Method Article
Label-Free Immunoprecipitation Mass Spectrometry Workflow for Large-scale Nuclear Interactome Profiling
In This Article
Summary
Described is a proteomics workflow for identifying protein interaction partners from a nuclear subcellular fraction using immunoaffinity enrichment of a given protein of interest and label-free mass spectrometry. The workflow includes subcellular fractionation, immunoprecipitation, filter aided sample preparation, offline cleanup, mass spectrometry, and a downstream bioinformatics pipeline.
Abstract
Immunoaffinity purification mass spectrometry (IP-MS) has emerged as a robust quantitative method of identifying protein-protein interactions. This publication presents a complete interaction proteomics workflow designed for identifying low abundance protein-protein interactions from the nucleus that could also be applied to other subcellular compartments. This workflow includes subcellular fractionation, immunoprecipitation, sample preparation, offline cleanup, single-shot label-free mass spectrometry, and downstream computational analysis and data visualization. Our protocol is optimized for detecting compartmentalized, low abundance interactions that are difficult to identify from whole cell lysates (e.g., transcription factor interactions in the nucleus) by immunoprecipitation of endogenous proteins from fractionated subcellular compartments. The sample preparation pipeline outlined here provides detailed instructions for the preparation of HeLa cell nuclear extract, immunoaffinity purification of endogenous bait protein, and quantitative mass spectrometry analysis. We also discuss methodological considerations for performing large-scale immunoprecipitation in mass spectrometry-based interaction profiling experiments and provide guidelines for evaluating data quality to distinguish true positive protein interactions from nonspecific interactions. This approach is demonstrated here by investigating the nuclear interactome of the CMGC kinase, DYRK1A, a low abundance protein kinase with poorly defined interactions within the nucleus.
Introduction
The human proteome exhibits vast structural and biochemical diversity through the formation of stable multisubunit complexes and transient protein-protein interactions. Accordingly, the identification of interaction partners for a protein of interest is commonly required in investigations to unravel molecular mechanism. Recent advances in affinity purification protocols and the advent of high-resolution fast-scanning mass spectrometry instrumentation have enabled easy mapping of protein interaction landscapes in a single unbiased experiment.
Protein interaction protocols commonly employ ectopic expression systems with affinity-tagged fusion constructs to identify protein interactions without requiring high-quality antibodies recognizing a protein of interest1,2. However, epitope tag-based methods have several drawbacks. Physical interactions with the epitope may lead to the detection of nonspecific copurifying proteins3. Additionally, fusion of these epitope tags to the N- or C-terminal of a protein may block native protein-protein interactions, or disrupt protein folding to promote non-physiological conformations4. Furthermore, ectopic expression systems typically overexpress the bait protein at supraphysiological concentrations, which can result in the identification of artifactual protein interactions, particularly for dosage-sensitive genes5. To circumvent these issues, the endogenous bait protein can be immunoprecipitated along with associated interacting prey proteins, assuming availability of a high-quality antibody that recognizes the native protein.
Provided here is an interaction proteomics workflow for detecting the nuclear interactome of an endogenous protein using the CMGC protein kinase DYRK1A as an example. Disruption of DYRK1A copy number, activity level, or expression can cause severe intellectual disability in humans, and embryonic lethality in mice6,7,8,9. DYRK1A exhibits dynamic spatiotemporal regulation10, and compartmentalized protein interactions11,12, requiring approaches capable of detecting low abundance interaction partners specific to different subcellular compartments.
This protocol employs cellular fractionation of human HeLa cells into cytosol and nucleoplasm fractions, immunoprecipitation, sample preparation for mass spectrometry, and an overview of a bioinformatic pipeline for evaluating data quality and visualizing results, with R scripts provided for analysis and visualization (Figure 1). Proteomics software packages used in this workflow are all freely available for download or can be accessed through a web interface. For additional information on software and computational methods, in-depth tutorials and instruction are available at the links provided.
Protocol
NOTE: All buffer compositions and protease mixtures are outlined in Table 1.
1. Preparation of cells
NOTE: A starting material of 1−10 mg nuclear lysate per replicate is desired for this immunoprecipitation mass spectrometry (IP-MS) approach. Cell quantities will be given for 1 mg of nuclear immunoprecipitations in triplicate plus triplicate controls.
- If using an adherent cell line, grow the cells to 90% confluency in 3 x 15 cm dishes per replicate prior to harvesting.
NOTE: It is recommended to perform a minimum of three replicate immunoprecipitations for bait and control conditions. This protocol will assume the use of ‘beads only’ controls, which control for abundant nonspecific interactions with the beads, starting at section 4. Other types of controls may be useful. These are described in depth in the discussion section.- Wash plates 2x with phosphate buffered saline (PBS) and trypsinize cells using 3 mL of 0.25% trypsin per 15 cm plate. Spin down the cells at 1,200 x g for 5 min and decant the trypsin.
- For suspension cells, grow to a similar scale/density to achieve 70−80 mg of total protein.
- Pellet cells at 1,200 x g and 4 °C for 5 min. Decant the media carefully.
NOTE: Pellets can be combined during this step to enable efficient processing using the large-scale subcellular fractionation outlined in section 2.
- Pellet cells at 1,200 x g and 4 °C for 5 min. Decant the media carefully.
- Wash the cell pellet 2x with PBS + 5 mM MgCl2 supplemented with protease inhibitors (PIs) and phosphatase inhibitors (PhIs) (see Table 1).
NOTE: Cell pellets may be flash frozen in liquid nitrogen and stored at -80 °C until ready for fractionation.
2. Preparation of nuclear extract
NOTE: Protease and phosphatase inhibitors should be added to the fractionation buffers within 30 min of use.
- If frozen, thaw the cell pellets for 15 min in 1x pellet volume of cold Buffer A + PIs/ PhIs. Place the cell pellet on a nutator at 4 °C to aid in resuspension while thawing. Otherwise, resuspend the cell pellet from step 1.3 into 1x pellet volume Buffer A + PIs /PhIs.
- Pellet at 2,000 x g and 4 °C for 10 min. Decant the buffer.
- Suspend the cells with 5x the packed cell volume with Buffer A and incubate on ice for 20 min.
- Pellet at 2,000 x g and 4 °C for 10 min. Decant the buffer and resuspend with 2x original packed cell volume Buffer A + PIs/ PhIs and dounce ~7x with “A”/loose pestle.
- Centrifuge the lysate for 10 min at 2,000 x g and 4 °C.
- Carefully pipette off the supernatant and flash freeze with liquid nitrogen. Store the lysate at -80 °C. The supernatant from this step is the cytosolic subcellular fraction.
NOTE: The nuclear pellet can be saved during this step by flash freezing with liquid nitrogen and storing at -80 °C - Resuspend the pellet with 0.9x pellet volume of Buffer B + PIs/ PhIs and mix on a nutator for 5 min at 4 °C.
- To lyse the nuclei, dounce 20x with a tighter pestle “B”.
- Mix the nuclear lysate on a nutator for 30 min at 4 °C so that it is homogenous.
- Centrifuge the nuclear lysate for 30 min at 21,000 x g at 4 °C. Pipette off the supernatant and save as a soluble nuclear protein extract.
NOTE: Nuclease treatment of the resulting nuclear pellet allows for the recovery of a chromatin-associated protein fraction. - Dialyze the soluble nuclear extract against Buffer C + PIs for 3 h at 4 °C.
- Cut an appropriate length of 24 mm width dialysis tubing with an 8 kDa molecular weight cut off. Clamp one side of the tubing and load nucleoplasm into the tube. After loading the lysate, clamp the other end and submerge into a clean glass container containing Buffer C + PIs.
- Centrifuge the dialyzed nuclear extract/nucleoplasm at 21,000 x g at 4 °C for 30 min. Aliquot 3x 20 µL volumes of nuclear extract for fractionation validation by western blot. The nuclear extract used for IP-MS analysis can be aliquoted and flash frozen in liquid nitrogen and stored at -80 °C, if needed.
3. Validation of subcellular fractionation
- Complete a protein assay to determine the protein concentration of the nuclear lysate. A bicinchoninic acid protein assay provides sufficient sensitivity for downstream application.
- Load 20 µg of both the cytosolic and nuclear fractions on an SDS-PAGE gel for the western blot analysis as previously described13. Skip lanes when loading to avoid mischaracterization of a sample.
- Probe the western blot for p84 (THOC1) as a nuclear marker, and GAPDH as a cytosolic marker. Determine the extent of fractionation by the ratio of cytosolic marker in the nuclear fraction and vice versa.
NOTE: Antibodies for other nuclear and cytosolic markers may be used.
4. Immunoprecipitation of endogenous nuclear bait protein
NOTE: It is recommended to use low retention tubes from this point on. This will reduce the nonspecific binding to the tubes during sample handling and avoid unnecessary loss of sample. Additionally, ensure that LCMS grade H2O is used to prepare buffers for the remaining steps.
- Prepare a protein A/G bead mixture for each replicate by combining 12.5 µL of bead volume for both protein-A and protein-G in microcentrifuge tubes. Store the bead stocks as a slurry containing 20% ethanol. Determine the concentration of beads within the slurry %(v/v) and pipette the necessary volume using a pipette tip that has been cut on the tip to ensure that the beads can enter the tip.
- Wash the protein A/G bead mixture 2x with 300 µL of IP Buffer 1. Spin the beads at 1,500 x g at 4 °C for 1 min and decant buffer.
- Prepare the antibody-protein A/G beads: To bind the antibody to the beads, add 300 µL of IP Buffer 1 and 10 µg of the desired antibody. Allow the bead/antibody mixture to rock on a nutator at 4 °C overnight. For bead-only controls, do not add any antibody.
NOTE: A total of 10 µg of antibody per replicate can be used as a starting point, but the exact amount will need to be optimized for each individual antibody and scale of the lysate used in the experiment - Thaw the nuclear lysates from step 2.10 in a water bath and aliquot appropriate volumes into low retention microcentrifuge tubes for 1 mg protein input per replicate.
- Spin the lysate at 16,000 x g for 30 min and transfer the supernatant to a new tube.
- Add 1 µL of benzonase (250 units/µL) per 1 mg of the nuclear lysate and rock on a nutator at 4 °C for 10−15 min.
- Prepare beads for preclearing the lysate. Add 12.5 µL of each protein A and protein G beads to 1.5 mL low retention tubes as in step 4.1. Wash 2x with IP Wash Buffer 1 + PIs and decant the buffer.
- Add 1 mg of the nuclear lysate to the beads from step 4.5. Incubate while rocking on a nutator for 1 h at 4 °C.
- Centrifuge precleared lysates at 1,500 x g and 4 °C for 1 min.
- While nuclear lysates are incubating with beads in step 4.5.1, wash the antibody-protein A/G beads 2x with IP Buffer 1 + PIs. Centrifuge at 1,500 x g and 4 °C for 1 min and decant the buffer.
- Transfer the precleared nuclear lysate from step 4.6.1 onto the antibody-protein A/G beads. Incubate while rocking on a nutator at 4 °C for 4 h. Centrifuge following the incubation at 1,500 x g and 4 °C for 1 min.
- Transfer the supernatant into tubes labeled as the flow through for each replicate.
- Wash the antibody-protein A/G beads with 1 mL of IP Buffer 2 + PIs. Centrifuge at 1,500 x g and 4 °C for 1 min, decant buffer, and repeat for a total of 3x.
- Wash the beads 2x with 1 mL of IP Buffer 1+ PIs centrifuging as in the previous step. Ensure that all buffer is removed after the last wash.
- Elute 2x with 20 µL of 0.1 M glycine (pH 2.75) for 30 min each. Ensure that the tubes is rocking during the incubation with the elution buffer. Spin at 750 x g and 4 °C for 1 min and pipette off the supernatant after each 30 min incubation.
NOTE: While the low pH glycine method described here elutes most bait proteins, some antibody-antigen interactions require more stringent buffer conditions. - Flash freeze eluates in liquid nitrogen and store at -80 °C.
5. Sample preparation
NOTE: Insulin spiked into the immunoprecipitation elution samples aids in the recovery of proteins during trichloroacetic acid (TCA) precipitation and sample processing, which is important for low abundance endogenous bait proteins.
- Thaw the eluates at room temperature if frozen.
- Place the samples on ice and add 10 µL of 1.0 mg/mL insulin for every 100 µL of eluate. Vortex and then immediately add 10 µL of 1% sodium deoxycholate. Vortex the sample again and add 30 µL of 20% TCA followed by one final vortex.
- Incubate the samples on ice for 20 min, then centrifuge at 21,000 x g at 4 °C for 30 min.
- Aspirate the supernatant and add 0.5 mL of acetone that has been prechilled to -20 °C. Vortex and then spin at 21,000 x g and 4 °C for 30 min. Repeat this step.
- Aspirate the supernatant and air dry the pellet remaining in the bottom of the tube.
- Prepare the sample for mass spectrometry using a modified Filter Aided Sample Prep (FASP) method, optimized for reducing sample handling, as outlined below14.
- Resuspend the protein pellet from step 5.1.4 with 30 µL of SDS Alkylation Buffer (see Table 1). Incubate the sample on a 95 °C heat block for 5 min. Let it cool at room temperature for 15 min before preceding to the next step.
- Add 300 µL of UA solution and 30 µL of 100 mM TCEP to each sample. Load this solution onto a 30k centrifugal filter. Spin the centrifugal filter at 21,000 x g at room temperature for 10 min.
NOTE: The bait protein and its putative interactors should be bound to the filter at this point. However, the flow through may be kept, in case there is a problem with the filter. - Wash the filter with 250 µL of UA and centrifuge at 21,000 x g for 10 min. Decant the flow through and repeat for a total of 3x.
- Wash the filter with 100 µL of 100 mM Tris pH 8.5 and centrifuge at 21,000 x g for 10 min. Decant the flow through and repeat for a total of 3x.
- Add 3 µL of 1 µg/µL Lys C resuspended in 0.1 M Tris pH 8.5. Fill the filters up to the 100 µL mark and allow to digest for 1 h at 37 °C while rocking on a nutator.
- Add 1 µL of 1 µg/ µL MS grade trypsin. Mix gently and allow for the trypsin to incubate with the sample overnight at 37 °C while rocking on a nutator.
- Centrifuge at 21,000 x g for 20 min to elute the peptide from the filter.
NOTE: Multiple rounds of centrifugation may be required to recover all the eluate. If this is not done, there is a potential for severe sample loss.
- Desalt the peptides using C18 spin columns. Follow the protocol provided by the manufacturer.
- Resuspend the lyophilized peptide in 7 µL of 0.1% TFA in 5% acetonitrile. Sonicate the sample for 3 min to ensure that the peptides have been resuspended. Spin down at 14,000 x g for 10 min.
- Transfer the resuspended peptide into an appropriate sample vial to for loading onto the liquid chromatography–mass spectrometry (LC/MS) system.
6. LC/MS system suitability
NOTE: Due to the small scale and generally lower abundance of protein from affinity-purified samples, it is critical that the LC/MS platform operates at a maximal sensitivity and robustness.
- Add 1 mL of LC/MS grade formic acid to 1 L of LC/MS grade water for mobile phase A, and add 1 mL of LC/MS grade formic acid to 1 L of LC/MS grade acetonitrile for mobile phase B.
- Prepare or install a 75 µm fused-silica capillary column packed with <2 µm reversed-phase C18 resin that is ≥250 mm in length. Best results will be had with a direct injection of samples into the column.
- Purge the Ultra Performance Liquid Chromatography (UPLC) system with fresh mobile phases. With a C18 column installed, establish a stable flow rate and electrospray with a suitable emitter (i.e., 20 µm id x 360 µm od pulled to a 10 µm tip). Maintain the column at 40−60 °C.
- Test the overall LC/MS system performance by injecting a complex quality control standard, such as 100−200ng of a HeLa whole cell lysate tryptic digest. Elute with a suitable gradient for a complex sample (i.e., 2−3 h gradient elution time). Establish a baseline system performance of the peptide and protein identifications.
NOTE: For best results, 3,000−5,000 or more protein identifications from 20,000−35,000 unique peptides will provide optimal performance for experimental samples. - For routine LC/MS system suitability, inject 100−200 fmol or less of a single protein digest standard, such as Bovine Serum Albumin (BSA). Elute with a short gradient (i.e., 20−30 min).
NOTE: Multiple injections of a protein digest will help establish baseline LC/MS system performance, and repeat injection after each IP-MS sample provides a measure of system performance throughout the experiment and allows for the detection of instrument drift, which can bias label-free experiments. A baseline of the select individual peak intensities and peak shapes will inform on MS, LC, and column performance. - To avoid overloading the analytical column, load a small portion (15−30% of the total) of an experimental sample onto the column and separate using a gradient suitable for complex samples (i.e., 2−3 h). If the number of protein identifications is unsatisfactory, load all of the sample onto the column.
- Run a single protein digest standard in between samples to monitor LC/MS system performance and sample carryover. Multiple standards may be required to reduce sample carryover depending on your samples.
7. Data Processing
- Download the proteomics software package MaxQuant found at https://www.maxquant.org/.
NOTE: This will be used to process the RAW MS data file from step 6.6 into data tables of protein IDs, gene names, and quantitative values associated with the identification of these for downstream analysis.- Select Load within the Input Data subheader of the Raw Data tab. Open the file location where the MS raw files are stored and select raw files for each MS/MS run.
- Click on the Group-Specific tab and select Digestion. Within the enzyme list select LysC and click on the right arrow to add this enzyme into the list that will be used in the search. Next, select Instrument and ensure that the correct instrument type appears in the drop-down list at the top of the screen. Leave other group-specific search parameters on standard settings.
- Click on the Global Parameters tab and select Sequences. Add the appropriate FASTA file for the taxonomy that will be used in this search. Peptides will not be assigned properly if this is not done. For the human proteome, download the FASTA file from UniProt at https://www.uniprot.org/help/human_proteome.
- Within the Global Parameters tab, click on Protein Quantification. Within the Peptides for Quantification drop-down menu, select Unique + Razor.
NOTE: MaxQuant offers alternate quantitation of proteins through intensity-based absolute quantification (iBAQ) and label-free quantitation (LFQ). However, peptide count information is sufficient for the downstream analysis in this protocol15. - In the bottom left of the MaxQuant interface, select the number of processors to be used for the search. This will directly affect the length of time required for the run, so select as many as possible for this). Click Start in the bottom left of the screen to start the run. Select the Performance tab at the top of the screen to view the progress of the search.
- When the run has completed, open the proteingroups.txt file in Perseus, a proteomics computation platform, or other spreadsheet program to view the data16.
- Use Perseus to remove common contaminants and hits to reversed protein sequences. Follow the detailed Perseus documentation at http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
NOTE: Opening the proteingroups.txt file in analysis software (e.g., Excel) will automatically corrupt certain gene and protein names.
- Use Perseus to remove common contaminants and hits to reversed protein sequences. Follow the detailed Perseus documentation at http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
- Analyze experimental data using the Contaminant Repository for Affinity Purification (CRAPome). Register an account at this repository http://crapome.org/ and follow the tutorial as needed17,18.
- Use the Analyze Your Data workflow found on the CRAPome homepage. Select external controls that correspond to the affinity purification system used in this interaction experiment.
NOTE: These controls can be used to calculate a second fold change enrichment that is useful for detecting common contaminants. - Generate an input file from the proteingroups.txt output from MaxQuant using Perseus or a suitable spreadsheet application. Details for manual formatting can be found at http://crapome.org/?q=fileformatting. Alternatively, use the provided R script “export_CRAPomeSAINT_Input_File.R” to generate the SAINT/CRAPome input file. See README.txt in the Supplemental Coding Files.
- Run an analysis to determine fold-change enrichment and SAINT (Significance Analysis of INTeractome) probability for each bait protein in the immunoprecipitation. Ensure that 'User Controls’ is selected in the drop-down menu under FC-A, ‘CRAPome controls’ or ‘All Controls’ are selected in the FC-B drop-down and Probability Score is selected to generate SAINT probabilities. When the run has concluded, view the output that is available under ‘Analysis Results’ along with a job ID. Download the data matrix from the ‘Analysis Results’ for future plotting and data visualization.
- Use the Analyze Your Data workflow found on the CRAPome homepage. Select external controls that correspond to the affinity purification system used in this interaction experiment.
- Plot proteins as a function of FC-A (IPs vs. user controls) and SAINT probability by following the R-Scripts as provided in Supplemental Coding Files.
NOTE: A set of R scripts is provided for producing plots of FC-A vs. SAINT probability and iBAQ vs. log2 (protein abundance), colored by the adjusted p value range from empirical Bayes analysis of the label-free intensities. The details of the differential statistical analysis and plotting are found in README.txt and the R script “main_differential_analysis.R” in the Supplemental Coding Files. - Evaluate where known interacting proteins of the bait protein are ranked by FC-A and SAINT. Make a cutoff of FC-A > 3.00 and SAINT > 0.7 for single bait experiments in triplicate as a starting point.
NOTE: Selection of cutoffs for a “high-confidence” interactor and a “low-confidence” interactor must be informed by biological information.
8. Data visualization
NOTE: There are many programs that can effectively visualize proteomics data (e.g., R, Perseus, Cytoscape, STRING-DB). Analyzing the connectivity between high-confidence hits, and functional enrichment of these interactors can be a useful strategy for prioritizing hits for further validation and functional characterization.
- Download Cytoscape, an open source network visualization tool at https://cytoscape.org/download.html19.
- Prepare an input file for interaction data as a tab delimited file formatted with three columns: bait (source node), prey (target node), type of interaction (edge type). This can be done in Perseus or any spreadsheet program of your choice.
- Select the Import Table from File icon towards the top left of the program (designated by a downwards arrow and a matrix in the icon). Cytoscape will auto-populate the interaction data into a network ready for custom formatting and design.
- Select the Style tab on the control panel for Cytoscape and click on the squares in the Def column to adjust the attribute for the entire network. Select specific nodes or edges in the network and then select the square within the Byp. column of the style menu to bypass the default settings and adjust only selected objects. Alternatively, click on the drop-down menu at the top of the style menu to view the preset network formats.
NOTE: STRING-db protein-protein interaction data may be integrated into this network at this time either manually through the input file or through various enrichment tools available as plug-ins in Cytoscape, http://apps.cytoscape.org/20. A recommended cytoscape plug-in for enrichment analysis is found at http://apps.cytoscape.org/apps/cluego21. - To increase the confidence in the dataset generated in this workflow, perform reciprocal IP-MS or IP-western experiments that target prey proteins of interest as the bait.
Results
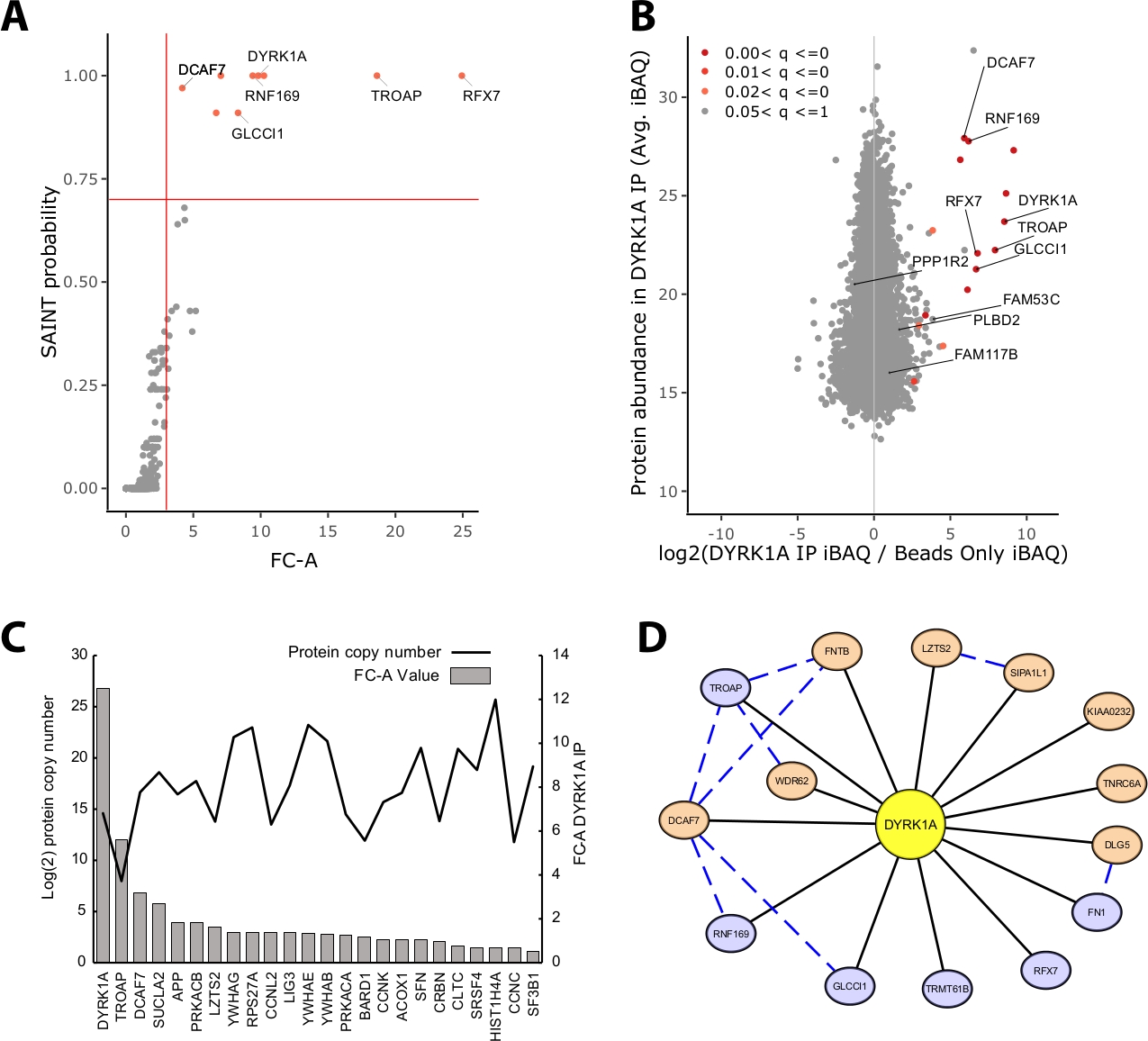
The majority of protein mass identified in an IP-MS experiment consists of nonspecific proteins. Thus, one of the key challenges of an IP-MS experiment is the interpretation of which proteins are high-confidence interactors vs. nonspecific interactors. To demonstrate the crucial parameters used in the evaluation of data quality the study analyzed triplicate immunoprecipitations from 5 mg of HeLa nuclear extract utilizing a bead only control. The first internal check to ensure that an IP-MS experiment is reliable is whether the bait protein ranks as one of the highest enriched proteins identified by both fold-change over control and SAINT probability. In this case, the bait DYRK1A ranked among the top three enriched proteins over the control (Figure 2A,B). In a nuclear interactome study of DYRK1A utilizing four independent antibodies, an FC-A cutoff of >3.00 and SAINT probability cutoff >0.7 provided a stringent cutoff for identification of both novel and previously validated interactors22. When applied to this experiment, a clear separation could be seen between the high-confidence interactors and >95% of copurified proteins identified as nonspecific (Figure 2A,B). Applying both a fold change enrichment and probability threshold increases stringency by requiring a consistently high enrichment of protein IDs across biological replicates.
In addition to statistical scoring, the CRAPome analysis workflow also maps previously reported interactions onto bait-prey data23. While this mapping can be useful for thresholding high and low-confidence interactions, previously reported interactions can score poorly by FC-A and SAINT probabilities, potentially indicating that many known interactions of a given bait may exist only in specific cell types, contexts, or organelles. For the example DYRK1A dataset, iREF interactor FC-A values were as low as 0.45, representing a very low enrichment over control (Figure 2C). To avoid inflation of false positives, statistical thresholding should be performed in a manner that prioritizes stringency over reduction of false negatives. It should be noted that the detection of these interactions was independent of protein abundance (Figure 2C). Calculated absolute copy number of each iREF interaction within HeLa cells showed no correlation to the detection levels of an interaction partner by IP-MS24.
Cytoscape serves as an effective tool for visualizing multiple layers of interaction data19. In the DYRK1A immunoprecipitation experiment described here, the combined use of FC-A > 3.0 and SAINT > 0.9 reduced the list of high-confidence interactors to six proteins (Figure 2D). However, when applying an FC-A cutoff of > 3.0 in isolation, eight additional proteins were added to the network. These additional protein interactors have high connectivity with the interactors already in the network, suggesting they are associated in similar complexes or functional roles. To this end, evidence from the STRING-DB of protein-protein interactions was integrated into this network as blue dashed lines20. While this single-bait, triplicate experiment provides a limited sample of the full DYRK1A interaction network, the use of additional baits, replicates, and integration of large public data sets can be used to expand the network of high-confidence interactions. The statistical cutoffs will thus be specific to each individual experiment and will need to be evaluated thoroughly.

Figure 1: Representative proteomics workflow for subcellular IP-MS. Cells are grown in either 4 L round bottom flasks or 15 cm tissue culture dishes and harvested at the same time for subcellular fractionation. Cells are fractionated into a cytosolic, nuclear, and a nuclear pellet, and immunoprecipitations are done from 1−10 mg of nuclear lysate using one or multiple antibodies recognizing the same bait. Filter aided sample prep (FASP) and offline sample cleanup are performed prior to single shot mass spectrometry. A downstream computational pipeline is used to process data into interpretable interaction data. Please click here to view a larger version of this figure.
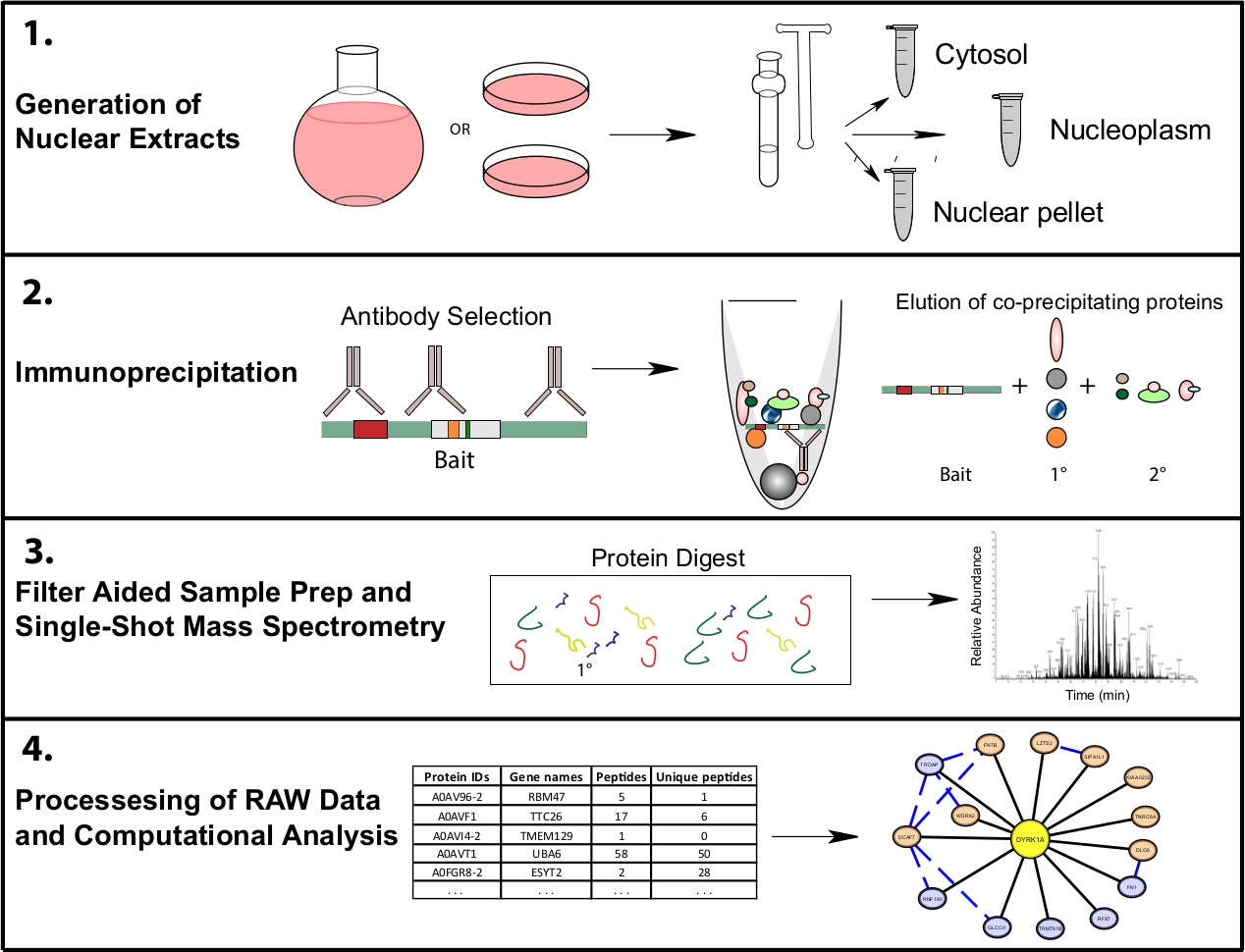{kind=link}

Figure 2: Representative data for a single-bait single-antibody IP-MS experiment. (A) FC-A and SAINT probability output from CRAPome analysis workflow for an optimal experiment using a single antibody for the kinase DYRK1A (n = 3). Beads-only controls were used for comparison. Red solid lines represent cutoffs set at FC-A > 3.00 and SAINT > 0.7. (B) MaxQuant protein abundance estimates (iBAQ) output vs. log2 ratio of protein abundance in DYRK1A IP to control, colored by the adjusted p value range from empirical Bayes analysis of the label-free intensities. (C) FC-A and estimated copy number of proteins listed as interacting proteins in the iRef database23,24. (D) Cytoscape network visualization of DYRK1A interactors. Blue nodes = FC-A > 3.00, SAINT > 0.7. Orange nodes = FC-A > 3.00. Black edges = proteins identified as interactors in IPMS experiment. Blue dashed edge = SAINT interaction between prey protein (confidence > .150). Please click here to view a larger version of this figure.
{kind=link}
| Protease inhibitor (PI) mixture | |
| Reagent | Final Concentration |
| Sodium Metabisulfite | 1 mM |
| Benzamidine | 1 mM |
| Dithiothreitol (DTT) | 1 mM |
| Phenylmethanesulfonyl fluoride (PMSF) | 0.25 mM |
| Phosphatase Inhibitor (PhI) mixture | |
| Reagent | Final Concentration |
| Microcystin LR | 1 µM |
| Sodium Orthovanadate | 0.1 mM |
| Sodium fluoride | 5 mM |
| Subcellular fractionation Buffers: | |
| Buffer A pH 7.9 | |
| Reagent | Final Concentration |
| HEPES | 10 mM |
| MgCl2 | 1.5 mM |
| KCl | 10 mM |
| Buffer B pH 7.9 | |
| Reagent | Final Concentration |
| HEPES | 20 mM |
| MgCl2 | 1.5 mM |
| NaCl | 420 mM |
| Ethylenediaminetetraacetic acid (EDTA) | 0.4 mM |
| Glycerol | 25% (v/v) |
| Buffer C pH 7.9 | |
| Reagent | Final Concentration |
| HEPES | 20 mM |
| MgCl2 | 2 mM |
| KCl | 100 mM |
| Ethylenediaminetetraacetic acid (EDTA) | 0.4 mM |
| Glycerol | 20% (v/v) |
| Immunoprecipitation Buffers: | |
| IP Buffer 1 | |
| Reagent | Final Concentration |
| HEPES | 20 mM |
| KCl | 150 mM |
| EDTA | 0.1 mM |
| NP-40 | 0.1% (v/v) |
| Glycerol | 10% (v/v) |
| IP Buffer 2 | |
| Reagent | Final Concentration |
| HEPES | 20 mM |
| KCl | 500 mM |
| EDTA | 0.1 mM |
| NP-40 | 0.1% (v/v) |
| Glycerol | 10% (v/v) |
| SDS Alkylation Buffer pH 8.5 | |
| Reagent | Final Concentration |
| SDS | 4% (v/v) |
| Chloroacetamide | 40 mM |
| TCEP | 10 mM |
| Tris | 100 mM |
| UA pH 8.5 | |
| Reagent | Final Concentration |
| Urea | 8 M |
| Tris | 0.1 M |
| * use HPLC grade H2O | |
Table 1: Buffer compositions
Supplemental Coding Files. Please click here to download this file.
Discussion
The proteomics workflow outlined here provides an effective method for identifying high-confidence protein interactors for a protein of interest. This approach decreases the sample complexity through subcellular fraction and focuses on increasing the identification interaction partners through robust sample preparation, offline sample clean up, and stringent quality control of the LC-MS system. The downstream data analysis described here allows for a simple statistical evaluation of the proteins identified as copurifying with the bait. However, due to a high number of experimental variables (scale, cell line, antibody choice), each experiment requires different cutoffs and considerations regarding data visualization and enrichment.
The first design consideration in an IP-MS experiment is the selection of antibodies that will be used for copurification of the protein of interest along with its interacting partners. While the availability of commercial antibodies has expanded to cover larger portions of the human proteome over the past several decades, there are still many proteins for which reagents are limited. Furthermore, antibodies that have been validated for applications such as western blot detection may be incapable of selective enrichment of the target protein in an immunoprecipitation experiment. Prior to conducting a large-scale interaction proteomics experiment, it is suggested to complete an IP from a 90% confluent 10 cm dish, or equivalent cell number, and probe for the target protein of interest by western blotting. If more than a single antibody is available for immunoprecipitation, it is additionally suggested to select multiple antibodies recognizing epitopes within different portions of the protein. The binding of an antibody to a bait protein can occlude the necessary binding interface for putative interacting partners. Selection of a secondary epitope for the bait protein will increase the coverage of the interaction profile identified by a mass spectrometry-based experiment.
A second major consideration lies in the selection of the appropriate control for distinguishing high-confidence interactions from low-confidence or nonspecific interactions from those identified as copurifying with the bait. The most stringent control for an IP-MS experiment is to complete the immunoprecipitation from a CRISPR KO cell line of the bait. Such a control enables identification and filtering out of nonspecific proteins that bind directly to the antibody rather than the bait protein. In cases where generating a CRISPR KO cell line of each bait protein is not feasible, an IgG-bead control of the same isotype of the bait antibody can be used. In experiments employing a panel of antibodies representing multiple species, the use of a beads only control can be appropriate but will increase the rate of false positives identified as high-confidence interactors.
Selection of the cell line used in an IP-MS experiment is dependent on several key factors. Protein expression and localization are largely dependent on cell type. While RNA expression estimates can be found for most genes in many commonly used cell lines, protein expression is poorly correlated with RNA expression and must be determined experimentally25. Cell lines in which a bait protein is expressed in very low copy number should be avoided to circumvent problems associated with drastic increases in cell culture scale that may be required. It should be noted, however, that sample preparation can be optimized for the detection of very low abundance proteins. The filter aided sample prep (FASP) method, while robust, can cause a more than 50% loss of peptide in a sample. The Single-Pot Solid-Phase-enhanced Sample Preparation (SP3) is an efficient method of generating samples for mass spectrometry analysis that minimizes sample loss26. The increased recovery enabled by the SP3 method of sample preparation can be a useful alternative in this workflow for quantification of proteins that fall near the limit of detection.
This proteomics workflow has been applied across many nuclear baits, including kinases, E3 ubiquitin ligases, and scaffolding members of multisubunit complexes. Assuming proper validation of antibody reagents, successful execution of this workflow will result in detection of high-confidence protein nuclear interaction partners for a protein of interest.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by a Grand Challenge grant to W.M.O. from the Linda Crnic Institute for Down syndrome and by a DARPA cooperative agreement 13-34-RTA-FP-007. We would like to thank Jesse Kurland and Kira Cozzolino for their contributions in reading and commenting on the manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin, 0.1% EDTA | Thermo Fisher Scientific | 25200056 | |
| 1.5 ml low-rention microcentrifuge tubes | Fisher Scientific | 02-681-320 | |
| 4-20% Mini PROTEAN TGX Precast Protein Gels | Bio-Rad | 4561096 | |
| acetone (HPLC) | Thermo Fisher Scientific | A949SK-4 | |
| Amicon Ultra 0.5 ml 30k filter column | Millipore Sigma | UFC503096 | |
| Benzamidine | Sigma-Aldrich | 12072 | |
| benzonase | Sigma-Aldrich | E1014 | |
| Chloroacetamide | Sigma-Aldrich | C0267 | |
| Dialysis tubing closure | Caroline Biological Supply Company | 684239 | |
| DTT | Sigma-Aldrich | 10197777001 | |
| EDTA | Sigma-Aldrich | EDS | |
| GAPDH antibody | Santa Cruz Biotechnology | Sc-47724 | |
| Glycerol | Fisher Scientific | 887845 | |
| Glycine | Sigma-Aldrich | G8898 | |
| HeLa QC tryptic digest | Pierce | 88329 | |
| HEPES | Fisher Scientific | AAJ1692630 | |
| insulin | Thermo Fisher Scientific | 12585014 | |
| iodoacetamide | Sigma-Aldrich | I1149 | |
| KONTES Dounce homogenizer 7 ml | VWR | KT885300-0007 | |
| Large Clearance pestle 7ml | VWR | KT885301-0007 | |
| Lysyl endopeptidase C | VWR | 125-05061 | |
| Magnesium Chloride | Sigma-Aldrich | 208337 | |
| Microcystin | enzo life sciences | ALX-350-012-C100 | |
| Nonidet P 40 Substitute solution | Sigma-Aldrich | 98379 | |
| p84 antibody | GeneTex | GTX70220 | |
| Phosphate Buffered Saline | |||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 | |
| Pierce BSA Protein Digest, MS grade | Thermo Fisher Scientific | 88341 | LCMS QC |
| Pierce C18 spin columns | Thermo Fisher Scientific | PI-89873 | |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | 90057 | For mass spectrometry sample prep |
| PMSF | Sigma-Aldrich | P7626 | |
| Potassium Chloride | Sigma-Aldrich | P9541 | |
| Protein A Sepharose CL-4B | GE Healthcare Bio-Sciences | 17-0780-01 | |
| Protein G Sepharose 4 Fast Flow | GE Healthcare Bio-Sciences | 17-0618-01 | |
| SDS | Sigma-Aldrich | L3771 | |
| Silica emitter tip | Pico TIP | FS360-20-10 | |
| Small Clearance pestle 7ml | VWR | KT885302-0007 | |
| Sodium Chloride | Sigma-Aldrich | S3014 | |
| Sodium Fluoride | Sigma-Aldrich | 201154 | |
| Sodium metabisulfite | Sigma-Aldrich | 31448 | |
| Sodium orthovanadate | Sigma-Aldrich | S6508 | |
| Spectra/ Por 8 kDa 24 mm dialysis tubing | Thomas Scientific | 3787K17 | |
| TC Dish 150, Standard | Sarstedt | 83.3903 | Tissue culture dish for adherent cells |
| TCA | Sigma-Aldrich | T9159 | |
| TCEP | Thermo Scientific | PG82080 | |
| TFA | Thermo Fisher Scientific | 28904 | |
| Thermo Scientific Orbitrap Fusion MS | Thermo Fisher Scientific | ||
| Trizma Base | Sigma-Aldrich | T6066 | |
| Urea | Thermo Fisher Scientific | 29700 | |
| Waters ACQUITY M-Class UPLC | Waters | ||
| Waters ACQUITY UPLC M-Class Column Reversed-Phase 1.7µm Spherical Hybrid (1.7 µm, 75 µm x 250 mm) | Waters | 186007484 | nanoflow C18 column |
References
- Varjosalo, M., et al. The protein interaction landscape of the human CMGC kinase group. Cell Reports. 3, 1306-1320 (2013).
- Kimple, M. E., Brill, A. L., Pasker, R. L. Overview of Affinity Tags for Protein Purification. Current Protocols in Protein Science. 73, (2013).
- Mahmood, N., Xie, J. An endogenous 'nonspecific' protein detected by a His-tag antibody is human transcription regulator YY1. Data in Brief. 2, 52 (2015).
- Zordan, R. E., Beliveau, B. J., Trow, J. A., Craig, N. L., Cormack, B. P. Avoiding the ends: internal epitope tagging of proteins using transposon Tn7. Genetics. 200, 47-59 (2015).
- Gibson, T. J., Seiler, M., Veitia, R. A. The transience of transient overexpression. Nature Methods. 10, 715-721 (2013).
- Bronicki, L. M., et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. European Journal of Human Genetics. 23, 1482-1487 (2015).
- Antonarakis, S. E. Down syndrome and the complexity of genome dosage imbalance. Nature Reviews Genetics. , (2016).
- Dowjat, W. K., et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neuroscience Letters. 413, 77-81 (2007).
- Fotaki, V., et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Molecular and Cellular Biology. 22, 6636-6647 (2002).
- Hämmerle, B., Elizalde, C., Tejedor, F. J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. European Journal of Neuroscience. 27, 1061-1074 (2008).
- Funakoshi, E., et al. Overexpression of the human MNB/DYRK1A gene induces formation of multinucleate cells through overduplication of the centrosome. BMC Molecular and Cell Biology. 4, 12 (2003).
- Yu, D., Cattoglio, C., Xue, Y., Zhou, Q. A complex between DYRK1A and DCAF7 phosphorylates the C-terminal domain of RNA polymerase II to promote myogenesis. Nucleic Acids Research. , 1-14 (2019).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 76, 4350-4354 (1979).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6, 359-362 (2009).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11, 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13, 731-740 (2016).
- Mellacheruvu, D., et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10, 730-736 (2013).
- Choi, H., et al. SAINT: Probabilistic scoring of affinity purificationg-mass spectrometry data. Nature Methods. 8, 70-73 (2011).
- Shannon, P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 13, 2498-2504 (2003).
- Szklarczyk, D., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. 45, 362-368 (2017).
- Bindea, G., et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 25, 1091-1093 (2009).
- Guard, S. E., et al. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Scientific Reports. 9, 6539 (2019).
- Razick, S., Magklaras, G., Donaldson, I. M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics. 9, 405 (2008).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11, 319-324 (2014).
- Liu, Y., Beyer, A., Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 165, 535-550 (2016).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10, 757 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved