina CorExplorer per il cancro ovarico TCGA RNA-seq.
- Una volta che la pagina è stata caricata, si noti dalla finestra di sopravvivenza che il fattore con il più grande differenziale di sopravvivenza per diversi strati è 114.
- Nella parte superiore della finestra del grafico dei fattori selezionare 'Layer1: 114' dall'elenco a discesa Fattore.
- Afferrare il cursore del peso del collegamento con il mouse e spostarlo fino a 0,5. Si noti che il gran numero di geni nel fattore 114 (1609), con nessuno che ha peso >0,35, indica un clustering relativamente debole.
- Successivamente, espandere l'elenco dei fattori nella finestra di sopravvivenza e selezionare il fattore migliore successivo nell'elenco a discesa della finestra di sopravvivenza, fattore 39, per mostrare le curve di sopravvivenza associate.
- Selezionare il fattore 39 nella finestra di annotazione facendo clic su di esso. Vengono visualizzate le annotazioni GO e KEGG significative.
- Per comprendere meglio il ruolo biologico dei geni nel fattore 39, interpretare i fattori utilizzando le informazioni di annotazione di vicinato come segue.
- Nella parte superiore della finestra del grafico dei fattori, selezionare il fattore 'Layer1: 39' nell'elenco a discesa del fattore. Quindi, spostare il mouse sulla finestra del grafico dei fattori e eseguire lo zoom indietro per visualizzare l'intero cluster L2_14 con 6 fattori: 14, 32, 39, 42, 52 e 82 (illustrato nella Figura 3).
- Per comprendere il significato relativo dei fattori legati al nodo L2_14, iniziare visualizzando i differenziali di sopravvivenza per ciascuno dei fattori L2_14. Deselezionare Ordina per p-val nella finestra di sopravvivenza e quindi fare clic su ciascuno dei numeri dei fattori in successione. In questo modo, si noti che solo i fattori 14, 32 e 39 mostrano un'associazione di sopravvivenza.
- Ora dalla barra dei menu in alto, selezionare PPI dal menu a discesa Aggiungi finestra ancora una volta. Premere Aggiungi per aggiungere una finestra del grafico PPI all'area di visualizzazione. Nella finestra del grafico PPI, selezionare il fattore 'Layer1: 52' per mostrare le interazioni proteina-proteina che sono significative. Un esempio di layout di finestre a questo punto è illustrato nella Figura 3.
- Fare clic sul collegamento Visualizza in corrispondenza di StringDB nella parte inferiore della finestra PPI per collegarsi al database online StringDB. Fare clic su Continua dalla prima schermata, quindi selezionare la scheda Analisi sotto il grafico di rete come prima per ottenere un'analisi GO online per i geni della rete PPI. Il componente cellulare superiore è 'Complesso proteico di classe MHC II.'
- Tornare alla scheda CorExplorer e alla finestra PPI e selezionare il fattore 32, questa volta dall'elenco a discesa del fattore. Fare clic sul collegamento Visualizza in StringDB all'analisi StringDB. Il componente cellulare superiore è 'MHC classe I complesso proteico,' in contrasto con la classe II per il fattore 52 nel passaggio precedente!
- Infine, torna alla finestra PPI e seleziona "Layer1: 39" dal menu a discesa del fattore in alto. Fare clic sul collegamento Visualizza in StringDB per creare un collegamento all'analisi StringDB.
- Fare clic su Continua dalla prima schermata, quindi selezionare la scheda Analisi sotto il grafico di rete per ottenere un'analisi GO online per i geni della rete PPI. Osservare che la funzione molecolare superiore è 'CXCR3 chemokine receptor binding.'
3. Utilizzo di sopravvivenza e annotazioni di database per cercare combinazioni terapeutiche promettenti
- Passare al melanoma TCGA CorExplorer selezionando TCGA_SKCM dal menu a discesa Dataset.
- Si noti che il fattore con il più grande differenziale di sopravvivenza è il fattore 171. Esaminare le annotazioni del fattore 171 scorrendo e notando che la "risposta immunitaria" e la "via di segnalazione mediata dalla citochina" sono vicine alla cima (come per il fattore ovarico superiore).
- Per trovare un fattore complementare, esaminare i principali fattori associati alla sopravvivenza insieme ai termini di annotazione principali. A tale scopo, fare clic sul collegamento Panoramica set di dati nella barra dei menu superiore per aprire una scheda separata contenente una tabella con i dettagli di elaborazione del set di dati, nonché un riepilogo dei fattori principali in base al valore p del differenziale di sopravvivenza. Si noti che il primo fattore non immunitario è 88.
- Tornare alla scheda del browser TCGA_SKCM.
- Selezionare il fattore 88 nelle finestre di sopravvivenza, annotazione e grafico. I termini successivi del GO sono correlati all'"elaborazione del rRNA" e all'"organizzazione dei mitocondri", confermandola come distinta dai fattori legati al sistema immunitario.
- Nella finestra di sopravvivenza, nell'elenco a discesa dei fattori accoppiati, selezionare '88_171' per vedere come viene migliorata la sopravvivenza per i pazienti nello strato intermedio per i fattori di espressione combinati 171 e 88. I confronti di annotazione e sopravvivenza sono illustrati nella Figura 4.
4. Trovare punti in comune e differenze di variazione dell'espressione genica tra i tipi di tumore utilizzando la pagina di ricerca
- Fare clic sull'intestazione CorExplorer per tornare alla prima pagina.
- Fare clic su Cerca nella barra dei menu superiore per passare a una pagina che consente la ricerca su tutti i set di dati sul sito CorExplorer.
- Nella casella di ricerca Gene, immettere 'FLT1' (VEGFR1) e premere Invio o premere Cerca. FLT1 si trova con un peso relativamente elevato nei seguenti fattori: OVCA - 76, LUAD - 162, SKCM - 195 e SKCM - 184, così come COAD - 112 e COAD - 74.
- In alternativa, cercare un termine GO correlato in tutti i set di dati. Prova questo nella casella 'GO Search' digitando 'angiogenesi' e premendo Invio o premendo Cerca. Tutti i fattori FLT1, ad eccezione di SKCM-195, sono elencati come statisticamente arricchiti per i geni "angiogenesi": il fattore 195 ha, infatti, l'annotazione, ma al di sotto della soglia predefinita di 10-8. I risultati della ricerca per questo e il passaggio precedente sono illustrati nella Figura 5.
- Come ulteriore esempio, nella casella di ricerca GO, primo tipo 'recettore del fattore di crescita epidermico.' Solo LUAD è arricchito per questo termine, un fattore di stratificazione ben noto per il cancro del polmone. Successivamente, digitare 'mesenchymal' nella casella di ricerca. Questo termine è arricchito in gruppi di espressione genica per ovCA, dove è un fattore di stratificazione ben studiato.
Risultati
La ricerca del gene "BRCA1" nel set di dati sul cancro del polmone rivela che è più fortemente associato al fattore CorEx 26 (Figura 2). GO arricchimento termine per questo fattore è visto per essere estremamente elevato, con la riparazione del DNA che mostra un FDR di soli 1 x 10-19. La selezione richiama inoltre l'attenzione sul cluster di secondo livello L2_8 che ha sei fattori strettamente correlati come elementi figlio. Selezionando "Riparazione del DNA" nelle annotazioni del termine GO o nel menu a discesa arricchito GO del grafico del fattore, i geni associati evidenziano i geni associati in ciascuno dei fattori, con il fattore 26 che ha di gran lunga il più importante, come previsto11. La rete di interazione proteina-proteina è fortemente connessa, supportando ulteriormente la funzionalità strettamente collegata dei geni nel fattore 26. Il grafico di sopravvivenza associato suggerisce una possibile associazione con la sopravvivenza del paziente, ma questo dovrebbe essere confermato in un set di dati più grande.
Iniziare con la sopravvivenza può consentire la dissezione delle ragioni per una migliore sopravvivenza associate a particolari gruppi di espressione genica. Ad esempio, il fattore superiore che influenza la sopravvivenza del cancro ovarico è visto come il numero 39, che è fortemente arricchito per i geni associati al sistema immunitario (Figura 3). Sono indicati anche altri cinque fattori associati allo stesso nodo di livello 2, tuttavia l'impatto di sopravvivenza sembra essere fortemente variabile tra di loro, con 39 che sono il più alto e 52 il più basso. L'aggiunta di una finestra di interazione proteina-proteina per un fattore mostra la rete di interazione immediata e consente di collegarsi al sito Web StringDB12 per interrogare vari arricchimenti per i geni della rete PPI. In questo modo per ciascuno dei fattori L2_14 a sua volta, si scopre che gli arricchimenti StringDB per i geni di rete PPI suggeriscono la seguente possibile spiegazione per le associazioni con la sopravvivenza. Il fattore 32 contiene geni che costituiscono il complesso proteico di classe I del complesso di istocompatibilità principale (MHC), riconosciuto dai linfociti T citotossici. Il fattore 39 corrisponde alla segnalazione delle citochine e al legame del recettore CXCR3, relativo ai linfociti T CD8. Entrambi questi fattori sembrano conferire un significativo vantaggio di sopravvivenza per i pazienti che presentano un'espressione relativamente elevata dei geni corrispondenti. I linfociti citotossici CD8'T sono principalmente responsabili dell'immunità antitumorale. Il fattore 52, d'altra parte, è costituito da geni che codificano per le proteine nel complesso MHC di classe II, che sono riconosciuti principalmente dalle cellule helper T CD4, piuttosto che direttamente dai linfociti T citotossici. I restanti fattori L2_14 riflettono l'attivazione generalizzata del sistema immunitario che non differenzia i due tipi di popolazioni di linfociti. Un'associazione di sopravvivenza specifica per il riconoscimento citotossico dei linfociti T degli antigeni cellulari di classe I è coerente con la nostra comprensione dell'immunità antitumorale in generale e da altri tumori come il melanoma13,14.
Il portale web supporta la scoperta di coppie di fattori con funzioni complementari che possono suggerire terapie di combinazione efficaci specifiche per il tumore. La panoramica del set di dati può essere analizzata alla ricerca di fattori che mostrano una correlazione con la sopravvivenza ma hanno arricchiti GO distinti. Per melanoma (TCGA_SKCM; Figura 4), si è visto che il fattore di sopravvivenza superiore 171 è immune correlato, mentre fattore 88 in basso nell'elenco mostra l'arricchimento per i geni legati all'organizzazione del mitocondrio. Infatti, questo è stato suggerito come un obiettivo nel melanoma15. L'aggiunta di finestre di sopravvivenza alla pagina CorExplorer permette di confrontare la stratificazione usando la coppia di fattori con quella di ogni fattore individualmente, dimostrando che i modelli di espressione genica favorevoli di entrambi i gruppi mostrano una tendenza di sopravvivenza migliore di quella per entrambi solo fattore. Lo strato superiore non sembra essere migliorato tuttavia, suggerendo che solo l'immunoterapia può essere l'opzione migliore per alcuni pazienti.
I punti in comune e le differenze tra i tumori possono essere visti cercando geni o termini GO tra i set di dati (Figura 5). Ad esempio, FLT1 (noto anche come VEGFR1) è un indicatore pro-angiogenico ben studiato16,17. Quando viene messo nella barra di ricerca, tutti i tumori hanno fattori in cui FLT1 svolge un ruolo importante. Al contrario, quando il termine GO "angiogenesi" è inserito nella pagina di ricerca, 5 su 6 dei gruppi FLT1 appaiono con tale arricchimento. Tutti i fattori FLT1, ad eccezione di SKCM-195, sono elencati come arricchiti statisticamente per i geni dell'"angiogenesi". Il sesto fattore ha, infatti, l'annotazione, ma al di sotto della soglia di default 10-8. Quando la ponderazione all'interno dell'elenco dei fattori viene utilizzata in un calcolatore di arricchimento alternativo, ad esempio Gene Set Enrichment Analysis (GSEA)18, il sesto fattore risulta essere notevolmente arricchito anche per i geni di "angiogenesi".
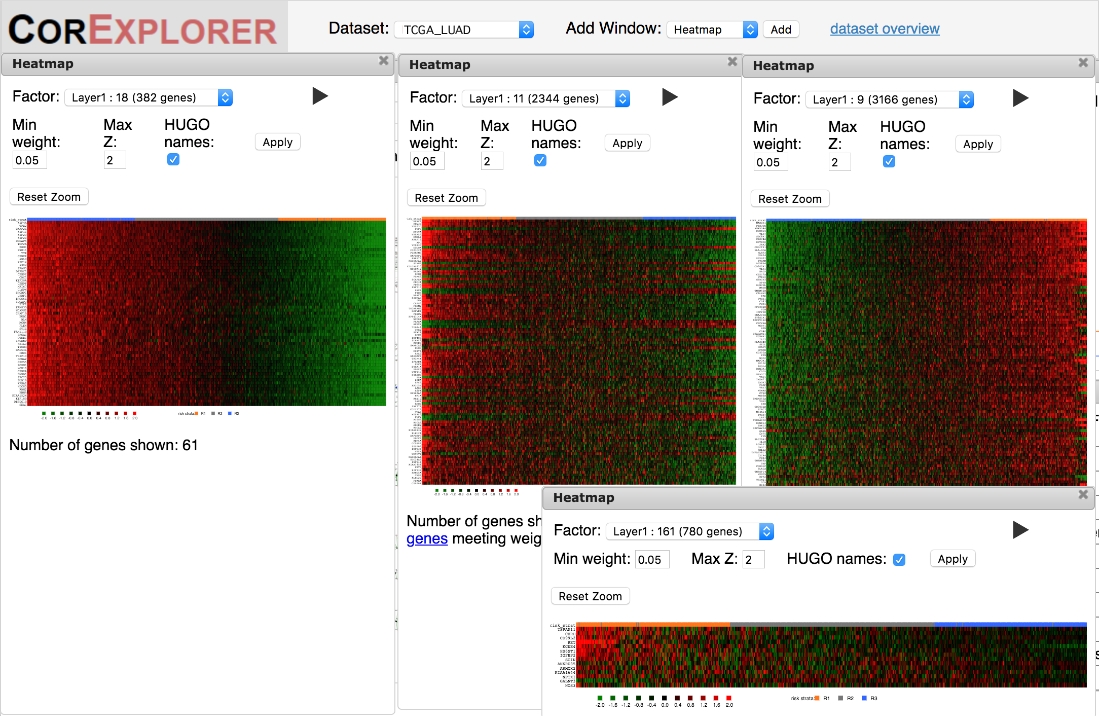
È importante controllare le mappe di calore per assicurarsi che il modello di espressione genica sia di qualità adeguata per supportare le interpretazioni biologiche. Le mappe di calore che mostrano una forte variazione chiara possono mostrare un'espressione coordinata dei geni del fattore che vanno da modelli bassi a quelli alti o più complessi, con alcuni geni a bassa espressione correlati con altri che hanno alto(Figura 6). Un marcatore chiave di un raggruppamento di alta qualità è la presenza di diversi geni con una variazione uniforme nell'espressione in funzione del punteggio del fattore. Le mappe di calore del fattore mostrano campioni ordinati in base al punteggio del fattore, quindi dovrebbe esserci un gradiente uniforme che si muove da sinistra a destra. Tuttavia, questo può non accadere in almeno due modi diversi. Più comunemente, le correlazioni possono essere estremamente rumorose (Figura 5C), mettendo in discussione la robustezza e l'utilità di eventuali deduzioni riguardanti la sopravvivenza e/o la funzione biologica. Inoltre, i modelli che si verificano solo in una piccola minoranza di campioni potrebbero non essere conformi al modello di tre stati di espressione assunti dall'algoritmo CorEx, determinando una classificazione fuorviante dei campioni (lato destro della figura 5D).

Figura 1: pagina iniziale di CorExplorer. Dopo aver cliccato su - accanto a Cancro ovarico sotto Collegamenti rapidi, vengono visualizzati i dettagli del grafico dei fattori. Il modello gerarchico CorEx è costituito da variabili di input (espressione genica in questo caso) sullo strato inferiore e fattori latenti dedotti negli strati superiori. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Utilizzo di un nome genetico per guidare l'esplorazione. La figura mostra una serie di screenshot che illustrano l'esplorazione dei fattori tumorali polmonari CorEx fortemente correlati al BRCA1. In primo luogo, selezionando 'BRCA1' nella casella di riepilogo a discesa Gene per il grafico dei fattori, la visualizzazione del grafico ingrandisce il fattore per il quale BRCA1 ha il peso maggiore. Lo zoom di un bit del livello a due nodi L2_8 che collega tale fattore ad altri elementi correlati. La sopravvivenza e le annotazioni possono essere confrontate: cliccando sul termine GO la riparazione del DNA evidenzia i geni annotati. Viene aggiunta una finestra PPI per mostrare le interazioni di rete per i geni nel fattore. L'uso del pulsante Aggiungi finestra per aggiungere una mappa termica mostra l'associazione dei modelli di espressione con la sopravvivenza, suggerendo che una maggiore espressione dei geni di riparazione del DNA può essere associata a una diminuzione della sopravvivenza. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Utilizzo di dati clinici (sopravvivenza) per guidare l'esplorazione. Esplorare il fattore di sopravvivenza superiore (39) per il cancro ovarico rivela interessanti relazioni tra i fattori vicini. Dopo aver selezionato il fattore 39 nel grafico dei fattori e aver eseguito un po' lo zoom indietro, si ritiene che il fattore due legato al fattore 39 abbia altri cinque fattori associati. Una finestra di sopravvivenza aggiuntiva consente di confrontare direttamente i differenziali di sopravvivenza associati. I fattori 39 e 32 mostrano entrambi una correlazione di sopravvivenza positiva, a differenza del fattore 52, che non lo fa. Le reti di interazione proteina-proteina sono tutte ben definite. Il collegamento a StringDB consente il confronto delle annotazioni GO (non mostrato): il fattore 39 è associato a una rete di segnalazione citochina relativa all'attivazione citototocilata dei linfociti CD8 e T e il fattore 32 è dominato dall'antigene MHC di classe I che presenta proteine che presentano proteine che presentano proteine che innescare il riconoscimento da tali linfociti; i fattori vicini, tuttavia, sono dominati da altri componenti del sistema immunitario come le cellule T helper CD4 e non mostrano alcuna correlazione di sopravvivenza. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Esplorare i principali fattori di sopravvivenza suggerisce potenziali combinazioni terapeutiche. Il collegamento "Set di dati" nella barra dei menu della home page porta a una tabella concisa dei fattori di sopravvivenza ordinati per valore p, insieme all'annotazione GO superiore (non mostrata). Utilizzando queste informazioni per il melanoma, la combinazione del fattore 171 per la funzione immunitaria con il fattore 88 per l'organizzazione dei mitocondri appare complementare. La figura mostra le finestre di annotazione per ognuno dei fattori affiancati per contrastarli. Le curve di sopravvivenza per i pazienti stratificate dai due fattori singolarmente o insieme indicano che la combinazione aumenta il differenziale di sopravvivenza rispetto a entrambi i fattori da solo. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: La pagina Di ricerca facilita l'analisi pan-cancro. I termini del processo biologico dei geni o del GO possono essere cercati in tutti i set di dati utilizzando il collegamento Cerca dalla home page. La figura mostra i risultati della ricerca per il gene FLT1 e il termine GO "angiogenesi". I risultati mostrano la presenza di FLT1 in fattori annotati con il termine "angiogenesi" tra i tumori. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Le mappe di calore possono essere utilizzate per valutare qualitativamente le correlazioni tra geni e campioni in base al punteggio del fattore. Le relazioni di espressione genica di alta qualità sono mostrate dalla gradazione uniforme quando i pazienti sono ordinati in base al punteggio del fattore nelle mappe di calore. La mappa di calore più a sinistra per il fattore 18 è un esempio. I modelli possono anche comprendere firme complesse di espressione su e giù come nella mappa di calore di grandi dimensioni come nella mappa di calore di grandi dimensioni per il fattore 11. Modelli di qualità più bassi a volte mostrano bruschi cambiamenti di espressione per un sottogruppo di pazienti come nel fattore 9 heatmap a destra o semplici correlazioni molto rumorose come nel fattore 161 heatmap in basso a destra. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Abbiamo presentato il sito CorExplorer, un server web accessibile pubblicamente per l'esplorazione interattiva dei fattori di espressione genica maggiormente correlati appresi dal tumore RNA-seq dall'algoritmo CorEx. Abbiamo dimostrato come il sito web può essere utilizzato per stratificare i pazienti secondo l'espressione genica del tumore, e come tale stratificazione corrisponde alla funzione biologica e alla sopravvivenza.
Sono stati costruiti altri webserver per l'analisi RNA-seq. L'analisi differenziale e co-espressione per i tumori può essere esaminata e integrata con altri tipi di dati in cbioPortal19,20. I server GenePattern21, Mev22e Morpheus23incorporano tecniche di clustering consolidate, ad esempio l'analisi dei componenti principali (PCA), i kmeans o le mappe auto-organizzanti (SOM). Gli sforzi più innovativi includono CamurWeb24, basato su un classificatore automatico di generazione di regole, e TACCO25, che implementa classificatori e lazo casuali di foreste. L'algoritmo CorEx qui utilizzato ottimizza le informazioni multivariate per trovare una gerarchia di fattori che spiegano i modelli nei dati. L'apprendimento dei fattori non lineari e gerarchici sembra produrre una migliore interpretabilità rispetto ai fattori lineari globali riscontrati tramite PCA4. Inoltre, l'analisi a grana fine della tecnica dei segnali campione consente confronti precisi del tumore nei sottotipi ampi più comunemente utilizzati. Questa combinazione di analisi dei fattori sovrapposti e gerarchici distingue CorExplorer dalla maggior parte degli altri approcci e richiede nuovi strumenti per la visualizzazione e il riepilogo.
Una parte fondamentale dell'analisi dei fattori di CorExplorer è la capacità di esplorare non solo diversi, ma oltre 100 fattori con modelli genici informativi che vengono posizionati all'interno di una gerarchia sovrapposta. Il CorExplorer facilita l'estrazione di queste miriadi di fattori per le associazioni biologiche e cliniche e consente una caratterizzazione eccezionalmente dettagliata dei singoli tumori. L'apprendimento senza supervisione di un numero così elevato di fattori significa che non tutti saranno rilevanti per la biologia della malattia. In tal caso, è essenziale utilizzare annotazioni o geni noti per estrarre i fattori di interesse o cercare fattori associati a dati clinici come la sopravvivenza. Pertanto, il CorExplorer consente agli utenti di implementare questo passaggio di filtro molto importante. La presenza di modelli genici fattoriali in un tumore può anche suggerire un approccio al trattamento oncologico personalizzato. Inoltre, la molteplicità dei punteggi dei fattori per ogni tumore che consente la scoperta di combinazioni terapeutiche potenzialmente utili.
A volte accade che non vengano presenti annotazioni GO significative per fattori altamente correlati alla sopravvivenza. Anche se ciò può verificarsi a causa di dati rumorosi o sottoposti a campioni, ci sono altre possibili cause come una dimensione del cluster che è troppo piccola per registrare punteggi significativi di arricchimento o il gruppo è un "carrello" di singoli geni da percorsi diversi senza linee biologiche coerenti associazione. Inoltre, una categoria di annotazione diversa dal processo biologico KEGG e GO, ad esempio il compartimento cellulare, può essere appropriata. È possibile accedervi mediante il collegamento a StringDB come illustrato nel protocollo. L'analisi dell'arricchimento dell'ontologia genica sul sito di CorExplorer attualmente non tiene conto della ponderazione genica in un fattore, anche se questo sarà probabilmente recuperato nel prossimo futuro. Si noti che un'opzione di elenco dei geni è disponibile in "Aggiungi finestra" che consente di scaricare l'elenco completo dei geni del fattore per un'ulteriore analisi con strumenti esterni.
Ai fini del sito Web, CorEx è stata eseguita su ciascuno dei set di dati cinque volte e l'esecuzione che ha comportato la massima correlazione globale della total correlazione è stata mantenuta. Avere una rappresentazione statistica dei risultati di più esecuzioni può essere più informativo ed è un obiettivo per il lavoro futuro. Inoltre, l'insieme di tipi di tumore disponibili sul server è piuttosto piccolo, ma ci aspettiamo che questo si espanda nel tempo in base all'interesse dell'utente.
Come descritto in precedenza, CorExplorer visualizza le relazioni del fattore RNA-seq CorEx insieme alle informazioni cliniche e di database, consentendo così una varietà di diverse modalità di interrogatorio. Siamo fiduciosi che questo strumento porterà a ulteriori lavori per utilizzare la potenza dell'analisi RNA-seq per la scoperta e l'applicazione clinica in oncologia.
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Riconoscimenti
GV è stato supportato dal premio DARPA W911NF-16-0575.
Materiali
| Name | Company | Catalog Number | Comments |
| Public server for CorExplorer website | USC | http://corex.isi.edu | Intel Xeon E5-2690 4-core 2.6 GHz, 8GB RAM. Backend architecture is LAMP: Linux, Apache, MySQL, PHP. |
| Web browser | Google/Apple | Chrome/Safari | Verified web browsers. |
Riferimenti
- Petryszak, R., et al. The RNASeq-er API-a gateway to systematically updated analysis of public RNA-seq data. Bioinformatics. 33, 2218-2220 (2017).
- Steeg, G. V., Galstyan, A. Maximally Informative Hierarchical Representations of High-Dimensional Data. Proceedings of the Eighteenth International Conference on Artificial Intelligence and Statistics (AISTATS). , (2015).
- Ver Steeg, G., Galstyan, A. Discovering structure in high-dimensional data through correlation explanation. Advances in Neural Information Processing Systems. , (2014).
- Pepke, S., Ver Steeg, G. Comprehensive discovery of subsample gene expression components by information explanation: therapeutic implications in cancer. BMC medical Genomics. 10, 12 (2017).
- Byron, S. A., Van Keuren-Jensen, K. R., Engelthaler, D. M., Carpten, J. D., Craig, D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature Reviews Genetics. 17, 257 (2016).
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511, 543 (2014).
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330 (2012).
- Akbani, R., et al. Genomic classification of cutaneous melanoma. Cell. 161, 1681-1696 (2015).
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609 (2011).
- Grossman, R. L., et al. Toward a shared vision for cancer genomic data. New England Journal of Medicine. 375, 1109-1112 (2016).
- Moynahan, M. E., Chiu, J. W., Koller, B. H., Jasin, M. Brca1 controls homology-directed DNA repair. Molecular Cell. 4, 511-518 (1999).
- Szklarczyk, D., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 47, 607-613 (2018).
- Durgeau, A., Virk, Y., Corgnac, S., Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Frontiers in Immunology. 9, 14 (2018).
- Sato, E., et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 102, 18538-18543 (2005).
- De Moura, M. B., et al. Mitochondrial respiration-an important therapeutic target in melanoma. PLoS One. 7, 40690 (2012).
- Folkman, J., Merler, E., Abernathy, C., Williams, G. Isolation of a tumor factor responsible for angiogenesis. Journal of Experimental Medicine. 133, 275-288 (1971).
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biological and Pharmaceutical Bulletin. 34, 1785-1788 (2011).
- Subramanian, A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 102, 15545-15550 (2005).
- Cerami, E., et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2, 401-404 (2012).
- Gao, J., et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signalling. 6, 1 (2013).
- Reich, M., et al. GenePattern 2.0. Nature Genetics. 38, 500 (2006).
- Wang, Y. E., Kutnetsov, L., Partensky, A., Farid, J., Quackenbush, J. WebMeV: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. Cancer Research. 77, 11-14 (2017).
- . Morpheus Available from: https://software.broadinstitute.org/morpheus (2019)
- Weitschek, E., Lauro, S. D., Cappelli, E., Bertolazzi, P., Felici, G. CamurWeb: a classification software and a large knowledge base for gene expression data of cancer. BMC Bioinformatics. 19, 354 (2018).
- Chou, P. -. H., et al. tACCo, a Database Connecting transcriptome Alterations, pathway Alterations and Clinical outcomes in Cancers. Scientific Reports. 9, 3877 (2019).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati