
Method Article
Chromatographischer Fingerabdruck durch Template-Matching für Daten, die durch umfassende zweidimensionale Gaschromatographie gesammelt werden
In diesem Artikel
Zusammenfassung
Dieses Protokoll stellt einen Ansatz für Den Fingerabdruck dar und untersucht mehrdimensionale Daten, die durch eine umfassende zweidimensionale Gaschromatographie in Verbindung mit der Massenspektrometrie gesammelt werden. Spezielle Mustererkennungsalgorithmen (Template Matching) werden angewendet, um die chemischen Informationen zu untersuchen, die in der flüchtigen Fraktion des nativen Olivenöls extra (d. H. Volatilom) verschlüsselt sind.
Zusammenfassung
Datenverarbeitung und -auswertung sind kritische Schritte der umfassenden zweidimensionalen Gaschromatographie (GCxGC), insbesondere in Verbindung mit der Massenspektrometrie. Die reichhaltigen Informationen, die in den Daten verschlüsselt sind, können sehr wertvoll sein, aber schwer effizient zugänglich sein. Datendichte und -komplexität können zu langen Ausarbeitungszeiten führen und erfordern mühsame, analystenabhängige Verfahren. Effektive und dennoch zugängliche Datenverarbeitungswerkzeuge sind daher der Schlüssel, um die Verbreitung und Akzeptanz dieser fortschrittlichen mehrdimensionalen Technik in Laboren für den täglichen Gebrauch zu ermöglichen. Das in dieser Arbeit vorgestellte Datenanalyseprotokoll verwendet chromatographische Fingerabdrücke und Vorlagenabgleich, um das Ziel der hochautomatisierten Dekonstruktion komplexer zweidimensionaler Chromatogramme in einzelne chemische Merkmale zu erreichen, um informative Muster innerhalb einzelner Chromatogramme und über Chromatogramme hinweg zu erkennen. Das Protokoll bietet eine hohe Konsistenz und Zuverlässigkeit mit wenig Eingriffen. Gleichzeitig ist die Analystenaufsicht in einer Vielzahl von Einstellungen und Einschränkungsfunktionen möglich, die angepasst werden können, um Flexibilität und Anpassungsfähigkeit an unterschiedliche Bedürfnisse und Ziele zu bieten. Der Vorlagenabgleich wird hier als leistungsstarker Ansatz zur Erforschung des nativen Olivenölvolatiloms extra gezeigt. Die Kreuzausrichtung von Peaks wird nicht nur für bekannte Targets, sondern auch für ungezielte Verbindungen durchgeführt, was die Charakterisierungsleistung für eine Vielzahl von Anwendungen signifikant erhöht. Es werden Beispiele vorgestellt, um die Leistung für die Klassifizierung und den Vergleich chromatographischer Muster aus Probensätzen zu belegen, die unter ähnlichen Bedingungen analysiert wurden.
Einleitung
Die umfassende zweidimensionale Gaschromatographie in Kombination mit der flugzeitspektrometrischen Detektion (GC×GC-TOF MS) ist heute der informativste analytische Ansatz zur chemischen Charakterisierung komplexer Proben1,2,3,4,5. Bei GC×GC werden Säulen seriell verbunden und durch einen Modulator (z. B. eine thermische oder ventilbasierte Fokussierschnittstelle) verbunden und verbunden, der eluierende Komponenten aus der ersten Dimension(1D) -Säule einfängt, bevor sie wieder in die zweite Dimension(2D) -Säule eingesiebt werden. Diese Operation wird innerhalb einer festen Modulationszeitspannperiode (PM) durchgeführt, die im Allgemeinen zwischen 0,5 und 8 s liegt. Durch thermische Modulation umfasst der Prozess kryo-trapping und Fokussierung des Eluierungsbandes mit einigen Vorteilen für die gesamte Trennleistung.
Obwohl GC×GC eine zweidimensionale Trenntechnik ist, erzeugt der Prozess sequenzielle Datenwerte. Der Analog-Digital-Wandler (A/D) des Detektors erhält den chromatographischen Signalausgang bei einer bestimmten Frequenz. Dann werden die Daten in bestimmten proprietären Formaten gespeichert, die nicht nur die digitalisierten Daten, sondern auch zugehörige Metadaten (Informationen über die Daten) enthalten. Der in GC×GC-Systemen verwendete A/D-Wandler hilft dabei, die Intensität des chromatographischen Signals auf eine digitale Zahl (DN) als Funktion der Zeit in den beiden analytischen Dimensionen abzubilden. Einkanaldetektoren (z. B. Flammenionisationsdetektor (FID), Elektroneneinfangdetektor (ECD), Schwefelchemilumineszenzdetektor (SCD) usw.) erzeugen Einzelwerte pro Probenahmezeit, während Mehrkanaldetektoren (z. B. Massenspektrometrischer Detektor (MS)) mehrere Werte (typischerweise über einen Spektralbereich) pro Probenahmezeit entlang des Analyselaufs erzeugen.
Um 2D-Datenzu visualisieren, beginnt die Ausarbeitung mit der Rasterung von Datenwerten einer einzelnen Modulationsperiode (oder eines Zyklus) als Pixelspalte (Bildelemente, die Detektorereignissen entsprechen). Entlang der Ordinate (Y-Achse, von unten nach oben) wird die 2D-Trennzeit visualisiert. Pixelspalten werden sequenziell verarbeitet, so dass die Abszisse (X-Achse, von links nach rechts) eine Trennzeitvon 1 D meldet. Diese Reihenfolge stellt die 2-D-Daten in einemrechtshändigen kartesischen Koordinatensystem dar, wobei die 1-D-Retentionsordinalzahl als erster Index in das Array aufgenommen wird.
Die Datenverarbeitung von 2D-Chromatogrammen ermöglicht den Zugriff auf ein höheres Informationsniveau als Rohdaten und ermöglicht die 2D-Peak-Erkennung,Peak-Identifizierung, Extraktion von Antwortdaten für quantitative Analysen und cross-vergleichende Analysen.
Die 2-D-Peakmuster können als einzigartiger Fingerabdruck der Probe und nachgewiesene Verbindungen als Minutienmerkmale für eine effektive crossvergleichende Analyse behandelt werden. Dieser Ansatz, bekannt als template-based fingerprinting6,7, wurde vom biometrischen Fingerabdruck6inspiriert. Automatische biometrische Fingerabdruck-Verifizierungssysteme basieren in der Tat auf einzigartigen Fingerspitzeneigenschaften: Kammverzweigungen und -enden, lokalisiert und extrahiert aus eingefärbten Abdrücken oder detaillierten Bildern. Diese Merkmale, die als Minutien-Features bezeichnet werden, werden dann mit den verfügbaren gespeicherten Vorlagen8,9abgeglichen.
Wie oben erwähnt, besteht jedes GC×GC-Trennmuster aus 2D-Peaks, die rational über eine zweidimensionale Ebene verteilt sind. Jeder Peak entspricht einem einzelnen Analyten, hat sein informatives Potenzial und kann als einzelnes Merkmal für die vergleichende Musteranalyse behandelt werden.
Hier stellen wir einen effektiven Ansatz für den chemischen Fingerabdruck mittels GC×GC-TOF MS mit Tandemionisation vor. Ziel ist es, Features aus einer Reihe von Chromatogrammen umfassend und quantitativ zu katalogisieren.
Im Vergleich zu bestehender kommerzieller Software oder internen Routinen10,11, die einen Peak-Features-Ansatz verwenden, zeichnet sich vorlagenbasiertes Fingerprinting durch hohe Spezifität, Effizienz und begrenzte Rechenzeit aus. Darüber hinaus verfügt es über eine intrinsische Flexibilität, die die Kreuzausrichtung von Minutienmerkmalen (d. H. 2D-Peaks) zwischen stark falsch ausgerichteten Chromatogrammen ermöglicht, wie sie durch verschiedene Instrumente oder in Langzeitrahmenstudien erworben wurden12,13,14.
Die grundlegenden Operationen der vorgeschlagenen Methode werden kurz beschrieben, um den Leser zu einemguten Verständnis der 2D-Musterkomplexität und Informationskraft zu führen. Durch die Untersuchung der Ausgabedatenmatrix des Instruments wird dann eine chemische Identifizierung durchgeführt und bekannte Zielanalyten befinden sich über dem zweidimensionalen Raum. Die Vorlage der zielgerichteten Peaks wird dann erstellt und auf eine Reihe von Chromatogrammen angewendet, die innerhalb derselben analytischen Charge erfasst wurden. Metadaten in Bezug auf Retentionszeiten, spektrale Signaturen und Antworten (absolut und relativ) werden aus neu ausgerichteten Mustern von Zielpeaks extrahiert und übernommen, um Kompositorische Unterschiede im Probensatz aufzudecken.
Als zusätzlicher, einzigartiger Schritt des Prozesses wird auch ein kombinierter ungezielter und gezielter Fingerabdruck (UT) an prägezielten Chromatogrammen durchgeführt, um das Fingerabdruckpotenzial sowohl auf bekannte als auch auf unbekannte Analyten auszudehnen. Der Prozess erstellt eine UT-Vorlage für eine wirklich umfassende vergleichende Analyse, die weitgehend automatisiert werden kann.
In einem letzten Schritt führt das Verfahren die Kreuzausrichtung von Merkmalen in zwei parallelen Detektorsignalen durch, die mit hohen und niedrigen Elektronenionisationsenergien (70 und 12 eV) erzeugt werden.
Das Protokoll ist sehr flexibel bei der Unterstützung von Analysen eines einzelnen Chromatogramms oder einer Reihe von Chromatogrammen und mit variabler Chromatographie und / oder mehreren Detektoren. Hier wird das Protokoll mit einer handelsüblichen GC×GC Software Suite (siehe Materialtabelle)kombiniert mit einer MS Library und Suchsoftware (siehe Materialtabelle)demonstriert. Einige der notwendigen Werkzeuge sind in anderer Software vorhanden und ähnliche Tools könnten unabhängig von Beschreibungen in der Literatur von Reichenbach und Mitarbeitern15 , 16,17,18,19implementiert werden. Die Rohdaten für die Demonstration stammen aus einer Forschungsstudie zu nativem Olivenöl extra (EVO), die im Labor der Autoren durchgeführt wurde14. Insbesondere die flüchtige Fraktion (d. h. das Volatilom) italienischer EVO-Öle wird durch Headspace-Festphasenmikroextraktion (HS-SPME) beprobt und von GC×GC-TOF MS analysiert, um diagnostische Fingerabdrücke für die Qualität und sensorische Qualifizierung von Proben zu erfassen. Einzelheiten zu Proben, Probenahmebedingungen und analytischem Aufbau finden Sie in der Materialtabelle.
Die Schritte 1–6 beschreiben die Vorverarbeitung der Chromatogramme. Die Schritte 7–9 beschreiben die Verarbeitung und Analyse einzelner Chromatogramme. Die Schritte 10 bis 12 beschreiben die Vorlagenerstellung und den Abgleich, die die Grundlage für die stichprobenübergreifende Analyse sind. Die Schritte 13 bis 16 beschreiben die Anwendung des Protokolls auf eine Reihe von Chromatogrammen, wobei die Schritte 14 bis 16 für die UT-Analyse vorgesehen sind.
Protokoll
1. Rohdaten importieren
HINWEIS: Dadurch wird ein zweidimensionales Raster-Array für die Visualisierung und Verarbeitung erstellt.
- Starten Sie die Bildsoftware.
- Wählen Sie Datei | Importieren; Navigieren Sie zu der vom GC×GC-TOF MS-System erfassten Rohdatendatei mit dem Namen "VIOLIN 101.lsc"(Zusatzdatei 1); Klicken Sie dann auf Öffnen. Das Chromatogramm öffnet sich in dieser Software.
HINWEIS: Das Format der Rohdatendatei hängt vom Gerätehersteller ab. Die Software importiert eine Vielzahl von Dateiformaten, die im Benutzerhandbuch aufgeführt sind. - Setzen Sie im Dialogfeld Importieren die Modulationszeit (PM) auf 3,5 s; Klicken Sie dann auf OK.
HINWEIS: Einige Erfassungssoftware zeichnet den Modulationszeitraum möglicherweise nicht auf. - Wählen Sie Datei | Bild speichern unter; Navigieren Sie zum gewünschten Ordner; Geben Sie den Namen "Oil 1 RAW.gci" ein (Ergänzungsdatei 2); Klicken Sie dann auf Speichern.
2. Verschiebung der Modulationsphase
HINWEIS: Dadurch werden alle Peaks in jedem Modulationszyklus in dieselbe Bildspalte eingefügt, einschließlich der Peaks, die sich um das Ende der Modulationsperiode in die Leerzeit der nächsten Modulationsperiode20wickeln.
- Wählen Sie | Schichtphase.
- Legen Sie im Dialogfeld "Verschiebungsphase" den Verschiebungsbetrag auf -0,8 s fest. Klicken Sie dann auf OK.
3. Baseline-Korrektur21
- Grafik-| auswählen Rechteck zeichnen.
- Klicken und ziehen Sie, um ein Rechteck im Bild zu zeichnen, bei dem keine Spitzen erkannt werden.
- | "Werkzeuge" auswählen Visualisieren Sie Daten; Beachten Sie den Mittelwert und die Standardabweichung des Detektorsignals, hier 21.850 ± 1.455 SD-Einheiten-less Digital number (DN); Schließen Sie dann das Tool.
- Wählen Sie Verarbeitungs- | Korrekte Baseline.
4. Einfärben des chromatographischen Bildes mit einer Wertekarte und einer Farbkarte20
- Wählen Sie | anzeigen aus. Einfärben.
- Wählen Sie im Dialogfeld "Einfärben" die Registerkarte "Importieren/Exportieren". Wählen Sie die benutzerdefinierte Farbkarte #AAAA (Supplementary File 3) aus, die als ergänzendes Material bereitgestellt wird. Klicken Sie dann auf Importieren.
- Legen Sie für die Steuerelemente Wertzuordnung den Wertebereich auf die Minimal- und Maximalwerte fest. Klicken Sie dann auf OK.
5. 2D Peaks (d.h. Blobs) Nachweis für Analyten18
- Wählen Sie Verarbeitungs- | Erkennen von Blobs mit den Standardeinstellungen; Beachten Sie dann, dass einige Spitzen geteilt sind und es falsche Erkennungen gibt.
- Wählen Sie | konfigurieren aus. | Einstellungen Blob-Erkennung; Setzen Sie dann Glättung auf 0,1 für die erste Dimension und 2,0 für die zweite Dimension und legen Sie Minimum Volume (d. h. Schwellenwert für die summierten Werte) auf 1,00 E6 fest. Klicken Sie dann auf OK.
- Wählen Sie Verarbeitungs- | Erkennen von Blobs mit den neuen Einstellungen; Beobachten Sie dann die Verbesserungen.
6. 2D Peaks Filtration
HINWEIS: Dies geschieht, um bedeutungslose Erkennungen aufgrund von Spaltenblutungen entlang des 1D und Schlägen oder Abraumungen entlang des 2D automatisch zu entfernen.
- Wählen Sie Verarbeitungs- | Interaktive Blob-Erkennung.
- Beachten Sie die Bloberkennungseinstellungen. Klicken Sie dann auf Erkennen.
- Klicken Sie im Generator für erweiterte Filter auf Hinzufügen; Wählen Sie dann im Dialogfeld Neue Einschränkung die Option Aufbewahrung IIaus. Klicken Sie dann auf OK.
- Legen Sie in den Randbedingungsreglern die minimalen und maximalen 2D-Aufbewahrungszeiten für den Filter fest, um die Anzahl der falschen Spitzen zu reduzieren, ohne echte Spitzen zu verlieren.
- Klicken Sie auf Übernehmen; Klicken Sie dann auf Ja, um die Erkennungseinstellungen mit dem neuen Filter zu speichern.
HINWEIS: Möglicherweise sind erweiterte Tools erforderlich, um bestimmte Erkennungsprobleme zu lösen, z. B. Ionenspitzenerkennung oder Dekonvolution für Co-Elutions19.
7. Kalibrierung linearer Retentionsindizes
HINWEIS: Führen Sie diesen Schritt22 (IT) für die spezifischen Aufbewahrungszeiten über den Satz von Aufbewahrungsindexstandards (RI) (in der Regel n-Alkane) aus.
- Wählen Sie | konfigurieren aus. RI-| Retentionsindex (Spalte I).
- Klicken Sie im Dialogfeld RI-Tabellenkonfiguration auf Importieren; Wählen Sie dann die RI-Kalibrierungsdatei (im CSV-Format mit Name, Aufbewahrungszeit und Aufbewahrungsindex) mit dem Namen "LRI-Tabelle.csv" – (Zusatzdatei 4).
- Wählen Sie Datei | Speichern Sie Bild A. Navigieren Sie zum gewünschten Ordner. Geben Sie den Namen "Oil 1 LRI CALIBRATED.gci" ein (Ergänzungsdatei 5); Klicken Sie dann auf Speichern.
8. Suche nach den Peak-Spektren in der NIST17 MS Library23
- Wählen Sie | konfigurieren aus. | Einstellungen Bibliothek durchsuchen.
- Legen Sie im Dialogfeld Suchbibliothek den Spektrumstyp auf Spitzen-MS, den Intensitätsschwellenwert auf 100, den NIST-Suchtyp auf Einfach (Ähnlichkeit), den NIST-RI-Spaltentyp auf Standardpolar und die NIST-RI-Toleranz auf 10fest. Klicken Sie dann auf OK. NIST MS Search bietet viele weitere Einstellungen, die hier auf die Standardeinstellungen eingestellt sind.
- Wählen Sie Verarbeitungs- | Bibliothek nach allen Blobs durchsuchen.
9. Überprüfung und Korrektur der Analytenidentifikation
- Legen Sie in der Werkzeugpalette den Cursormodus auf Blob-| Wählen Sie Blobsaus.
- Klicken Sie in der Bildansicht mit der rechten Maustaste auf den gewünschten Peak.
- Überprüfen Sie im Dialogfeld Blobeigenschaften die Blobeigenschaften. Klicken Sie dann auf Trefferliste.
- Überprüfen Sie die Trefferliste; Wenn die Identifizierung falsch ist, aktivieren Sie das Häkchen neben der richtigen Identifikation.
- Geben Sie im Dialogfeld Blobeigenschaften den Gruppennamen ein, um die chemische Klasse und alle anderen gewünschten Metadaten festzulegen. Klicken Sie dann auf OK.
- Wählen Sie Datei | Bild speichern unter; Navigieren Sie zum gewünschten Ordner; Geben Sie den Namen "Oil 1 COLORIZED for Template construction.gci" ein (Ergänzungsdatei 6); Klicken Sie dann auf Speichern.
HINWEIS: Diese Datei ist im ergänzenden Archiv enthalten, das für Schritt 10 geöffnet werden kann.
10. Erstellen Sie eine Vorlage mit gezielten Peaks15
- Wählen Sie in der Bildansicht (noch im Modus Blobs auswählen ab Schritt 9.1) mit einem Klick auf den ersten Peak und STRG+ Klick auf die zusätzlichen Peaks die gewünschten Peaks aus.
- Klicken Sie in der Werkzeugpalette auf die Schaltfläche Zur Vorlage hinzufügen.
- Wenn die Vorlage abgeschlossen ist, wählen Sie Datei | Vorlage speichern; Geben Sie den Ordner- und Dateinamen an. Klicken Sie dann auf Speichern.
- Wählen Sie Datei | Bild schließen.
HINWEIS: An dieser Stelle fahren diese Anweisungen mit der Vorlage fort, die erstellt wurde, um die gewünschten Zielspitzen einzuschließen, die als "Targeted tamplate.bt" (Ergänzungsdatei 7) verfügbar sind.
11. Passen Sie die Vorlage an und wenden Sie sie an
HINWEIS: Matching erkennt das Vorlagenmuster in den erkannten Peaks eines neuen Chromatogramms. Anwenden der übereinstimmenden Set-Identifikationen und anderer Metadaten im neuen Chromatogramm aus der Vorlage.
- Wählen Sie Datei | Bild öffnen; Navigieren Sie zur Chromatogrammdatei "Oil 2 COLORIZED.gci" (Supplementary File 8) (die vorverarbeitet ist) und wählen Sie sie aus. Klicken Sie dann auf Öffnen.
- Legen Sie in der Werkzeugpalette den Cursormodus auf Vorlagen-| Wählen Sie Objekteaus.
- | auswählen Vorlage laden.
- Klicken Sie im Dialogfeld Vorlage laden auf Durchsuchen; Navigieren Sie zur Vorlage für gezielte Peaks "Targeted template.bt" (Ergänzungsdatei 7); Klicken Sie dann auf Öffnen.
- Klicken Sie im Dialogfeld Vorlage laden auf Laden, und schließen Sie dann auf Schließen.
- Klicken Sie in der Bildansicht mit der rechten Maustaste auf eine Vorlagenspitze. Überprüfen Sie dann die Objekteigenschaften, einschließlich qCLIC und Referenz-MS.
- | auswählen Interaktive Match- und Transformationsvorlage.
- Klicken Sie in der Benutzeroberfläche für interaktive Übereinstimmung auf Alle abgleichen. Überprüfen Sie dann die Übereinstimmenden Ergebnisse sowohl in der Tabelle als auch im Bild, in dem jeder Vorlagenpeak mit ungefüllten Kreisen markiert ist und, wenn eine Übereinstimmung erzielt wird, ein Link zu einem gefüllten Kreis für den erkannten Peak besteht.
- Bearbeiten Sie die Übereinstimmungen wie gewünscht; Wenn Sie zufrieden sind, klicken Sie auf Übernehmen, um Metadaten von der Vorlage in das Chromatogramm zu übertragen.
HINWEIS: Übereinstimmende Einschränkungen, wie z. B. qCLIC, helfen dabei, das richtige Muster unter den erkannten Peaks des neuen Chromatogramms abzugleichen. Zu den Einschränkungsparametern gehören der Typ der MS-Signatur, die als Vorlagenreferenz verwendet wird(Peak MS oder Blob MS)und die Schwellenwerte für die spektrale Ähnlichkeit (Direct Match Factor (DMF) und Reverse Match Factor (RMF)). Hier werden Parameter basierend auf früheren Studien13,14 festgelegt, um falsch negative Übereinstimmungen zu begrenzen: Peak MS und DMF und RMF Ähnlichkeitsschwelle 700.
12. Transformieren Sie die Vorlage für eine wesentlich andere Chromatographie
HINWEIS: Dieser Schritt ist nicht erforderlich, es sei denn, die chromatographischen Bedingungen variieren erheblich, was dazu führt, dass die Vorlage mit einem neuen Chromatogramm falsch ausgerichtet wird, wie dies bei Langzeitstudien oder nach der Installation einer neuen Säule der Fall sein kann. In solchen Fällen kann die Schablone geometrisch in der chromatographischen Retentionszeitebene transformiert werden, um besser zum neuen Chromatogramm12,13zu passen. In diesem Beispiel sind die Peakmuster der Vorlage und des Chromatogramms ähnlich, unterscheiden sich jedoch in der Geometrie der Retentionszeiten, wie sie für verschiedene chromatographische Bedingungen zu sehen wäre.
- Wiederholen Sie die Schritte 11.2 bis 11.5, außer navigieren, auswählen und laden Sie die zielgerichtete Vorlage 2.bt (Zusatzdatei 9).
- | auswählen Interaktive Match-Vorlage; Klicken Sie dann auf Transformation bearbeiten.
- Variieren Sie in der Benutzeroberfläche "Vorlage transformieren" die Skalen, Übersetzungen und Scheren von 1D und 2D, um die Vorlage besser an den erkannten Spitzen auszurichten. Klicken Sie dann auf Vorlage transformieren.
- Klicken Sie mit der transformierten Vorlage auf Übereinstimmung bearbeiten. Wiederholen Sie dann die Schritte 11.8 bis 11.9.
13. Führen Sie kombinierte ungezielte und gezielte Analysen über eine Reihe von Chromatogrammen durch
HINWEIS: Eine kombinierte untargeted and targeted (UT) Vorlage, auch als Feature-Vorlage 24,25bezeichnet, stellt bei Übereinstimmung mit jedem satz von Chromatogrammen Übereinstimmungen zwischen ungezielten und gezielten Analyten her, dann werden konsistente Cross-Sample-Features zur Mustererkennung extrahiert.
- Führen Sie eine Vorverarbeitung (Schritte 1–6) und einen UT-Vorlagenabgleich (Schritte 11.1–11.9) für alle Chromatogramme im Set (d. h. 2D-Chromatogramme von Ölen) durch. Alternativ automatisieren Sie diesen Schritt mit Projektsoftware oder ähnlicher Software, die hier nicht beschrieben wird.
- Starten Sie die Investigator-Software.
- Wählen Sie Datei | Offene Analyse; wählen Sie dann und öffnen Sie "Feature Jove su 70 eV.gca" (Ergänzungsdatei 10).
- Klicken Sie auf OK, um die Ergebnisse zu öffnen und zu überprüfen.
- Klicken Sie auf die Registerkarte Verbindungen, um metrische Werte und Statistiken für bestimmte Analyten (d. h. zielgerichtete Analyten mit zugeordneten chemischen Namen) oder nicht zielgerichtete Analyten mit (#) Identifikatoren zu überprüfen, die über alle Chromatogramme hinweg ausgerichtet sind, und führen Sie dann die folgenden Schritte aus.
- Klicken Sie auf die Registerkarte Attribute, um Werte und Statistiken für bestimmte Metriken in Chromatogrammen zu überprüfen.
- Klicken Sie auf die Registerkarte Zusammenfassung, um die Zusammenfassungsstatistiken für Verbindungen und Merkmale anzuzeigen. Wenn die Chromatogramme aus verschiedenen Klassen stammen, wie in diesem Fall Öle, die aus Oliven hergestellt werden, die in zwei verschiedenen Regionen Italiens geerntet wurden, dann listet die Registerkarte Zusammenfassung die Fisher-Verhältnisstatistiken (F und FDR) auf, die Einblicke in Merkmale für die Unterscheidung zwischen Klassen geben.
- Zeigen Sie verschiedene Diagramme auf allen Registerkarten an und führen Sie auf Wunsch die Hauptkomponentenanalyse (Principal Component Analysis, PCA) auf der Registerkarte Attribute durch.
14. Ändern Sie die UT-Vorlage für die parallele MS-Analyse
HINWEIS: Die Analyse wurde sowohl mit 70 eV als auch mit 12 eV (d.h. hohen und niedrigen) Elektronenionisationsenergien26,27durchgeführt.
- Öffnen Sie eines der 12 eV-Chromatogramme, z.B. "Oil 1 12 eV RAW.gci" (Supplementary File 11), führen Sie eine Vorverarbeitung durch (Schritte 1–6) und laden Sie die UT-Vorlage "UT template 70 relaxed.bt" (Supplementary File 12) wie in den Schritten 11.1–11.6 beschrieben. Dateien werden als ergänzendes Material zur Verfügung gestellt.
- Passen Sie die Vorlage ggf. an die detektierten 12 eV-Peaks an, wie in Schritt 12 beschrieben. Hier gibt es keine signifikante Fehlausrichtung, da die Tandemsignale gemultiplext sind. Es sollte jedoch beachtet werden, dass, da die verschiedenen Ionisationseinstellungen unterschiedliche Fragmentierungen erzeugen, es notwendig ist, die Einschränkungen für die qCLIC-Einschränkungen für die spektrale Ähnlichkeit von DMF und RMF zu lockern (hier nicht gezeigt).
- Wählen Sie Datei | Vorlage speichern; Geben Sie den Ordner- und Dateinamen an, z. B. "UT-Vorlage 12.bt" (Zusatzdatei 13); Klicken Sie dann auf Speichern.
15. Führen Sie kombinierte ungezielte und zielgerichtete Analysen über 12 eV-Chromatogramme durch
- Wählen Sie Datei | Offene Analyse; wählen und öffnen Sie dann "Feature Jove su 12 eV.gca" - Ergänzende Datei 14 zur Verfügung gestellt.
- Klicken Sie auf OK, um die Ergebnisse zu öffnen und zu untersuchen.
- Klicken Sie auf die Registerkarte Verbindungen, um die metrischen Werte zu überprüfen, beziehen Sie sich auf 12 eV-Antworten und Statistiken für bestimmte Analyten (d. H. Gezielte Analyten mit zugeordneten chemischen Namen) oder nicht zielgerichtete Analyten mit (#) Identifikatoren, die über alle Chromatogramme hinweg ausgerichtet sind, und führen Sie dann die folgenden Schritte aus.
- Klicken Sie auf die Registerkarte Attribute, um Werte und Statistiken für bestimmte Metriken in Chromatogrammen zu überprüfen.
- Klicken Sie auf die Registerkarte Zusammenfassung, um die zusammenfassenden Statistiken für Verbindungen und Merkmale bei 12 eV anzuzeigen. Wenn die Chromatogramme aus verschiedenen Klassen stammen, wie in diesem Fall Öle, die aus Oliven hergestellt werden, die in zwei verschiedenen Regionen Italiens geerntet wurden, dann listet die Registerkarte Zusammenfassung die Fisher-Verhältnisstatistiken (F und FDR) auf, die Einblicke in Merkmale für die Unterscheidung zwischen Klassen geben.
- Zeigen Sie die verschiedenen Diagramme an, die auf allen Registerkarten verfügbar sind, und führen Sie auf Wunsch die Hauptkomponentenanalyse (Principal Component Analysis, PCA) auf der Registerkarte Attribute durch.
Ergebnisse
GC×GC-TOF MS-Muster von hochwertigem nativem Olivenöl extra zeigen etwa 500 2D-Peaks über einem Signal-Rausch-Verhältnis (SNR) -Schwellenwert von 100. Ein solcher Schwellenwert wurde in früheren Untersuchungen an flüchtigen Lebensmitteln14,27 als minimales relatives Signal über dem Schwellenwert definiert, um zuverlässige Spektren für die vergleichende Analyse zu erhalten. Komponenten werden über den chromatographischen Raum entsprechend ihrer relativen Retention in den beiden chromatographischen Dimensionen und speziell basierend auf ihrer Flüchtigkeit / Polarität im 1D und der Flüchtigkeit im 2D verteilt. Hier ist die Säulenkombination polar × semipolar (d.h. Carbowax 20M × OV1701).
Das 2D-Musterzeigt einen hohen Ordnungsgrad. Relative Retentionsmuster für homologe Reihen und Klassen sind in Abbildung 1A mit Anmerkungen (Grafiken für Gruppen und Blasen für Peaks) für lineare gesättigte Kohlenwasserstoffe (schwarz), ungesättigte Kohlenwasserstoffe (gelb), lineare gesättigte Aldehyde (blau), einfach ungesättigte Aldehyde (rot), mehrfach ungesättigte Aldehyde (Lachs), primäre Alkohole (grün) und kurzkettige Fettsäuren (Cyano) dargestellt.
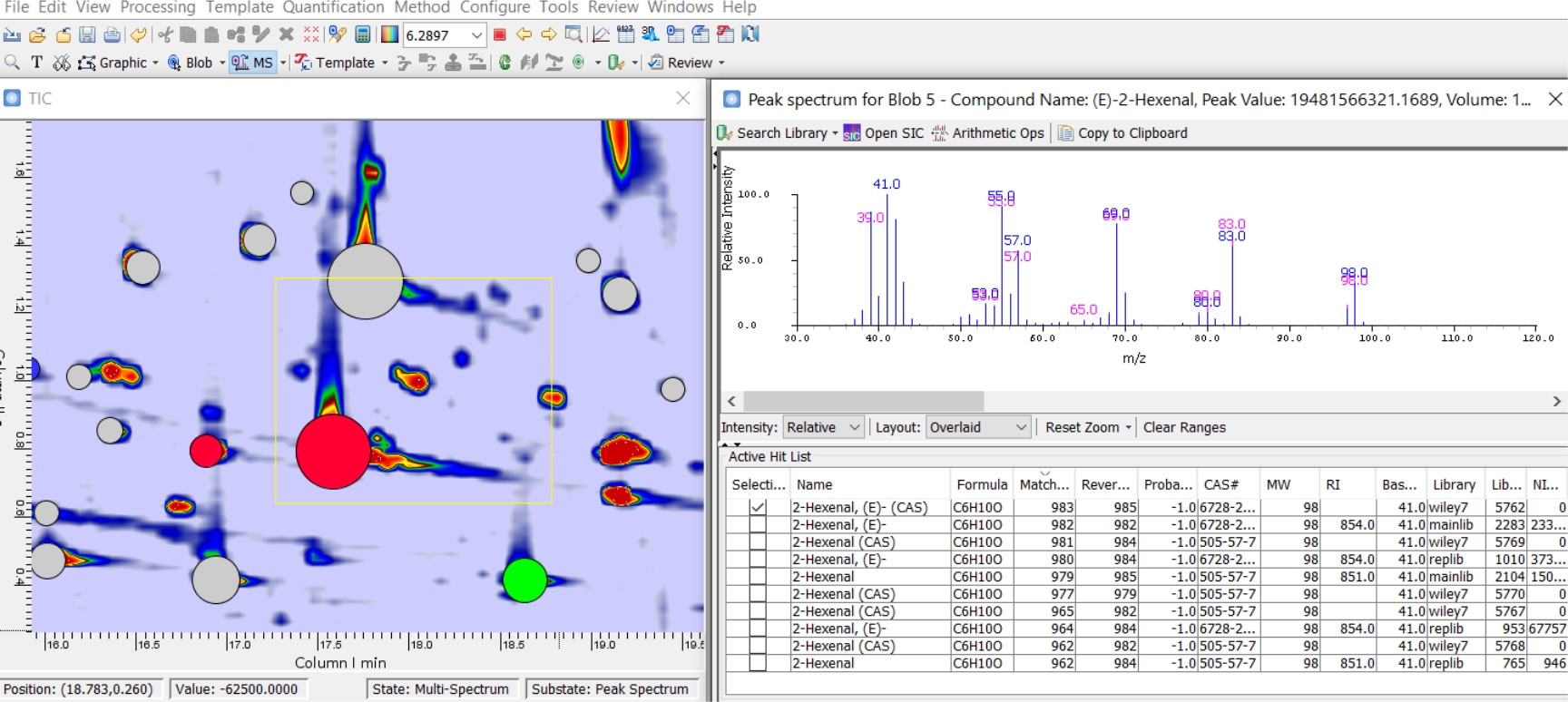
Detektierte 2-D-Peaks können dann identifiziert werden, indem das durchschnittliche MS-Spektrum aus dem gesamten 2-D-Peak(Blob-Spektrum) oder aus dem größten Spektrum(Apex-Spektrum) extrahiert wird. Abbildung 2 zeigt die Ausgabe der Apex-Spektrum-Suche nach Blob 5 und gibt eine Übereinstimmung mit hoher Ähnlichkeit (erste 10 Treffer) für (E)-2-Hexenal zurück. Untersuchte Datenbanken sind diejenigen, die vom Analysten in Schritt 8 der Methode vorausgewählt wurden.
Die Identifizierung wird durch aktive Retentionsindizierung validiert. Für die 2D Peaks wurde der experimentelle IT-Wert berechnet, so dass die Bibliothekssuche in diesem Stadium Ergebnisse mit kohärenten Werten der tabellarischen ITpriorisiert. Toleranzfenster können basierend auf Analystenerfahrung, Zuverlässigkeit der Referenzdatenbankwerte entsprechend der stationären Phase und angewendeten analytischen Bedingungen angepasst werden. Neue Werkzeuge zur intelligenten Kalibrierung linearer Retentionsindizes ohne experimentelle Kalibrierung mit n-Alkanen wurden kürzlich entwickelt und in einer Studie von Reichenbach et al19diskutiert.
Die Sammlung identifizierter 2D-Peaks(d. H. Zielpeaks) kann übernommen werden, um eine Vorlage von Zielpeaks zu erstellen, um umgehend zuverlässige Übereinstimmungen zwischen derselben Verbindung über alle Probenchromatogramme hinweg herzustellen. Die Auflistung der zielgerichteten Vorlagenspitzen ist in Abbildung 1Bdargestellt. Rote Kreise entsprechen den 196 Zielverbindungen, darunter zwei Interne Standards (IS), die mit Vorlagenspitzen mit Verbindungslinien verknüpft sind. IS werden für die Antwortnormalisierung verwendet und Verbindungsleitungen helfen zu visualisieren, welche der enthaltenen IS übernommen werden, um jede 2D Peak/ Blob-Antwort zu normalisieren.
In Abbildung 1Bzeigen gefüllte Kreise positive Übereinstimmungen zwischen dem Vorlagenpeak und dem tatsächlichen Muster an, während leere Kreise für Vorlagenspitzen gelten, für die die Übereinstimmung nicht überprüft wurde. Falsch negative Übereinstimmungen können durch geeignete Auswahl von Schwellenwertparametern, Referenzspektren und Randbedingungsfunktionen13,14,18,19begrenzt werden. Für komplexe Muster mit mehreren Co-Elutionen sind Ionenspitzen-Detektionsfunktionen, die auf spektraler Dekonvolution basieren, ratsam und könnten eine gültige Option sein19. Vorlagen-Peak-Metadaten sind im vergrößerten Bereich von Abbildung 1B für (E)-2-hexenal dargestellt.
Die Spezifität des Vorlagenabgleichs beruht auf der Möglichkeit, Einschränkungsfunktionen anzuwenden, die die positive Korrespondenz auf diejenigen Kandidatenspitzen beschränken, die innerhalb des Suchfensters des Algorithmus eine MS-spektrale Ähnlichkeit oberhalb eines bestimmten Schwellenwerts aufweisen. In diesem Fall wurden in Schritt 11 die Ähnlichkeitsschwellen23 auf 700 gemäß früheren Experimenten festgelegt, die darauf abzielten, optimale Parameter zu definieren, die falsch negative Übereinstimmungen begrenzen14. Hervorgehobene Bereiche der Vorlagenspitzeneigenschaften in Abbildung 1B zeigen die Informationen über die Referenz-MS-Spektrumzeichenfolge und die qCLIC-Einschränkungsfunktion (d. h. (Match("
Durch die Anwendung der Schablone auf alle Chromatogramme einer Menge könnte man auf herausfordernde Situationen stoßen, wie im Falle einer teilweisen Fehlausrichtung von Mustern. Dies kann auf Inkonsistenzen bei der Ofentemperatur, Trägergasströmungs-/Druckinstabilitäten oder auf einen manuellen Eingriff in das System zurückzuführen sein, wie im Falle einer Säulensubstitution oder eines Modulatorschleifenkapillarersatzes14,28. Abbildung 3 zeigt eine Situation einer teilweisen Fehlausrichtung zwischen der Zielschablone und dem tatsächlichen Chromatogramm. Für minimale Fehlausrichtungen können interaktive Vorlagentransformationen(Abbildung 3,Systemsteuerung) Vorlagenspitzen für eine bessere Anpassung neu positionieren. Nach der Neupositionierung kann die Vorlage abgeglichen werden, um Korrespondenzen herzustellen. Im Beispiel stimmen dieVorlagenspitzen (Abbildung 3,Schritt 12) korrekt mit dem tatsächlichen 2D-Musterüberein. Im Falle von schwerwiegenden Fehlausrichtungen, die hier nicht besprochen werden, kann die Wiederholung von Match-Transform-Update-Aktionen die Position der Vorlagenpeaks iterativ an das tatsächliche Peakmuster12,13,14anpassen.
Hier liefern die anvisierten Peaks (d.h. bekannte Analyten) etwa 40% des chromatographischen Ergebnisses (196 gezielte Peaks von durchschnittlich etwa 500 nachweisbaren Peaks). Die anderen 60% der Verbindungen werden zusammen mit den Informationen, die sie bringen, bei der gezielten Analyse nicht berücksichtigt. Um die Untersuchung wirklich umfassend zu gestalten, sollte auch eine konsistente Kreuzausrichtung von ungezielten 2-D-Peaks hergestellt werden. Die erste Anwendung, bei der der Template-Matching auf alle nachweisbaren Analyten ausgeweitet wurde, befasste sich mit dem komplexen Volatilom von geröstetem Kaffee7. Dieser Prozess wird mit einer Software (z. B. Investigator) automatisiert, die hier in den Schritten 14–15 gezeigt wird.
In diesem Prozess werden vorgefertigte Bilder des untersuchten Probensatzes (20 Proben) verwendet, um zuverlässige Peaks durch Cross-Matching aller Bildmuster zu definieren29. Anschließend wird ein zusammengesetztes Chromatogramm erstellt, aus dem man UT-zuverlässige Peaks und Peak-Regionen (d.h. 2D Peaks Footprint) im sogenannten Feature-Template17identifizieren kann.
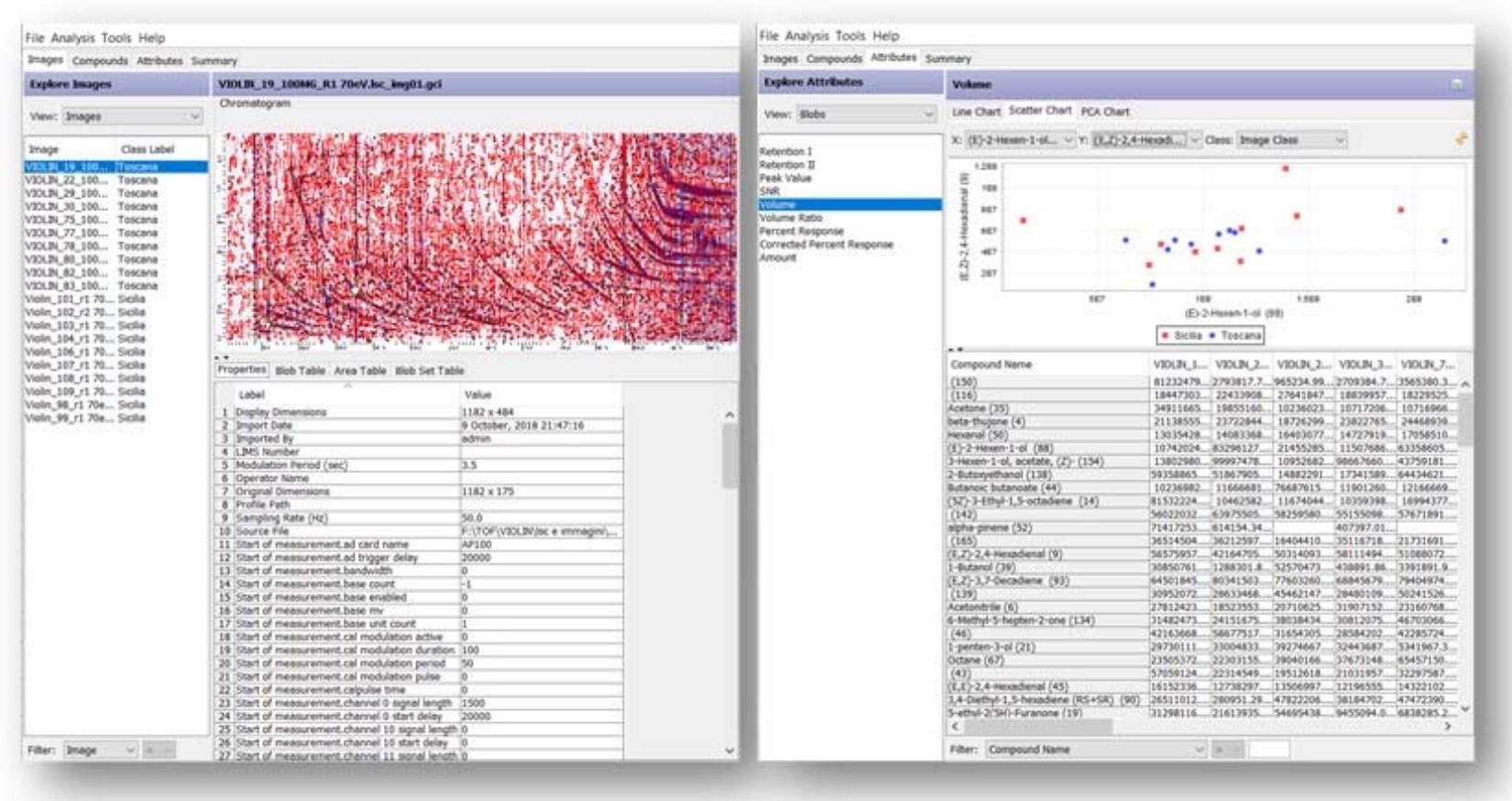
Für Analysen bei 70 eV ermittelte der Prozess 144 zuverlässige Peaks mit entspannter Zuverlässigkeit29, von denen 76 zur Liste der Zielpeaks gehören. Basierend auf diesen 144 zuverlässigen Peaks richtet der Prozess alle Chromatogramme konsistent an den durchschnittlichen Retentionszeiten der reliablen Peaks aus und kombiniert sie dann zu einem zusammengesetzten Chromatogramm. Abbildung 4 zeigt eine Liste aller Proben, die nach dem Produktionsbereich des Öls gekennzeichnet sind (links) und die Liste der zuverlässigen Peaks/Blob-Volumina in jeder Probe (rechts).
Die nicht zielgerichtete Feature-Vorlage besteht aus 2D-Peaks von Analyten, die im zusammengesetzten Chromatogramm detektiert wurden (siehe Abbildung 5A),die von der Vorlage für zuverlässige Peaks abgeglichen werden (n = 168 – rote Kreise für zielgerichtete Peaks und grüne Kreise für nicht zielgerichtete Peaks). Die Massenspektren der zusammengesetzten Peaks sowie deren Retentionszeiten werden inder Feature-Vorlage wie für ( Z)-3-Hexenolacetat im vergrößerten Bereich gezeigt erfasst. Peak-Bereiche sind in Abbildung 5B als rot eingefärbte Grafiken dargestellt. sie werden stattdessen durch die Umrisse aller 2D-Peaks definiert, die im zusammengesetzten Chromatogramm detektiert wurden (n = 3578).
Wenn die unbeaufsichtigte Mustererkennung durch die Hauptkomponentenanalyse auf die gezielte Peakverteilung innerhalb der 20 analysierten Proben angewendet wird, gruppieren sich sizilianische und toskanische Öle getrennt, was darauf hindeutet, dass pedoklimatische Bedingungen und Terroir die relative Prävalenz flüchtiger Stoffe beeinflussen. Die Ergebnisse sind in Abbildung 6A dargestellt, und die PCA-Ergebnisse aus der zuverlässigen Peaks-Verteilung sind in Abbildung 6B dargestellt. Die beiden Ansätze bestätigen, dass Öle aus verschiedenen geografischen Gebieten unterschiedliche chemische Signaturen aufweisen, unabhängig davon, ob es sich um gezielte oder ungezielte Verbindungen oder beides handelt.
Schließlich ermöglicht die Software eine schnelle und effektive Neuausrichtung von Mustern über parallele Erkennungskanäle hinweg. In dieser Anwendung wird die Neuausrichtung für Tandemionisationssignale vorgeschlagen. Die Ionenquelle der MS multiplexiert zwischen zwei Ionisationsenergien (d.h. 70 und 12 eV) bei einer Erfassungsfrequenz von 50 Hz pro Kanal30. Die beiden resultierenden chromatographischen Muster sind eng aufeinander abgestimmt, während Spektraldaten (d.h. spektrale Signaturen und Antworten) komplementäre Informationen mit unterschiedlichen dynamiken Antwortbereichen26,27bringen. Die ausgerichteten Muster ermöglichen das Extrahieren von Merkmalen(2D-Peaks und Peak-Regionen) mit eindeutigen IDs (d. H. Chemische Namen für Ziel-Peaks und eindeutige Nummerierung # für ungezielte Peaks und Peak-Regionen).
Der Vorlagenabgleich ermöglicht eine effektive Querausrichtung. In dieser Situation gibt es nicht viel Fehlausrichtung, aber MS-Einschränkungen müssen gelockert werden, um Übereinstimmungen für UT-Spitzen zu ermöglichen. Auf der anderen Seite werden vorgestellte UT-Peak-Regionen, die keine MS-Einschränkungen haben, sofort ohne falsch negative Übereinstimmungen abgeglichen. Abbildung 5C zeigt einen vergrößerten Bereich eines 12-eV-Chromatogramms, in dem die aus 70 eV-Daten erstellte Feature-Vorlage abgeglichen wird. Zuverlässige UT-Peaks werden aufgrund der gesenkten qCLIC-Einschränkungen (z. B. DMF-Schwellenwert bei 600) positiv aufeinander abgestimmt. Zu beachten ist, dass bei 12 eV aufgrund der begrenzten Fragmentierung, die durch niedrige Ionisationsenergie induziert wird, weniger Peaks detektiert werden.

Abbildung 1: Zweidimensionales Konturdiagramm und Zielvorlage. (A) Konturdiagramm der flüchtigen Fraktion eines nativen Olivenöls extra aus der Toskana. Geordnete Muster von Homolog-Serien und -Klassen werden mit verschiedenen Farben und Linien hervorgehoben: lineare gesättigte Kohlenwasserstoffe (schwarze Linie und 2D-Konturen), ungesättigte Kohlenwasserstoffe (gelb), lineare gesättigte Aldehyde (blau), einfach ungesättigte Aldehyde (rot), mehrfach ungesättigte Aldehyde (Lachs), primäre Alkohole (grün) und kurzkettige Fettsäuren (Cyano). (B) Überlagerte zielgerichtete Vorlage bekannter Analyten (rote Kreise) mit Verbindungslinien, die interne Standards (ISs) verbinden. In den Bereichen werden 2D-Peak-/Blob-Eigenschaften, Metadaten (Decanal) oder Template-Peak-Eigenschaften angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Apex MS-Suche. Ausgabe der Apex-MS-Suche nach Blob 5. Liste der Datenbankeinträge mit der höchsten Ähnlichkeit und zugehörige Metadaten, die in der Bibliothek verfügbar sind. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Neuausrichtung der Vorlage. Workflow zur Veranschaulichung der Schritte, die eine Neuausrichtung der Vorlage durch Transformation ermöglichen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: GC Investigator-Schnittstelle. Prüferpanel mit allen ausgewählten Bildern, die nach der Produktionsregion des Öls (links) und der Liste der zuverlässigen Peaks/Blob-Volumina in jeder Probe (rechts) gekennzeichnet sind. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Ziel- und UT-Vorlage. (A) Zuverlässige Spitzen, wie sie sich aus der automatisierten Verarbeitung in Schritt 11 ergeben; rote Kreise entsprechen bekannten Analyten, während grüne Kreise unbekannt sind. Im überlagerten Bedienfeld werden Vorlagenobjekteigenschaften für den (Z)-3-Hexenal angezeigt. (B) Vergrößerter Bereich, der die UT-Peaks (rote und grüne Kreise) und Peak-Regionen (rote Grafiken) der UT-Vorlage zeigt, die auf einem Probenöl mit 70 eV-Ionisationsenergie übereinstimmen. (C) UT-Vorlage, die auf ein Probenöl abgestimmt ist, das mit 12 eV Ionisationsenergie aufgenommen wurde. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: PCA-Ladediagramme. Sie zeigen die natürliche Konformation von Proben (Öle aus der Toskana und Sizilien), wie sie sich aus (A) gezielter Peakverteilung oder (B) UT Peaks Verteilung ergeben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergänzende Dateien. Bitte klicken Sie hier, um diese Dateien herunterzuladen.
Diskussion
Die Visualisierung von GC×GC-TOF MS-Daten ist ein grundlegender Schritt für ein angemessenes Verständnis der Ergebnisse, die durch umfassende zweidimensionale Trennungen erzielt werden. Bildplots mit kundenspezifischer Einfärbung ermöglichen es Analysten, Detektorreaktionsunterschiede und damit die differentielle Verteilung der Probenkomponenten zu schätzen. Dieser visuelle Ansatz verändert die Perspektive der Analysten auf die Interpretation und Ausarbeitung von Chromatogrammen völlig. Dieser erste Schritt, einmal verstanden und von Chromatographen souverän genutzt, eröffnet eine neue Perspektive in der weiteren Verarbeitung.
Ein weiterer grundlegender Aspekt der Datenverarbeitung ist die Zugänglichkeit der vollständigen Datenmatrix (d. H. MS-Spektraldaten und -antworten) für alle Probenpunkte, von denen jeder einem einzigen Detektorereignis entspricht. In dieser Hinsicht erreicht 2D Peaks Integration, so dass die Erfassung von Detektorereignissen, die einem einzelnen Analyten entsprechen, einen kritischen Schritt darstellt. Im aktuellen Protokoll basiert die 2-D-Peaks-Detektion auf dem Watershed-Algorithmus18 mit einigen Anpassungen, um die Detektionsempfindlichkeit bei partiellen co-eluierenden Verbindungen zu verbessern. Um diesen Prozess spezifischer zu gestalten, muss eine Dekonvolution durchgeführt und ausgefeiltere Verfahren eingeführt werden. Dies ist möglich, indem eine Ionenspitzenerkennung für MS-Daten durchführt; Der Algorithmus verarbeitet das Datenarray und isoliert die Antwort von einzelnen Analyten basierend auf Spektralprofilen19,31.
Ein wichtiger, aber kritischer Schritt des Protokolls und jedes GC×GC-MS-Dateninterpretationsprozesses bezieht sich auf die Identifizierung von Analyten. Dieses Verfahren, das in den Schritten 8 und 9 vorgeschlagen wird, in Ermangelung einer bestätigenden Analyse mit authentischen Standards, muss vom Analysten sorgfältig durchgeführt werden. Automatisierte Aktionen sind in jeder kommerziellen Software verfügbar; Dazu gehören die Bewertung der spektralen Signaturähnlichkeit der MS anhand der gesammelten Referenzspektren (d. h. Spektralbibliotheken) und die Bewertung der charakteristischen Verhältnisse zwischen Qualifizierer-/Quantifiziererionen. Es sind jedoch zusätzliche Bestätigungskriterien erforderlich, um die Identifizierung von Isomeren zu verdeutlichen. Das Protokoll schlägt die Einführung linearer Aufbewahrungsindizes vor, um die Liste der Kandidaten zu priorisieren; die Grenze bezieht sich hier auf die Verfügbarkeit von Aufbewahrungsdaten und deren Konsistenz.
Das Hauptmerkmal, das diesen Ansatz einzigartig macht, ist die Vorlage, die12,13,15,29entspricht. Der Vorlagenabgleich ermöglicht die 2D-Mustererkennungauf sehr effektive, spezifische und intuitive Weise. Es kann in Bezug auf Sensitivität und Spezifität durch Anwendung benutzerdefinierter Schwellenwerte und / oder Einschränkungsfunktionen festgelegt werden, während der Analyst das Verfahren durch aktive Interaktion mit Transformationsfunktionsparametern überwachen kann. Die Besonderheit dieses Verfahrens beruht auf der Möglichkeit, gezielte und ungezielte Peaks-Informationen zwischen Proben einer einheitlichen Charge, aber auch zwischen Proben, die trotz mittlerer bis schwerer Fehlausrichtung unter den gleichen Nennbedingungen aufgenommen wurden, quer auszurichten. Die Vorteile dieser Operation beziehen sich auf die Möglichkeit, alle gezielten Analytenidentifikationen zu erhalten, was für den Analysten eine zeitaufwändige Aufgabe ist, und alle Metadaten, die für gezielte und ungezielte Peaks aus früheren Ausarbeitungssitzungen gespeichert werden.
Der Vorlagenabgleich ist auch in Bezug auf die Rechenzeit sehr effektiv; MS-Datendateien mit niedriger Auflösung bestehen aus etwa 1-2 GB gepackten Daten, während hochauflösende MS-Analysen 10-15 Gb pro einzelnem Analyselauf erreichen können. Der Vorlagenabgleich verarbeitet nicht jedes Mal die vollständige Datenmatrix, sondern führt zunächst eine Retentionszeitausrichtung zwischen Chromatogrammen unter Verwendung von Vorlagenspitzen durch, verarbeitet dann Kandidatenspitzen innerhalb des Suchfensters für ihre Ähnlichkeitsübereinstimmung mit dem Verweis in der Vorlage. Im Falle einer schweren Fehlausrichtung, der schwierigsten Situation, schnitten globale Polynomtransformationen zweiter Ordnung besser ab als lokale Methoden, während die Rechenzeit reduziert wurde13.
Damit sich die GC×GC-Technik weit über akademische und Forschungslabors hinaus verbreiten kann, müssen Datenverarbeitungswerkzeuge grundlegende Operationen für die Visualisierung und Chromatogramminspektion erleichtern. Die Identifizierung von Analyten sollte die Möglichkeit bieten, standardisierte Algorithmen und Verfahren (z. B. NIST-Suchalgorithmus undI-T-Kalibrierung) zu übernehmen; und die crossvergleichende Analyse sollte intuitiv, effektiv und durch interaktive Tools unterstützt werden. Der vorgeschlagene Ansatz adressiert diese Anforderungen und bietet gleichzeitig erweiterte Optionen und Werkzeuge, um mit komplexen Situationen wie Analyten-Co-Elution, Mehrfachanalytkalibrierung, Gruppentypanalyse und paralleler Detektionsausrichtung umzugehen.
Die referenzierte Literatur deckt viele mögliche Szenarien ab, in denen GC×GC und ganz allgemein eine umfassende zweidimensionale Chromatographie einzigartige Lösungen und zuverlässige Ergebnisse bieten, die mit der 1-D-Chromatographie in der Einzellaufanalyse nicht erreicht werden können. 5,32,33 Obwohl GC×GC das leistungsstärkste Werkzeug ist, das die Trennkapazität und -empfindlichkeit erhöht, gibt es immer Einschränkungen bei der Trennkraft, der Empfindlichkeit und anderen systemischen Kapazitäten. Mit der Annäherung an diese systemischen Grenzen wird die Datenanalyse zunehmend schwieriger. Daher müssen Forschung und Entwicklung die uns zur Verfügung stehenden Analyseinstrumente weiter verbessern.
Offenlegungen
Prof. Stephen E. Reichenbach und Dr. Qingping Tao sind finanziell an GC Image, LLC interessiert. Dr. Daniela Peroni ist Mitarbeiterin von SRA Instruments, einem Distributor von GC Image in Italien und Frankreich. Dr. Federico Stilo, Prof. Chiara Cordero und Prof. Carlo Bicchi erklären keine Interessenkonflikte.
Danksagungen
Die Forschung wurde von Progetto Ager − Fondazioni in rete per la ricerca agroalimentare unterstützt. Projektakronym Violin - Valorisierung italienischer Olivenprodukte durch innovative Analysewerkzeuge (https://olivoeolio.progettoager.it/index.php/i-progetti-olio-e-olivo/violin-valorization-of-italian-olive-products-through-innovative-analytical-tools/violin-il-progetto). Die GC Image-Software ist für eine kostenlose Testversion für Leser verfügbar, die das Protokoll demonstrieren und testen möchten.
Materialien
| Name | Company | Catalog Number | Comments |
| 1D SolGel-Wax column (100% polyethylene glycol; 30 m × 0.25 mm dc × 0.25 μm df). Carrier gas helium at a constant nominal flow of 1.3 mL/min. | Trajan SGE Analytical Science, Ringwood, Australia | PN 054796 | Carrier gas helium at a constant nominal flow of 1.3 mL/min. Oven temperature programming set as follows: 40°C (2 min) to 240°C (10 min) at 3.5°C/min. |
| 2D OV1701 column (86% polydimethylsiloxane, 7% phenyl, 7% cyanopropyl; 1 m × 0.1 mm dc × 0.10 μm df) from . | Mega, Legnano, Milan, Italy | PN MEGA-1701 | |
| Automated system for sample preparation: SPR Autosampler for GC | SepSolve-Analytical, Llantrisant, UK | ||
| Extra Virgin Olive oils: Sicily and Tuscany, Italy | Project VIOLIN (Ager - Fondazioni in rete per la ricerca agroalimentare) | Samples (n=10) were collected during the production year 2018 within the "Violin" project sampling campaign. Oils were submitted to HS-SPME to sample volatiles according to a reference protocol validated in a previous study of Stilo et al.14 | |
| Gas chromatograph: Model 7890B GC | Agilent Technologies Wilmington DE, USA | ||
| GC Image GC×GC edition V 2.9 | GC Image LLC, Lincoln, Nebraska | https://www.gcimage.com/gcxgc/trial.html | |
| Image processing software | GC Image LLC, Lincoln, Nebraska | https://www.gcimage.com/gcxgc/trial.html | |
| Mass spectrometer: BenchTOF-Select | Markes International Llantrisant, UK | ||
| Methyl-2-octynoate (CAS 111-12-6) | Merck-Millipore/Supelco | PN: 68982 | |
| Modulator controller: Optimode v2.0 | SRA Intruments, Cernusco sul Naviglio, Milan, Italy | ||
| Modulator: KT 2004 loop type | Zoex Corporation Houston, TX, USA | ||
| MS library and search software: NIST Library V 2017, Software V 2.3 | National Institute of Standards and Technology (NIST), Gaithersburg MD | https://www.nist.gov/srd/nist-standard-reference-database-1a-v17 | |
| n-alkanes C8-C40 for retention indexing | Merck-Millipore/Supelco | PN: 40147-U | |
| n-hexane (CAS 110-54-3) gas chromatography MS SupraSolv | Merck-Millipore/Supelco | PN: 100795 | |
| Solid Phase Microextraction fiber | Merck-Millipore/Supelco | PN 57914-U | |
| α- /β-thujone (CAS 546-80-5) | Merck-Millipore/Sigma Aldrich | PN: 04314 |
Referenzen
- Tranchida, P. Q., et al. Potential of comprehensive chromatography in food analysis. Trends in Analytical Chemistry. 52, 186-205 (2013).
- Cordero, C., Kiefl, J., Reichenbach, S. E., Bicchi, C. Characterization of odorant patterns by comprehensive two-dimensional gas chromatography: A challenge in omic studies. Trends in Analytical Chemistry. 113, 364-378 (2019).
- Cordero, C., Kiefl, J., Schieberle, P., Reichenbach, S. E., Bicchi, C. Comprehensive two-dimensional gas chromatography and food sensory properties: potential and challenges. Analytical and Bioanalytical Chemistry. 407, 169-191 (2015).
- Adahchour, M., Beens, J., Vreuls, R. J. J., Brinkman, U. A. T. Recent developments in comprehensive two-dimensional gas chromatography (GC × GC) - Introduction and instrumental set-up. Trends in Analytical Chemistry. 25 (5), 438-454 (2006).
- Prebihalo, S. E., et al. Multidimensional gas chromatography: Advances in instrumentation, chemometrics, and applications. Analytical Chemistry. 90 (1), 505-532 (2018).
- Cordero, C., et al. Profiling food volatiles by comprehensive two-dimensional gas chromatography coupled with mass spectrometry: advanced fingerprinting approaches for comparative analysis of the volatile fraction of roasted hazelnuts (Corylus Avellana L.) from different origins. Journal of Chromatography A. 1217, 5848-5858 (2010).
- Cordero, C., et al. Targeted and non-targeted approaches for complex natural sample profiling by GC×GC-QMS. Journal of Chromatography Sciences. 48 (4), 251-261 (2010).
- Maio, D., Maltoni, D. Direct gray-scale minutiae detection in fingerprints. IEEE Transactions on Pattern Analysis and Machine Intelligence. 19 (1), 27-40 (1997).
- Jain, A. K., Hong, L., Pankanti, S., Bolle, R. An identity-authentication system using fingerprints. Proceedings of IEEE. 85 (9), 1365-1388 (1997).
- Parsons, B. A., et al. Tile-based fisher ratio analysis of comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry (GC × GC-TOFMS) data using a null distribution approach. Analytical Chemistry. 87 (7), 3812-3819 (2015).
- Pierce, K. M., Kehimkar, B., Marney, L. C., Hoggard, J. C., Synovec, R. E. Review of chemometric analysis techniques for comprehensive two dimensional separations data. Journal of Chromatography A. 1255, 3-11 (2012).
- Reichenbach, S. E., et al. Alignment for comprehensive two-dimensional gas chromatography with dual secondary columns and detectors. Analytical Chemistry. 87 (19), 10056-10063 (2015).
- Rempe, D. W., et al. Effectiveness of global, low-degree polynomial transformations for GCxGC data alignment. Analytical Chemistry. 88 (20), 10028-10035 (2016).
- Stilo, F., et al. Untargeted and targeted fingerprinting of extra virgin olive oil volatiles by comprehensive two-dimensional gas chromatography with mass spectrometry: challenges in long-term studies. Journal of Agricultural and Food Chemistry. 67 (18), 5289-5302 (2019).
- Reichenbach, S. E., Carr, P. W., Stoll, D. R., Tao, Q. Smart templates for peak pattern matching with comprehensive two-dimensional liquid chromatography. Journal of Chromatography A. 1216 (16), 3458-3466 (2009).
- Reichenbach, S. E., et al. Informatics for cross-sample analysis with comprehensive two-dimensional gas chromatography and high-resolution mass spectrometry (GCxGC-HRMS). Talanta. 83 (4), 1279-1288 (2011).
- Reichenbach, S. E., Tian, X., Cordero, C., Tao, Q. Features for non-targeted cross-sample analysis with comprehensive two-dimensional chromatography. Journal of Chromatography A. 1226, 140-148 (2012).
- Latha, I., Reichenbach, S. E., Tao, Q. Comparative analysis of peak-detection techniques for comprehensive two-dimensional chromatography. Journal of Chromatography A. 1218 (38), 6792-6798 (2011).
- Reichenbach, S. E., Tao, Q., Cordero, C., Bicchi, C. A data-challenge case study of analyte detection and identification with comprehensive two-dimensional gas chromatography with mass spectrometry (GC×GC-MS). Separations. 6 (3), 38 (2019).
- Reichenbach, S. E. Chapter 4 Data Acquisition, Visualization, and Analysis. Comprehensive Analytical Chemistry. , 77-106 (2009).
- Reichenbach, S. E., Ni, M., Zhang, D., Ledford, E. B. Image background removal in comprehensive two-dimensional gas chromatography. Journal of Chromatography A. 985 (1-2), 47-56 (2003).
- Kratz, P. A Generalization of the retention index system including linear temperature programmed gas-liquid partition chromatography. Journal of Chromatography A. 11, 463-471 (1963).
- NIST Mass Spectrometry Data Center. NIST Standard Reference Database 1A: NIST/EPA/NIH Mass Spectral Library (NIST 08) and NIST Mass Spectral Search Program (Version 2.0f). National Institute of Standards and Technology (NIST). , (2005).
- Magagna, F., et al. Combined untargeted and targeted fingerprinting with comprehensive two-dimensional chromatography for volatiles and ripening indicators in olive oil. Analytica Chimica Acta. 936, 245-258 (2016).
- Reichenbach, S. E., et al. Benchmarking machine learning methods for comprehensive chemical fingerprinting and pattern recognition. Journal of Chromatography A. 1595, 158-167 (2019).
- Cialiè Rosso, M., et al. Adding extra-dimensions to hazelnuts primary metabolome fingerprinting by comprehensive two-dimensional gas chromatography combined with time-of-flight mass spectrometry featuring tandem ionization: insights on the aroma potential. Journal of Chromatography A. 1614 (460739), 1-11 (2020).
- Cordero, C., et al. Comprehensive two-dimensional gas chromatography coupled with time of flight mass spectrometry featuring tandem ionization: challenges and opportunities for accurate fingerprinting studies. Journal of Chromatography A. 1597, 132-141 (2019).
- Ni, M., Reichenbach, S. E., Visvanathan, A., TerMaat, J., Ledford, E. B. Peak pattern variations related to comprehensive two-dimensional gas chromatography acquisition. Journal of Chromatography A. 1086, 165-170 (2005).
- Reichenbach, S. E., et al. Reliable peak selection for multisample analysis with comprehensive two-dimensional chromatography. Analytical Chemistry. 85 (10), 4974-4981 (2013).
- Markes International. Select-EV: The next Generation of Ion Source Technology. Technical Note. , (2016).
- Tao, Q., Reichenbach, S. E., Heble, C., Wu, Z. New investigator tools for finding unique and common components in multiple samples with comprehensive two-dimensional chromatography. Chromatography Today. , (2018).
- Seeley, J. V., Seeley, S. K. Multidimensional gas chromatography: fundamental advances and new applications. Analytical Chemistry. 85 (2), 557-578 (2013).
- Tranchida, P. Q., Aloisi, I., Giocastro, B., Mondello, L. Current state of comprehensive two-dimensional gas chromatography-mass spectrometry with focus on processes of ionization. Trends in Analytical Chemistry. 105, 360-366 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten