Method Article
Etikettenfreier Immunpräzipitations-Massenspektrometrie-Workflow für großangelegtes Nuklear-Interactome-Profiling
In diesem Artikel
Zusammenfassung
Described ist ein Proteomik-Workflow zur Identifizierung von Proteininteraktionspartnern aus einer nuklearen subzellulären Fraktion mithilfe der Immunaffinitätsanreicherung eines bestimmten Proteins von Interesse und der etikettenfreien Massenspektrometrie. Der Workflow umfasst subzelluläre Fraktionierung, Immunpräzipitation, filtergestützte Probenvorbereitung, Offline-Bereinigung, Massenspektrometrie und eine nachgelagerte Bioinformatik-Pipeline.
Zusammenfassung
Die Immunaffinitätsreinigungsmassenspektrometrie (IP-MS) hat sich als robuste quantitative Methode zur Identifizierung von Protein-Protein-Wechselwirkungen herausgebildet. Diese Publikation stellt einen vollständigen Workflow für Interaktionsproteomik dar, der entwickelt wurde, um proteinarme Protein-Protein-Wechselwirkungen aus dem Zellkern zu identifizieren, die auch auf andere subzelluläre Kompartimente angewendet werden könnten. Dieser Workflow umfasst subzelluläre Fraktionierung, Immunpräzipitation, Probenvorbereitung, Offline-Bereinigung, einschussfreie Massenspektrometrie sowie nachgeschaltete Rechenanalyse und Datenvisualisierung. Unser Protokoll ist für die Detektion von kompartifalisierten Wechselwirkungen mit geringer Häufigkeit optimiert, die von ganzen Zelllysaten (z. B. Transkriptionsfaktor-Wechselwirkungen im Zellkern) durch Immunpräzipation endogenes Proteine aus fraktionierte subzelluläre Fächer. Die hier skizzierte Probenvorbereitungspipeline enthält detaillierte Anweisungen für die Herstellung von HeLa-Zellkernextrakt, die Immunaffinitätsreinigung von endogenem Köderprotein und die quantitative Massenspektrometrieanalyse. Wir diskutieren auch methodische Überlegungen zur Durchführung großangelegter Immunpräzipitation in Massenspektrometrie-basierten Interaktionsprofilierungsexperimenten und geben Richtlinien für die Bewertung der Datenqualität, um echtes positives Protein zu unterscheiden. Wechselwirkungen aus unspezifischen Interaktionen. Dieser Ansatz wird hier demonstriert, indem das nukleare Interaktivom der CMGC-Kinase DYRK1A untersucht wird, eine Proteinkinase mit geringer Häufigkeit mit schlecht definierten Wechselwirkungen innerhalb des Kerns.
Einleitung
Das menschliche Proteom weist durch die Bildung stabiler Multisubunit-Komplexe und transienter Protein-Protein-Wechselwirkungen eine enorme strukturelle und biochemische Vielfalt auf. Dementsprechend ist die Identifizierung von Interaktionspartnern für ein Protein von Interesse in Untersuchungen häufig erforderlich, um molekulare Mechanismen zu entwirren. Jüngste Fortschritte bei Affinitätsreinigungsprotokollen und das Aufkommen hochauflösender Schnellscan-Massenspektrometrie-Instrumente haben eine einfache Kartierung von Proteininteraktionslandschaften in einem einzigen unvoreingenommenen Experiment ermöglicht.
Protein-Interaktionsprotokolle verwenden häufig ektopische Expressionssysteme mit Affinitäts-getaggten Fusionskonstrukten, um Protein-Wechselwirkungen zu identifizieren, ohne dass hochwertige Antikörper benötigt werden, die ein Protein von Interesse erkennen1,2. Epitop-Tag-basierte Methoden haben jedoch mehrere Nachteile. Physikalische Wechselwirkungen mit dem Epitop können zum Nachweis unspezifischer koreinigender Proteine führen3. Zusätzlich kann die Fusion dieser Epitop-Tags zum N- oder C-Terminal eines Proteins native Protein-Protein-Wechselwirkungen blockieren oder die Proteinfaltung stören, um nicht-physiologische Konformationen zu fördern4. Darüber hinaus überprimieren ektopische Expressionssysteme das Köderprotein in der Regel in supraphysiologischen Konzentrationen, was zur Identifizierung von artefaktischen Proteinwechselwirkungen führen kann, insbesondere bei dosisempfindlichen Genen5. Um diese Probleme zu umgehen, kann das endogene Köderprotein zusammen mit den damit verbundenen interagierenden Beuteproteinen immunpräzipiert werden, vorausgesetzt, es steht ein hochwertiger Antikörper zur Verfügung, der das native Protein erkennt.
Hier ist ein Interaktionsproteomik-Workflow zum Nachweis des nuklearen Interkolonms eines endogene Proteins am Beispiel der CMGC-Proteinkinase DYRK1A vorgesehen. Störung der DYRK1A Kopiernummer, Aktivitätsniveau oder Ausdruck kann schwere geistige Behinderung beim Menschen verursachen, und embryonale Letalität bei Mäusen6,7,8,9. DYRK1A weist eine dynamische raumzeitliche Regulation10und kompartalisierte Proteinwechselwirkungen11,12auf, die Ansätze erfordern, die in der Lage sind, Interaktionspartner mit geringer Häufigkeit zu erkennen, die für verschiedene subzelluläre Kompartimente spezifisch sind.
Dieses Protokoll verwendet die zelluläre Fraktionierung menschlicher HeLa-Zellen in Zytosol- und Nukleoplasmafraktionen, Immunpräzipitation, Probenvorbereitung für die Massenspektrometrie und einen Überblick über eine bioinformatische Pipeline zur Auswertung der Datenqualität und zur Visualisierung von Ergebnissen, wobei R-Skripte für die Analyse und Visualisierung bereitgestellt werden (Abbildung 1). Proteomics-Softwarepakete, die in diesem Workflow verwendet werden, stehen alle frei zum Download zur Verfügung oder können über eine Web-Oberfläche aufgerufen werden. Weitere Informationen zu Software und Rechenmethoden finden Sie unter den bereitgestellten Links.
Protokoll
HINWEIS: Alle Pufferzusammensetzungen und Proteasemischungen sind in Tabelle 1beschrieben.
1. Vorbereitung der Zellen
HINWEIS: Für diesen Immunpräzipitations-Massenspektrometrie-Ansatz (IP-MS) wird ein Ausgangsmaterial von 1 bis 10 mg Kernlysat pro Replikation gewünscht. Zellmengen werden für 1 mg nukleare Immunpräzipitation in dreifacher plus dreifacher Kontrolle verabreicht.
- Wenn Sie eine anhehrende Zelllinie verwenden, wachsen Sie die Zellen vor der Ernte in 3 x 15 cm Schalen pro Replikation auf 90 % Konfluenz.
HINWEIS: Es wird empfohlen, mindestens drei replizierende Immunpräzipitationen für Köder- und Kontrollbedingungen durchzuführen. Dieses Protokoll wird die Verwendung von "nur Perlen"-Kontrollen annehmen, die ausgehend von Abschnitt 4 für reichlich unspezifische Wechselwirkungen mit den Perlen steuern. Andere Arten von Steuerelementen können nützlich sein. Diese werden im Diskussionsbereich ausführlich beschrieben.- Waschen Sie Platten 2x mit Phosphat gepufferter Kochsaline (PBS) und trypsinisieren Zellen mit 3 ml 0,25% Trypsin pro 15 cm Platte. Drehen Sie die Zellen bei 1.200 x g für 5 min und dekantieren Sie das Trypsin.
- Für Suspensionszellen, wachsen zu einer ähnlichen Skala / Dichte zu erreichen 70 x 80 mg Gesamtprotein.
- Pelletzellen bei 1.200 x g und 4 °C für 5 min. Dekantieren Sie die Medien sorgfältig.
HINWEIS: Pellets können in diesem Schritt kombiniert werden, um eine effiziente Verarbeitung mit der in Abschnitt 2 beschriebenen großangelegten subzellulären Fraktionierung zu ermöglichen.
- Pelletzellen bei 1.200 x g und 4 °C für 5 min. Dekantieren Sie die Medien sorgfältig.
- Waschen Sie das Zellpellet 2x mit PBS + 5 mM MgCl2, ergänzt mit Proteasehemmern (PIs) und Phosphatase-Inhibitoren (PhIs) (siehe Tabelle 1).
HINWEIS: Zellpellets können in flüssigem Stickstoff eingefroren und bei -80 °C gelagert werden, bis sie zur Fraktionierung bereit sind.
2. Herstellung des Kernextrakts
HINWEIS: Protease- und Phosphatase-Inhibitoren sollten den Fraktionierungspuffern innerhalb von 30 min nach Gebrauch zugesetzt werden.
- Wenn gefroren, tauen Sie die Zellpellets für 15 min in 1x PelletVolumen von kaltem Puffer A + PIs / PhIs auf. Legen Sie das Zellpellet auf einen Nutator bei 4 °C, um beim Auftauen bei der Resuspension zu helfen. Andernfalls setzen Sie das Zellpellet von Schritt 1.3 in 1x Pelletvolumen Buffer A + PIs /PhIs wieder aus.
- Pellet bei 2.000 x g und 4 °C für 10 min. Den Puffer dekantieren.
- Setzen Sie die Zellen mit dem 5-fachen der gepackten Zellvolumen mit Puffer A auf und brüten Sie 20 min auf Eis.
- Pellet bei 2.000 x g und 4 °C für 10 min. Den Puffer dekantieren und mit 2x original verpacktem Zellvolumen Puffer A + PIs/ PhIs wieder aufsetzen und mit "A"/losem Stößel 7x abstoßen.
- Zentrifugieren Sie das Lysat für 10 min bei 2.000 x g und 4 °C.
- Sorgfältig Pipetten aus dem Überstand und Blitz einfrieren mit flüssigem Stickstoff. Bewahren Sie das Lysat bei -80 °C auf. Der Überstand aus diesem Schritt ist die zytosolische subzelluläre Fraktion.
ANMERKUNG: Das Kernpellet kann in diesem Schritt durch Blitzeinfrieren mit flüssigem Stickstoff und Lagerung bei -80 °C - Das Pellet mit 0,9x Pelletvolumen von Buffer B + PIs/ PhIs aufsetzen und auf einem Nutator 5 min bei 4 °C mischen.
- Um die Kerne zu lysieren, 20x mit einem engeren Stößel "B" abprallen.
- Das Kernlysat auf einem Nutator 30 min bei 4 °C so mischen, dass es homogen ist.
- Zentrifugieren Sie das Kernlysat 30 min bei 21.000 x g bei 4 °C. Pipette aus dem Überstand und speichern als löslicher Kernproteinextrakt.
HINWEIS: Die Nuklease-Behandlung des resultierenden Kernpellets ermöglicht die Rückgewinnung einer chromatinassoziierten Proteinfraktion. - Dialysieren Sie den löslichen Kernextrakt gegen Puffer C + PIs für 3 h bei 4 °C.
- Schneiden Sie eine entsprechende Länge von 24 mm Breite Dialyseschläuche mit einem 8 kDa Molekulargewicht abgeschnitten. Klemmen Sie eine Seite des Schlauches und laden Sie Nukleoplasma in das Rohr. Nach dem Laden des Lysats das andere Ende einklemmen und in einen sauberen Glasbehälter mit Puffer C + PIs eintauchen.
- Zentrifugieren Sie den dialysierten Kernextrakt/Nukleoplasma bei 21.000 x g bei 4 °C für 30 min. Aliquot 3x 20 L Volumen kerniatischer Extrakt zur Fractionationsvalidierung durch Western Blot. Der für die IP-MS-Analyse verwendete Kernextrakt kann aliquoted und in flüssigem Stickstoff eingefroren und bei Bedarf bei -80 °C gelagert werden.
3. Validierung der subzellulären Fraktionierung
- Schließen Sie einen Proteintest ab, um die Proteinkonzentration des Kernlysats zu bestimmen. Ein Bicinchoninsäure-Protein-Assay bietet eine ausreichende Empfindlichkeit für die nachgeschaltete Anwendung.
- Für die Western-Blot-Analyse wie zuvor beschrieben13. Überspringen Sie Die Spuren beim Laden, um eine Fehlcharakterisierung einer Probe zu vermeiden.
- Sonden Sie den westlichen Blot für p84 (THOC1) als Kernmarker und GAPDH als zytosolischen Marker. Bestimmen Sie den Grad der Fraktionierung durch das Verhältnis des zytosolischen Markers in der Kernfraktion und umgekehrt.
HINWEIS: Es können Antikörper gegen andere kerntechnische und zytosolische Marker verwendet werden.
4. Immunpräzipitation von endogenem Kernköderprotein
HINWEIS: Es wird empfohlen, ab diesem Zeitpunkt niedrigretentionsarme Rohre zu verwenden. Dadurch wird die unspezifische Bindung an die Rohre während der Probenhandhabung reduziert und unnötiger Probenverlust vermieden. Stellen Sie außerdem sicher, dass LCMS-Klasse H2O verwendet wird, um Puffer für die verbleibenden Schritte vorzubereiten.
- Bereiten Sie ein Protein-A/G-Perlengemisch für jede Replikation vor, indem Sie 12,5 l Perlenvolumen für Protein-A und Protein-G in Mikrozentrifugenröhrchen kombinieren. Bewahren Sie die Perlenbestände als Gülle auf, die 20 % Ethanol enthält. Bestimmen Sie die Konzentration der Perlen innerhalb der Gülle %(v/v) und Pipette das notwendige Volumen mit einer Pipettenspitze, die auf der Spitze geschnitten wurde, um sicherzustellen, dass die Perlen in die Spitze gelangen können.
- Waschen Sie das Protein A/G Perlengemisch 2x mit 300 l IP-Puffer 1. Drehen Sie die Perlen bei 1.500 x g bei 4 °C für 1 min und Dekantpuffer.
- Bereiten Sie die Antikörper-Protein-A/G-Perlen vor: Um den Antikörper an die Perlen zu binden, fügen Sie 300 l IP-Puffer 1 und 10 g des gewünschten Antikörpers hinzu. Lassen Sie die Perlen-Antikörper-Mischung über Nacht bei 4 °C auf einem Nutator schaukeln. Fügen Sie bei nur Perlenkontrollelementen keinen Antikörper hinzu.
ANMERKUNG: Als Ausgangspunkt können insgesamt 10 g Antikörper pro Replikation verwendet werden, aber die genaue Menge muss für jeden einzelnen Antikörper und die Skala des im Experiment verwendeten Lysats optimiert werden. - Die Kernlysate von Schritt 2.10 in einem Wasserbad auftauen und geeignete Volumina in niedrigretentionsarme Mikrozentrifugenröhren für 1 mg Proteineinsatz pro Replikation auftauen.
- Drehen Sie das Lysat bei 16.000 x g für 30 min und übertragen Sie den Überstand auf ein neues Rohr.
- Fügen Sie 1 l Benzonase (250 Einheiten/L) pro 1 mg Kernlysat und Gestein auf einen Nutator bei 4 °C für 10 bis 15 min.
- Bereiten Sie Perlen für die Vorklärung des Lysats vor. Fügen Sie 12,5 l jedes Proteins A und Protein G Perlen zu 1,5 ml Retentionsröhrchen wie in Schritt 4.1 hinzu. 2x mit IP Wash Buffer 1 + PIs waschen und den Puffer dekantisieren.
- Fügen Sie 1 mg des Kernlysats zu den Perlen ab Schritt 4.5 hinzu. Beim Schaukeln auf einem Nutator 1 h bei 4 °C inkubieren.
- Zentrifuge vorgereinigte Lysate bei 1.500 x g und 4 °C für 1 min.
- Während Kernlysate in Schritt 4.5.1 mit Perlen inkubieren, waschen Sie das Antikörper-Protein A/G-Perlen 2x mit IP-Puffer 1 + PIs. Zentrifuge bei 1.500 x g und 4 °C für 1 min und dekantieren den Puffer.
- Übertragen Sie das vorgereinigte Kernlysat von Schritt 4.6.1 auf die Antikörper-Protein-A/G-Perlen. Beim Schaukeln auf einem Nutator bei 4 °C für 4 h inkubieren. Zentrifuge nach der Inkubation bei 1.500 x g und 4 °C für 1 min.
- Übertragen Sie den Überstand in Rohre, die als Durchfluss für jede Replikation gekennzeichnet sind.
- Waschen Sie die Antikörper-Protein-A/G-Perlen mit 1 ml IP-Puffer 2 + PIs. Zentrifuge bei 1.500 x g und 4 °C für 1 min, Dekanatspuffer, und insgesamt 3x wiederholen.
- Waschen Sie die Perlen 2x mit 1 ml IP Buffer 1+ PIs Zentrifugieren wie im vorherigen Schritt. Stellen Sie sicher, dass der gesamte Puffer nach der letzten Wäsche entfernt wird.
- Elute 2x mit 20 l von 0,1 m Glycin (pH 2,75) für je 30 min. Stellen Sie sicher, dass die Rohre während der Inkubation mit dem Elutionspuffer schaukeln. Drehen Sie bei 750 x g und 4 °C für 1 min und Pipette aus dem Überstand nach jeder 30 min Inkubation.
HINWEIS: Während die hier beschriebene Niedrig-pH-Glykol-Methode die meisten Köderproteine eluiert, erfordern einige Antikörper-Antigen-Wechselwirkungen strengere Pufferbedingungen. - Flash Freeze eluatiert in flüssigem Stickstoff und lagern bei -80 °C.
5. Probenvorbereitung
HINWEIS: Insulin, das in die Immunpräzipations-Elutionsproben eingegliedert wird, hilft bei der Wiederherstellung von Proteinen während der Trichloressigsäure (TCA) Ausfällung und Probenverarbeitung, was für endogene Köderproteine mit geringer Häufigkeit wichtig ist.
- Die Eluates bei Raumtemperatur auftauen, wenn sie gefroren sind.
- Legen Sie die Proben auf Eis und fügen Sie 10 l 1,0 mg/ml Insulin pro 100 l Eluat hinzu. Wirbel und dann sofort 10 l Von 1% Natriumdeoxycholat hinzufügen. Wirbeln Sie die Probe erneut und fügen Sie 30 l von 20% TCA gefolgt von einem letzten Wirbel hinzu.
- Die Proben 20 min auf Eis bebrüten, dann bei 21.000 x g bei 4 °C 30 min zentrieren.
- Den Überstand ansaugen und 0,5 ml Aceton hinzufügen, das auf -20 °C vorgechillt wurde. Wirbel und drehen Sie dann bei 21.000 x g und 4 °C für 30 min. Wiederholen Sie diesen Schritt.
- Aspirieren Sie den Überstand und trocknen Sie die Luft das Pellet, das im Boden des Rohres verbleibt.
- Bereiten Sie die Probe für die Massenspektrometrie mit einer modifizierten FASP-Methode (Filter Aided Sample Prep) vor, die für die Reduzierung der Probenhandhabung optimiert ist, wie unter14beschrieben.
- Setzen Sie das Proteinpellet aus Schritt 5.1.4 mit 30 l SDS-Alkylierungspuffer aus (siehe Tabelle 1). Inkubieren Sie die Probe auf einem 95 °C-Wärmeblock für 5 min. Lassen Sie es bei Raumtemperatur für 15 min abkühlen, bevor Sie zum nächsten Schritt gehen.
- Fügen Sie 300 L UA-Lösung und 30 l von 100 mM TCEP zu jeder Probe hinzu. Laden Sie diese Lösung auf einen 30k Zentrifugalfilter. Drehen Sie den Zentrifugalfilter bei 21.000 x g bei Raumtemperatur für 10 min.
HINWEIS: Das Köderprotein und seine vermeintlichen Interaktoren sollten an dieser Stelle an den Filter gebunden werden. Der Durchfluss kann jedoch beibehalten werden, falls ein Problem mit dem Filter auftritt. - Waschen Sie den Filter mit 250 l UA und Zentrifuge bei 21.000 x g für 10 min. Den Durchfluss dekantieren und insgesamt 3x wiederholen.
- Waschen Sie den Filter mit 100 l von 100 mM Tris pH 8,5 und Zentrifuge bei 21.000 x g für 10 min. Den Durchfluss dekantieren und insgesamt 3x wiederholen.
- Fügen Sie 3 l von 1 g/l Lys C in 0,1 M Tris pH 8,5 auf. Füllen Sie die Filter bis zur 100-L-Marke und lassen Sie 1 h bei 37 °C verdauen, während Sie auf einem Nutator schaukeln.
- Fügen Sie 1 L von 1 g / L MS-Grade-Trypsin hinzu. Sanft mischen und das Trypsin bei 37 °C über Nacht mit der Probe bebrüten lassen, während es auf einem Nutator schaukelt.
- Zentrifuge bei 21.000 x g für 20 min, um das Peptid aus dem Filter zu löschen.
HINWEIS: Es können mehrere Zentrifugationsrunden erforderlich sein, um das gesamte Eluat wiederherzustellen. Wenn dies nicht geschieht, besteht ein Risiko für einen schweren Probenverlust.
- Entaltieren Sie die Peptide mit C18-Spin-Spalten. Befolgen Sie das vom Hersteller bereitgestellte Protokoll.
- Setzen Sie das lyophilisierte Peptid in 7 l von 0,1% TFA in 5% Acetonitril aus. Beschallen Sie die Probe für 3 min, um sicherzustellen, dass die Peptide resuspendiert wurden. Drehen Sie bei 14.000 x g für 10 min.
- Übertragen Sie das resuspendierte Peptid in eine geeignete Probendurchstechflasche zum Laden auf das Flüssigchromatographie-Massenspektrometrie-System (LC/MS).
6. LC/MS-Systemtauglichkeit
HINWEIS: Aufgrund des geringen Umfangs und der allgemein geringeren Proteinmenge aus affinitätsgereinigten Proben ist es entscheidend, dass die LC/MS-Plattform mit maximaler Empfindlichkeit und Robustheit arbeitet.
- Fügen Sie 1 ml LC/MS-ameisensäure auf 1 L LC/MS-Wasser für die mobile Phase A hinzu und fügen Sie 1 ml LC/MS-ameisensäuregrad 1 L LC/MS-Acetonititytrile für mobile Phase B hinzu.
- Bereiten oder installieren Sie eine 75-mm-Kapillarsäule mit 75 mm Geschmolzen-Silica-Kapillare, die mit <2 m Umkehrphase C18-Harz verpackt ist, das 250 mm lang ist. Die besten Ergebnisse werden mit einer direkten Injektion von Proben in die Säule erzielt.
- Reinigen Sie das Ultra Performance Liquid Chromatography (UPLC)-System mit frischen mobilen Phasen. Mit einer installierten C18-Säule können Sie eine stabile Durchflussrate und elektrospray mit einem geeigneten Emitter (d.h. 20 m id x 360 m od auf eine 10'm-Spitze) ziehen. Halten Sie die Säule bei 40 bis 60 °C.
- Testen Sie die Gesamtleistung des LC/MS-Systems, indem Sie einen komplexen Qualitätskontrollstandard injizieren, z. B. 100 bis 200 ng eines HeLa-Vollzelllysat-Tryptic-Digests. Elute mit einem geeigneten Gradienten für eine komplexe Probe (d. h. 2 x 3 h Gradientenelutionszeit). Legen Sie eine Grundlegende Systemleistung der Peptid- und Proteinidentifikationen fest.
HINWEIS: Für beste Ergebnisse bieten 3.000 bis 5.000 oder mehr Protein-Identifikationen von 20.000 bis 35.000 einzigartigen Peptiden eine optimale Leistung für experimentelle Proben. - Für die routinemäßige Eignung des LC/MS-Systems können Sie 100 bis 200 Fmol oder weniger eines einzelnen Protein-Digest-Standards injizieren, z. B. Bovine Serum Albumin (BSA). Elute mit einem kurzen Farbverlauf (d.h. 20 bis 30 min).
HINWEIS: Mehrere Injektionen eines Protein-Digests helfen dabei, die Grundlegende LC/MS-Systemleistung zu ermitteln, und die wiederholte Injektion nach jeder IP-MS-Probe liefert ein Maß für die Systemleistung während des gesamten Experiments und ermöglicht die Detektion von Instrumentendrift, was etikettenfreie Experimente beeinträchtigen kann. Eine Baseline der ausgewählten einzelnen Spitzenintensitäten und Spitzenformen informiert über die MS-, LC- und Spaltenleistung. - Um eine Überlastung der analytischen Säule zu vermeiden, laden Sie einen kleinen Teil (15 bis 30 % der Gesamtmenge) einer Versuchsprobe auf die Säule und trennen Sie mit einem Gradienten, der für komplexe Proben geeignet ist (d. h. 2 x 3 h). Wenn die Anzahl der Proteinidentifikationen unbefriedigend ist, laden Sie die gesamte Probe auf die Spalte.
- Führen Sie einen einzelnen Protein-Digest-Standard zwischen den Proben aus, um die Leistung des LC/MS-Systems und die Probenübertragung zu überwachen. Es können mehrere Standards erforderlich sein, um die Probenübertragung je nach Ihren Proben zu reduzieren.
7. Datenverarbeitung
- Laden Sie das proteomics Softwarepaket MaxQuant herunter, das sie auf https://www.maxquant.org/ gefunden hat.
HINWEIS: Dies wird verwendet, um die RAW-MS-Datendatei aus Schritt 6.6 in Datentabellen von Protein-IDs, Gennamen und quantitativen Werten zu verarbeiten, die mit der Identifizierung dieser Daten für die nachgelagerte Analyse verknüpft sind.- Wählen Sie Laden innerhalb des Unterheaders Eingabedaten der Registerkarte Rohdaten. Öffnen Sie den Dateispeicherort, an dem die MS-Rohdateien gespeichert sind, und wählen Sie Rohdateien für jede MS/MS-Ausführung aus.
- Klicken Sie auf die Registerkarte Gruppenspezifisch und wählen Sie Verdauungaus. Wählen Sie innerhalb der Enzymliste LysC aus und klicken Sie auf den rechten Pfeil, um dieses Enzym in die Liste aufzunehmen, die bei der Suche verwendet wird. Wählen Sie als Nächstes Instrument aus, und stellen Sie sicher, dass der richtige Instrumententyp in der Dropdown-Liste oben auf dem Bildschirm angezeigt wird. Lassen Sie andere gruppenspezifische Suchparameter in den Standardeinstellungen.
- Klicken Sie auf die Registerkarte Globale Parameter und wählen Sie Sequenzenaus. Fügen Sie die entsprechende FASTA-Datei für die Taxonomie hinzu, die bei dieser Suche verwendet wird. Peptide werden nicht ordnungsgemäß zugewiesen, wenn dies nicht erfolgt. Für das humane Proteom laden Sie die FASTA-Datei von UniProt unter https://www.uniprot.org/help/human_proteome herunter.
- Klicken Sie auf der Registerkarte Globale Parameter auf Protein quantification. Wählen Sie im Dropdown-Menü Peptide for Quantification Die Option Unique + Razoraus.
HINWEIS: MaxQuant bietet eine alternative Quantifizierung von Proteinen durch intensitätsbasierte absolute Quantifizierung (iBAQ) und etikettenfreie Quantifizierung (LFQ). Für die nachgelagerte Analyse in diesem Protokoll15reichen jedoch Peptidanzahlinformationen aus. - Wählen Sie unten links in der MaxQuant-Schnittstelle die Anzahl der Prozessoren aus, die für die Suche verwendet werden sollen. Dies wirkt sich direkt auf die für den Lauf benötigte Zeiteinwirkung aus, also wählen Sie dafür so viele wie möglich aus). Klicken Sie unten links auf dem Bildschirm auf Start, um die Ausführung zu starten. Wählen Sie die Registerkarte Leistung oben auf dem Bildschirm aus, um den Fortschritt der Suche anzuzeigen.
- Wenn der Lauf abgeschlossen ist, öffnen Sie die Datei proteingroups.txt in Perseus, einer Proteomik-Berechnungsplattform oder einem anderen Tabellenkalkulationsprogramm, um die Daten16anzuzeigen.
- Verwenden Sie Perseus, um häufige Verunreinigungen und Treffer zu umgekehrten Proteinsequenzen zu entfernen. Folgen Sie der ausführlichen Perseus-Dokumentation unter http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
HINWEIS: Das Öffnen der Datei proteingroups.txt in der Analysesoftware (z. B. Excel) beschädigt automatisch bestimmte Gen- und Proteinnamen.
- Verwenden Sie Perseus, um häufige Verunreinigungen und Treffer zu umgekehrten Proteinsequenzen zu entfernen. Folgen Sie der ausführlichen Perseus-Dokumentation unter http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
- Analysieren Sie experimentelle Daten mit dem Contaminant Repository for Affinity Purification (CRAPome). Registrieren Sie ein Konto in diesem Repository http://crapome.org/ und folgen Sie dem Tutorial nach Bedarf17,18.
- Verwenden Sie den Workflow Analysieren Ihrer Daten auf der CRAPome-Homepage. Wählen Sie externe Steuerelemente aus, die dem In diesem Interaktionsexperiment verwendeten Affinitätsreinigungssystem entsprechen.
HINWEIS: Diese Steuerelemente können verwendet werden, um eine zweite Faltwechselanreicherung zu berechnen, die für die Erkennung von häufigen Verunreinigungen nützlich ist. - Generieren Sie eine Eingabedatei aus der Proteingroups.txt-Ausgabe von MaxQuant mit Perseus oder einer geeigneten Tabellenkalkulationsanwendung. Details zur manuellen Formatierung finden Sie unter http://crapome.org/?q=fileformatting. Alternativ können Sie das bereitgestellte R-Skript "export_CRAPomeSAINT_Input_File.R" verwenden, um die SAINT/CRAPome-Eingabedatei zu generieren. Siehe README.txt in den Supplemental Coding Files.
- Führen Sie eine Analyse durch, um die Faltenveränderungsanreicherung und die SAINT-Wahrscheinlichkeit (Significance Analysis of INTeractome) für jedes Köderprotein in der Immunpräzipitation zu bestimmen. Stellen Sie sicher, dass im Dropdown-Menü unter FC-A 'User Controls' 'User Controls'ausgewählt ist, 'CRAPome-Steuerelemente' oder 'Alle Steuerelemente' in der DROPdown-Liste FC-B ausgewählt sind und wahrscheinlicher Score ausgewählt ist, um SAINT-Wahrscheinlichkeiten zu generieren. Wenn der Lauf abgeschlossen ist, zeigen Sie die Ausgabe an, die unter "Analyseergebnisse" zusammen mit einer Auftrags-ID verfügbar ist. Laden Sie die Datenmatrix aus den 'Analyseergebnissen' für zukünftiges Plotten und Datenvisualisierung herunter.
- Verwenden Sie den Workflow Analysieren Ihrer Daten auf der CRAPome-Homepage. Wählen Sie externe Steuerelemente aus, die dem In diesem Interaktionsexperiment verwendeten Affinitätsreinigungssystem entsprechen.
- Plotten Sie Proteine als Funktion von FC-A (IPs vs. Benutzersteuerungen) und SAINT-Wahrscheinlichkeit, indem Sie den R-Scripts folgen, wie in Supplemental Coding Filesangegeben.
HINWEIS: Für die Erstellung von Plots mit FC-A vs. SAINT-Wahrscheinlichkeit und iBAQ vs. Log2 (Protein-Überfluss) wird ein Satz von R-Skripten bereitgestellt, die durch den angepassten p-Wertbereich aus der empirischen Bayes-Analyse der etikettenfreien Intensitäten gefärbt sind. Die Details der differenziellen statistischen Analyse und Darstellung finden Sie in README.txt und dem R-Skript "main_differential_analysis. R" in den ergänzenden Codierungsdateien. - Bewerten Sie, wo bekannte interagierende Proteine des Köderproteins nach FC-A und SAINT eingestuft werden. Machen Sie einen Cutoff von FC-A > 3.00 und SAINT > 0.7 für einzelne Köderexperimente in Dreifacharbeit als Ausgangspunkt.
HINWEIS: Die Auswahl von Cutoffs für einen "hochvertrauensigen" Interaktor und einen "low-confidence"-Interaktor muss durch biologische Informationen informiert werden.
8. Datenvisualisierung
HINWEIS: Es gibt viele Programme, die Proteomics-Daten effektiv visualisieren können (z. B. R, Perseus, Cytoscape, STRING-DB). Die Analyse der Konnektivität zwischen hochkonfidenden Getroffenen und der funktionalen Bereicherung dieser Interaktoren kann eine nützliche Strategie für die Priorisierung von Treffern für die weitere Validierung und funktionale Charakterisierung sein.
- Laden Sie Cytoscape herunter, ein Open-Source-Netzwerk-Visualisierungstool für https://cytoscape.org/download.html19.
- Bereiten Sie eine Eingabedatei für Interaktionsdaten als eine durch Tabund getrennte Datei vor, die mit drei Spalten formatiert ist: Köder (Quellknoten), Beute (Zielknoten), Interaktionstyp (Kantentyp). Dies kann in Perseus oder einem beliebigen Tabellenkalkulationsprogramm Ihrer Wahl erfolgen.
- Wählen Sie das Symbol Tabelle aus Datei importieren oben links des Programms (gekennzeichnet durch einen Pfeil nach unten und eine Matrix im Symbol). Cytoscape füllt die Interaktionsdaten automatisch in ein Netzwerk, das für benutzerdefinierte Formatierungen und Designs bereit ist.
- Wählen Sie die Registerkarte Stil im Bedienfeld für Cytoscape aus, und klicken Sie auf die Quadrate in der Spalte Def, um das Attribut für das gesamte Netzwerk anzupassen. Wählen Sie bestimmte Knoten oder Kanten im Netzwerk aus, und wählen Sie dann das Quadrat in der Spalte Byp. des Stilmenüs aus, um die Standardeinstellungen zu umgehen und nur ausgewählte Objekte anzupassen. Alternativ klicken Sie auf das Dropdown-Menü oben im Stilmenü, um die voreingestellten Netzwerkformate anzuzeigen.
HINWEIS: STRING-db Protein-Protein-Interaktionsdaten können zu diesem Zeitpunkt entweder manuell über die Eingabedatei oder über verschiedene Anreicherungswerkzeuge, die als Plug-Ins in Cytoscape, http://apps.cytoscape.org/20, zur Verfügung stehen, in dieses Netzwerk integriert werden. Ein empfohlenes Cytoscape-Plug-in für die Anreicherungsanalyse finden Sie unter http://apps.cytoscape.org/apps/cluego21. - Um das Vertrauen in den in diesem Workflow generierten Datensatz zu erhöhen, führen Sie reziprokierte IP-MS- oder IP-Western-Experimente durch, die auf Beuteproteine abzielen, die als Köder von Interesse sind.
Ergebnisse
Der Großteil der in einem IP-MS-Experiment identifizierten Proteinmasse besteht aus unspezifischen Proteinen. Eine der wichtigsten Herausforderungen eines IP-MS-Experiments ist daher die Interpretation, welche Proteine vertrauensvolle Interaktoren im Vergleich zu unspezifischen Interaktoren sind. Um die entscheidenden Parameter zu demonstrieren, die bei der Bewertung der Datenqualität verwendet werden, analysierte die Studie dreifache Immunpräzipitationen aus 5 mg HeLa-Kernextrakt unter Verwendung einer reinen Perlenkontrolle. Die erste interne Überprüfung, um sicherzustellen, dass ein IP-MS-Experiment zuverlässig ist, ist, ob das Köderprotein zu den am höchsten angereicherten Proteinen zählt, die sowohl durch Faltenwechsel als auch durch SAINT-Wahrscheinlichkeit identifiziert werden. In diesem Fall rangierte der Köder DYRK1A unter den drei topierierten Proteinen über die Kontrolle (Abbildung 2A,B). In einer nuklearen Interactome-Studie mit DYRK1A unter Verwendung von vier unabhängigen Antikörpern bot ein FC-A-Cutoff von >3,00 und SAINT-Wahrscheinlichkeitsschnitt >0,7 einen strengen Cutoff zur Identifizierung sowohl neuartiger als auch zuvor validierter Interaktoren22. Bei anwendung auf dieses Experiment konnte eine klare Trennung zwischen den hochkonfidenkigen Interaktoren und >95% der als unspezifisch identifizierten korifizierten Proteine gesehen werden (Abbildung 2A,B). Die Anwendung sowohl einer Faltenänderungsanreicherung als auch der Wahrscheinlichkeitsschwelle erhöht die Stringenz, da eine gleichbleibend hohe Anreicherung von Protein-IDs über biologische Replikationen hinweg erforderlich ist.
Neben der statistischen Bewertung bildet der CRAPome-Analyse-Workflow auch zuvor gemeldete Interaktionen auf Köder-Beute-Datenab 23. Während diese Zuordnung für die Schwelle von Interaktionen mit hohem und niedrigem Vertrauen nützlich sein kann, können zuvor gemeldete Interaktionen durch FC-A- und SAINT-Wahrscheinlichkeiten schlecht bewertet werden, was möglicherweise darauf hindeutet, dass viele bekannte Wechselwirkungen eines bestimmten Köders nur in bestimmten Zelltypen, Kontexten oder Organellen vorhanden sein können. Für das Beispiel-DYRK1A-Dataset waren die FC-A-Werte des iREF-Interagierenden nur 0,45, was eine sehr geringe Anreicherung über die Steuerung darstellt (Abbildung 2C). Um eine Inflation falscher Positivmeldungen zu vermeiden, sollten statistische Schwellenwerte in einer Weise durchgeführt werden, die die Strenge vorrang vor der Reduzierung falscher Negatives priorisiert. Es sei darauf hingewiesen, dass der Nachweis dieser Wechselwirkungen unabhängig von Proteinüberfluss war (Abbildung 2C). Die berechnete absolute Kopiernummer jeder iREF-Interaktion innerhalb von HeLa-Zellen zeigte keine Korrelation mit den Nachweisstufen eines Interaktionspartners durch IP-MS24.
Cytoscape dient als effektives Werkzeug zur Visualisierung mehrerer Ebenen von Interaktionsdaten19. Im hier beschriebenen DYRK1A-Immunpräzipitationsexperiment reduzierte die kombinierte Verwendung von FC-A > 3.0 und SAINT > 0,9 die Liste der hochkonfidenzvertrauten Interaktoren auf sechs Proteine (Abbildung 2D). Bei der isolierten Anwendung eines FC-A-Cutoffs von > 3.0 wurden dem Netzwerk jedoch acht zusätzliche Proteine zugesetzt. Diese zusätzlichen Proteininteraktoren verfügen über eine hohe Konnektivität mit den Interaktoren, die sich bereits im Netzwerk befinden, was darauf hindeutet, dass sie in ähnlichen Komplexen oder funktionalen Rollen assoziiert sind. Zu diesem Zweck wurden Beweise aus dem STRING-DB von Protein-Protein-Wechselwirkungen als blaue gestrichelte Linien20in dieses Netzwerk integriert. Während dieses dreifache Einzelbait-Experiment eine begrenzte Stichprobe des vollständigen DYRK1A-Interaktionsnetzwerks bietet, kann die Verwendung zusätzlicher Köder, Replikationen und integrationen großer öffentlicher Datensätze verwendet werden, um das Netzwerk hochvertrauensstarker Interaktionen zu erweitern. Die statistischen Abschnitte sind daher für jedes einzelne Experiment spezifisch und müssen gründlich bewertet werden.

Abbildung 1: Repräsentativer Proteomik-Workflow für subzelluläre suläre IP-MS. Die Zellen werden entweder in 4 L runden Bodenkolben oder 15 cm Gewebekulturschalen angebaut und gleichzeitig zur subzellulären Fraktionierung geerntet. Die Zellen werden in ein zytosolisches, nukleares und ein nukleares Pellet fraktioniert, und Immunpräzipitationen werden aus 1bis 10 mg Kernlysat mit einem oder mehreren Antikörpern durchgeführt, die denselben Köder erkennen. Filtergestützte Probenvorbereitung (FASP) und Offline-Probenbereinigung werden vor der Einzelaufnahme-Massenspektrometrie durchgeführt. Eine nachgelagerte Berechnungspipeline wird verwendet, um Daten in interpretierbare Interaktionsdaten zu verarbeiten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
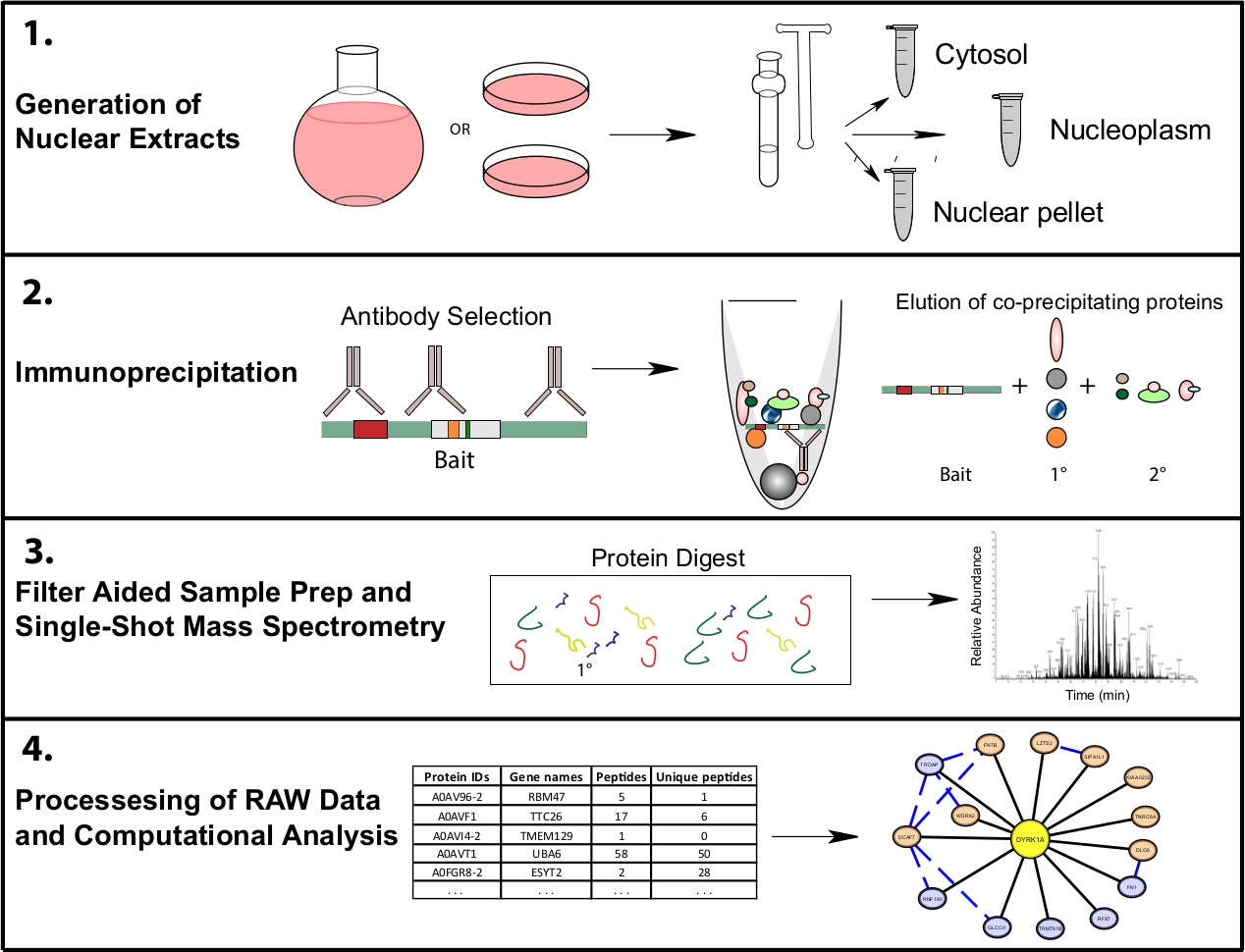{kind=link}

Abbildung 2: Repräsentative Daten für ein IP-MS-Experiment mit einem Einzigen-Köder-Antikörper. (A) FC-A und SAINT Wahrscheinlichkeitsausgabe aus dem CRAPome-Analyse-Workflow für ein optimales Experiment mit einem einzigen Antikörper für die Kinase DYRK1A (n = 3). Für den Vergleich wurden nur Beads-Kontrollen verwendet. Rote durchgezogene Linien stellen Trennlinien dar, die auf FC-A > 3.00 und SAINT > 0.7 gesetzt sind. (B) MaxQuant Proteinabundance estimates (iBAQ) output vs. log2 ratio of protein abundance in DYRK1A IP to control, colored by the adjusted p value range from empirical Bayes analysis of the label-free intensities. (C) FC-A und geschätzte Kopierzahl der Proteine, die in der iRef-Datenbank als interagierende Proteine aufgeführt sind23,24. (D) Visualisierung des Cytoscape-Netzwerks von DYRK1A-Interaktoren. Blaue Knoten = FC-A > 3.00, SAINT > 0.7. Orange Knoten = FC-A > 3.00. Schwarze Kanten = Proteine, die im IPMS-Experiment als Interaktoren identifiziert wurden. Blau gestrichelte Kante = SAINT-Interaktion zwischen Beuteprotein (Vertrauen > .150). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
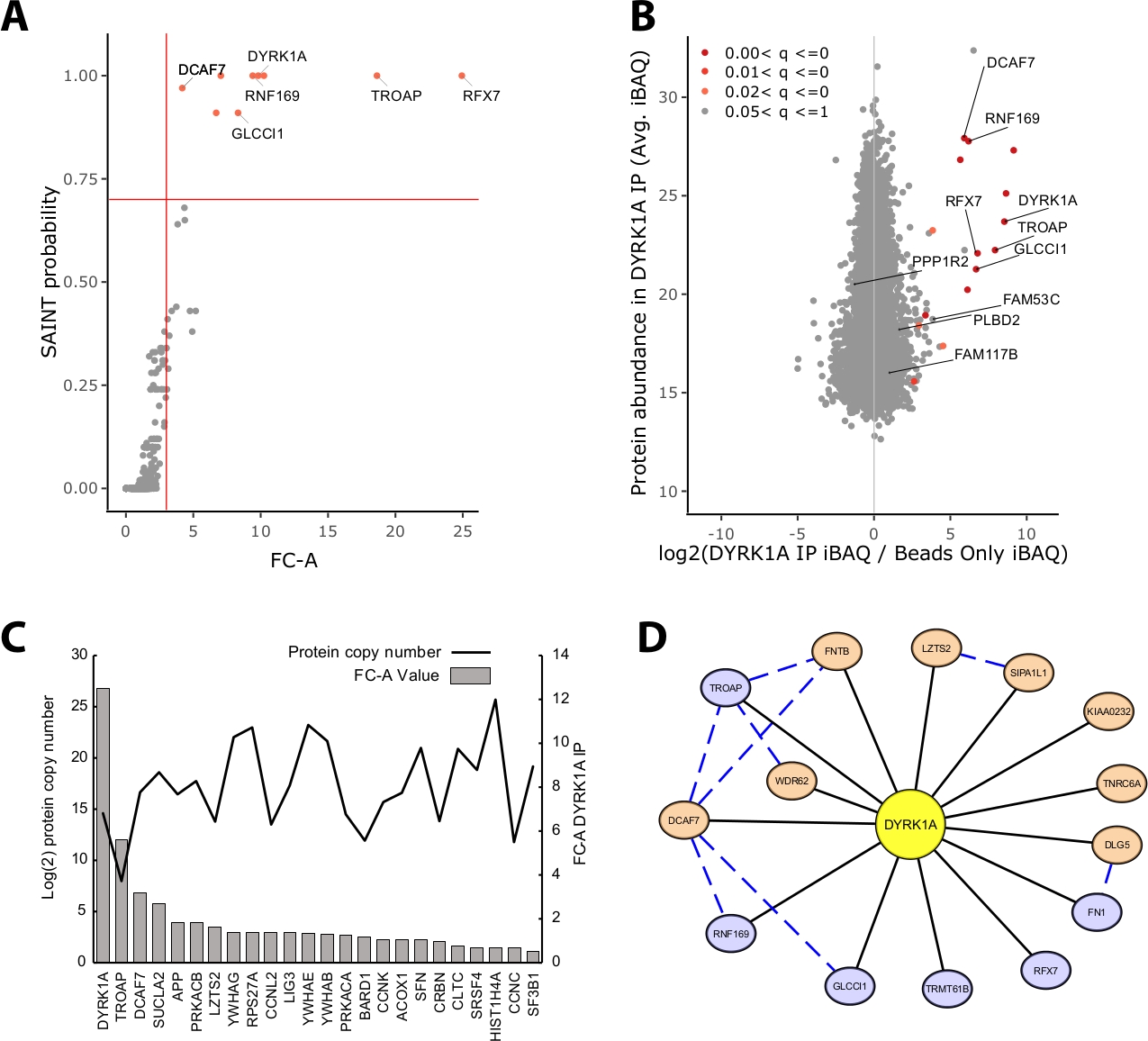{kind=link}
| Protease-Inhibitor (PI)-Mischung | |
| Reagenz | Endgültige Konzentration |
| Natriummetabisulfit | 1 mM |
| Benzamidin | 1 mM |
| Dithiothreitol (DTT) | 1 mM |
| Phenylmethansulfonylfluorid (PMSF) | 0,25 mM |
| Phosphatase-Inhibitor (PhI)-Mischung | |
| Reagenz | Endgültige Konzentration |
| Mikrocystin LR | 1 M |
| Natrium-Orthovanadate | 0,1 mM |
| Natriumfluorid | 5 mM |
| Subzelluläre Fraktionierungspuffer: | |
| Puffer A pH 7,9 | |
| Reagenz | Endgültige Konzentration |
| HEPES | 10 mM |
| MgCl2 | 1,5 mM |
| Kcl | 10 mM |
| Puffer B pH 7,9 | |
| Reagenz | Endgültige Konzentration |
| HEPES | 20 mM |
| MgCl2 | 1,5 mM |
| Nacl | 420 mM |
| Ethylendiamintetraessigsäure (EDTA) | 0,4 mM |
| Glycerin | 25% (v/v) |
| Puffer C pH 7,9 | |
| Reagenz | Endgültige Konzentration |
| HEPES | 20 mM |
| MgCl2 | 2 mM |
| Kcl | 100 mM |
| Ethylendiamintetraessigsäure (EDTA) | 0,4 mM |
| Glycerin | 20% (v/v) |
| Immunpräzipitationspuffer: | |
| IP-Puffer 1 | |
| Reagenz | Endgültige Konzentration |
| HEPES | 20 mM |
| Kcl | 150 mM |
| Edta | 0,1 mM |
| NP-40 | 0,1% (v/v) |
| Glycerin | 10% (v/v) |
| IP-Puffer 2 | |
| Reagenz | Endgültige Konzentration |
| HEPES | 20 mM |
| Kcl | 500 mM |
| Edta | 0,1 mM |
| NP-40 | 0,1% (v/v) |
| Glycerin | 10% (v/v) |
| SDS Alkylierungspuffer pH 8,5 | |
| Reagenz | Endgültige Konzentration |
| Sds | 4% (v/v) |
| Chloracetamid | 40 mM |
| TCEP | 10 mM |
| Tris | 100 mM |
| UA pH 8,5 | |
| Reagenz | Endgültige Konzentration |
| Harnstoff | 8 M |
| Tris | 0,1 M |
| * HPLC-Grade H2O verwenden | |
Tabelle 1: Pufferzusammensetzungen
Ergänzende Codierungsdateien. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Der hier skizzierte Proteomik-Workflow bietet eine effektive Methode zur Identifizierung von proteinreichen Proteininteraktoren für ein Protein von Interesse. Dieser Ansatz verringert die Probenkomplexität durch subzelluläre Fraktion und konzentriert sich auf die Erhöhung der Identifikationsinteraktionspartner durch robuste Probenvorbereitung, Offline-Probenbereinigung und strenge Qualitätskontrolle des LC-MS-Systems. Die hier beschriebene nachgelagerte Datenanalyse ermöglicht eine einfache statistische Auswertung der Proteine, die als mit dem Köder koreinigend identifiziert wurden. Aufgrund einer hohen Anzahl experimenteller Variablen (Skala, Zelllinie, Antikörperwahl) erfordert jedes Experiment jedoch unterschiedliche Cutoffs und Überlegungen zur Datenvisualisierung und -anreicherung.
Die erste Design-Überlegung in einem IP-MS-Experiment ist die Auswahl von Antikörpern, die zusammen mit ihren interagierenden Partnern zur Koreinigung des von Interesse interessierten Proteins verwendet werden. Während die Verfügbarkeit kommerzieller Antikörper in den letzten Jahrzehnten auf größere Teile des menschlichen Proteoms ausgeweitet wurde, gibt es immer noch viele Proteine, für die Reagenzien begrenzt sind. Darüber hinaus können Antikörper, die für Anwendungen wie den Western-Blot-Nachweis validiert wurden, in einem Immunpräzipationsexperiment nicht in der Lage sein, das Zielprotein selektiv anzureichern. Vor der Durchführung eines groß angelegten Interaktionsproteomik-Experiments wird vorgeschlagen, eine IP von einer 90% konfluenten 10 cm Schale oder einer gleichwertigen Zellnummer abzuschließen und das Zielprotein von Interesse durch Western Blotting zu untersuchen. Wenn mehr als ein einziger Antikörper für Immunpräzipitation verfügbar ist, wird zusätzlich vorgeschlagen, mehrere Antikörper auszuwählen, die Epitope in verschiedenen Teilen des Proteins erkennen. Die Bindung eines Antikörpers an ein Köderprotein kann die notwendige Bindungsschnittstelle für vermeintlich eimierende Partner ausblenden. Die Auswahl eines sekundären Epitops für das Köderprotein erhöht die Abdeckung des Interaktionsprofils, das durch ein Experiment auf Derspektrometrie identifiziert wird.
Eine zweite wichtige Überlegung liegt in der Auswahl der geeigneten Kontrolle, um vertrauensbesonnene Wechselwirkungen von vertrauensarmen oder unspezifischen Wechselwirkungen von solchen zu unterscheiden, die als mit dem Köder koeiniger identifiziert wurden. Die strengste Kontrolle für ein IP-MS-Experiment besteht darin, die Immunpräzipitation aus einer CRISPR KO-Zelllinie des Köders abzuschließen. Eine solche Kontrolle ermöglicht die Identifizierung und Filterung von unspezifischen Proteinen, die direkt an den Antikörper und nicht an das Köderprotein binden. In Fällen, in denen die Erzeugung einer CRISPR KO-Zelllinie jedes Köderproteins nicht möglich ist, kann eine IgG-Perlenkontrolle desselben Isotyps des Köderantikörpers verwendet werden. In Experimenten, die ein Panel von Antikörpern verwenden, die mehrere Arten darstellen, kann die Verwendung einer Perlen-Only-Kontrolle angemessen sein, erhöht aber die Rate der falsch positiven Ergebnisse, die als hochvertrauensgefährdete Interaktoren identifiziert wurden.
Die Auswahl der in einem IP-MS-Experiment verwendeten Zelllinie hängt von mehreren Schlüsselfaktoren ab. Proteinexpression und Lokalisation sind weitgehend vom Zelltyp abhängig. Während für die meisten Gene in vielen häufig verwendeten Zelllinien RNA-Expressionsschätzungen gefunden werden können, ist die Proteinexpression schlecht mit der RNA-Expression korreliert und muss experimentell bestimmt werden25. Zelllinien, in denen ein Köderprotein in sehr geringer Kopierzahl exprimiert wird, sollten vermieden werden, um Probleme zu umgehen, die mit drastischen Erhöhungen der Zellkulturskala verbunden sind, die erforderlich sein können. Es sollte jedoch beachtet werden, dass die Probenvorbereitung für den Nachweis von Proteinen mit sehr geringer Häufigkeit optimiert werden kann. Die FASP-Methode (Filter Aided Sample Prep) ist robust, kann aber zu einem Peptidverlust von mehr als 50 % in einer Probe führen. Die Single-Pot Solid-Phase-Enhanced Sample Preparation (SP3) ist eine effiziente Methode zur Generierung von Proben für die Massenspektrometrieanalyse, die den Probenverlust minimiert26. Die erhöhte Rückgewinnung, die durch die SP3-Methode der Probenvorbereitung ermöglicht wird, kann eine nützliche Alternative in diesem Workflow für die Quantifizierung von Proteinen sein, die in die Nähe der Nachweisgrenze fallen.
Dieser Proteomik-Workflow wurde in vielen Kernködern angewendet, einschließlich Kiinasen, E3-Ubiquitin-Ligasen und Gerüstbestandteilen von Multisubunit-Komplexen. Unter der Annahme einer ordnungsgemäßen Validierung von Antikörperreagenzien führt die erfolgreiche Ausführung dieses Workflows zum Nachweis von proteinreichen Partnern für kernkraft für ein Protein von Interesse.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Arbeit wurde durch ein Grand Challenge-Stipendium an W.M.O. vom Linda Crnic Institute for Down-Syndrom und durch eine DARPA-Kooperationsvereinbarung 13-34-RTA-FP-007 unterstützt. Wir danken Jesse Kurland und Kira Cozzolino für ihre Beiträge zur Lektüre und Kommentierung des Manuskripts.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin, 0.1% EDTA | Thermo Fisher Scientific | 25200056 | |
| 1.5 ml low-rention microcentrifuge tubes | Fisher Scientific | 02-681-320 | |
| 4-20% Mini PROTEAN TGX Precast Protein Gels | Bio-Rad | 4561096 | |
| acetone (HPLC) | Thermo Fisher Scientific | A949SK-4 | |
| Amicon Ultra 0.5 ml 30k filter column | Millipore Sigma | UFC503096 | |
| Benzamidine | Sigma-Aldrich | 12072 | |
| benzonase | Sigma-Aldrich | E1014 | |
| Chloroacetamide | Sigma-Aldrich | C0267 | |
| Dialysis tubing closure | Caroline Biological Supply Company | 684239 | |
| DTT | Sigma-Aldrich | 10197777001 | |
| EDTA | Sigma-Aldrich | EDS | |
| GAPDH antibody | Santa Cruz Biotechnology | Sc-47724 | |
| Glycerol | Fisher Scientific | 887845 | |
| Glycine | Sigma-Aldrich | G8898 | |
| HeLa QC tryptic digest | Pierce | 88329 | |
| HEPES | Fisher Scientific | AAJ1692630 | |
| insulin | Thermo Fisher Scientific | 12585014 | |
| iodoacetamide | Sigma-Aldrich | I1149 | |
| KONTES Dounce homogenizer 7 ml | VWR | KT885300-0007 | |
| Large Clearance pestle 7ml | VWR | KT885301-0007 | |
| Lysyl endopeptidase C | VWR | 125-05061 | |
| Magnesium Chloride | Sigma-Aldrich | 208337 | |
| Microcystin | enzo life sciences | ALX-350-012-C100 | |
| Nonidet P 40 Substitute solution | Sigma-Aldrich | 98379 | |
| p84 antibody | GeneTex | GTX70220 | |
| Phosphate Buffered Saline | |||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 | |
| Pierce BSA Protein Digest, MS grade | Thermo Fisher Scientific | 88341 | LCMS QC |
| Pierce C18 spin columns | Thermo Fisher Scientific | PI-89873 | |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | 90057 | For mass spectrometry sample prep |
| PMSF | Sigma-Aldrich | P7626 | |
| Potassium Chloride | Sigma-Aldrich | P9541 | |
| Protein A Sepharose CL-4B | GE Healthcare Bio-Sciences | 17-0780-01 | |
| Protein G Sepharose 4 Fast Flow | GE Healthcare Bio-Sciences | 17-0618-01 | |
| SDS | Sigma-Aldrich | L3771 | |
| Silica emitter tip | Pico TIP | FS360-20-10 | |
| Small Clearance pestle 7ml | VWR | KT885302-0007 | |
| Sodium Chloride | Sigma-Aldrich | S3014 | |
| Sodium Fluoride | Sigma-Aldrich | 201154 | |
| Sodium metabisulfite | Sigma-Aldrich | 31448 | |
| Sodium orthovanadate | Sigma-Aldrich | S6508 | |
| Spectra/ Por 8 kDa 24 mm dialysis tubing | Thomas Scientific | 3787K17 | |
| TC Dish 150, Standard | Sarstedt | 83.3903 | Tissue culture dish for adherent cells |
| TCA | Sigma-Aldrich | T9159 | |
| TCEP | Thermo Scientific | PG82080 | |
| TFA | Thermo Fisher Scientific | 28904 | |
| Thermo Scientific Orbitrap Fusion MS | Thermo Fisher Scientific | ||
| Trizma Base | Sigma-Aldrich | T6066 | |
| Urea | Thermo Fisher Scientific | 29700 | |
| Waters ACQUITY M-Class UPLC | Waters | ||
| Waters ACQUITY UPLC M-Class Column Reversed-Phase 1.7µm Spherical Hybrid (1.7 µm, 75 µm x 250 mm) | Waters | 186007484 | nanoflow C18 column |
Referenzen
- Varjosalo, M., et al. The protein interaction landscape of the human CMGC kinase group. Cell Reports. 3, 1306-1320 (2013).
- Kimple, M. E., Brill, A. L., Pasker, R. L. Overview of Affinity Tags for Protein Purification. Current Protocols in Protein Science. 73, (2013).
- Mahmood, N., Xie, J. An endogenous 'nonspecific' protein detected by a His-tag antibody is human transcription regulator YY1. Data in Brief. 2, 52 (2015).
- Zordan, R. E., Beliveau, B. J., Trow, J. A., Craig, N. L., Cormack, B. P. Avoiding the ends: internal epitope tagging of proteins using transposon Tn7. Genetics. 200, 47-59 (2015).
- Gibson, T. J., Seiler, M., Veitia, R. A. The transience of transient overexpression. Nature Methods. 10, 715-721 (2013).
- Bronicki, L. M., et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. European Journal of Human Genetics. 23, 1482-1487 (2015).
- Antonarakis, S. E. Down syndrome and the complexity of genome dosage imbalance. Nature Reviews Genetics. , (2016).
- Dowjat, W. K., et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neuroscience Letters. 413, 77-81 (2007).
- Fotaki, V., et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Molecular and Cellular Biology. 22, 6636-6647 (2002).
- Hämmerle, B., Elizalde, C., Tejedor, F. J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. European Journal of Neuroscience. 27, 1061-1074 (2008).
- Funakoshi, E., et al. Overexpression of the human MNB/DYRK1A gene induces formation of multinucleate cells through overduplication of the centrosome. BMC Molecular and Cell Biology. 4, 12 (2003).
- Yu, D., Cattoglio, C., Xue, Y., Zhou, Q. A complex between DYRK1A and DCAF7 phosphorylates the C-terminal domain of RNA polymerase II to promote myogenesis. Nucleic Acids Research. , 1-14 (2019).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 76, 4350-4354 (1979).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6, 359-362 (2009).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11, 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13, 731-740 (2016).
- Mellacheruvu, D., et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10, 730-736 (2013).
- Choi, H., et al. SAINT: Probabilistic scoring of affinity purificationg-mass spectrometry data. Nature Methods. 8, 70-73 (2011).
- Shannon, P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 13, 2498-2504 (2003).
- Szklarczyk, D., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. 45, 362-368 (2017).
- Bindea, G., et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 25, 1091-1093 (2009).
- Guard, S. E., et al. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Scientific Reports. 9, 6539 (2019).
- Razick, S., Magklaras, G., Donaldson, I. M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics. 9, 405 (2008).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11, 319-324 (2014).
- Liu, Y., Beyer, A., Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 165, 535-550 (2016).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10, 757 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten