
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Isolierung von Zellkernen aus humanem intermuskulärem Fettgewebe und nachgeschaltete Einzelkern-RNA-Sequenzierung
In diesem Artikel
Zusammenfassung
Die Biologie des intermuskulären Fettgewebes (IMAT) ist aufgrund der begrenzten Zugänglichkeit von menschlichem Gewebe weitgehend unerforscht. Hier präsentieren wir ein detailliertes Protokoll für die Zellkernisolierung und die Bibliotheksvorbereitung von gefrorenen humanen IMAT für die Einzelkern-RNA-Sequenzierung, um die zelluläre Zusammensetzung dieses einzigartigen Fettdepots zu identifizieren.
Zusammenfassung
Intermuskuläres Fettgewebe (IMAT) ist ein relativ wenig untersuchtes Fettdepot, das sich zwischen Muskelfasern befindet. Der IMAT-Gehalt steigt mit dem Alter und dem BMI und wird mit Stoffwechsel- und Muskeldegenerativen Erkrankungen in Verbindung gebracht. Das Verständnis der biologischen Eigenschaften von IMAT und seines Zusammenspiels mit den umgebenden Muskelfasern fehlt jedoch erheblich. In den letzten Jahren haben uns die Einzelzell- und Zellkern-RNA-Sequenzierung zelltypspezifische Atlanten verschiedener menschlicher Gewebe geliefert. Die zelluläre Zusammensetzung der humanen IMAT ist jedoch aufgrund der inhärenten Herausforderungen ihrer Zugänglichkeit aus der Biopsieentnahme beim Menschen weitgehend unerforscht. Neben der begrenzten Menge an entnommenem Gewebe ist die Verarbeitung der menschlichen IMAT aufgrund ihrer Nähe zu Skelettmuskelgewebe und Faszien kompliziert. Die lipidbeladene Natur der Adipozyten macht sie mit der Einzelzellisolierung unvereinbar. Daher ist die Einzelkern-RNA-Sequenzierung optimal für die Erlangung hochdimensionaler Transkriptomik mit Einzelzellauflösung und bietet das Potenzial, die Biologie dieses Depots aufzudecken, einschließlich der genauen zellulären Zusammensetzung von IMAT. Hier stellen wir ein detailliertes Protokoll für die Zellkernisolierung und die Bibliotheksvorbereitung von gefrorenen humanen IMAT für die Einzelkern-RNA-Sequenzierung vor. Dieses Protokoll ermöglicht die Profilierung von Tausenden von Zellkernen unter Verwendung eines tröpfchenbasierten Ansatzes und bietet so die Möglichkeit, seltene und wenig häufige Zelltypen zu erkennen.
Einleitung
Intermuskuläres Fettgewebe (IMAT) ist ein ektopisches Fettdepot, das sich zwischen und um die Muskelfasern befindet1. Wie in einer kürzlich erschienenen Übersichtsarbeit von Goodpaster et al. ausführlich beschrieben, kann IMAT mittels hochauflösender Computertomographie (CT) und Magnetresonanztomographie (MRT) nachgewiesen werden (Abbildung 1A,B) und befindet sich um und in Muskelfasern im gesamten Körper1. Die Menge der IMAT variiert stark zwischen den Individuen und wird durch BMI, Alter, Geschlecht, Rasse und Bewegungsmangel beeinflusst 2,3,4. Darüber hinaus wird die IMAT-Ablagerung häufig bei pathologischen Zuständen beobachtet, die mit Muskeldegeneration verbunden sind5, und zahlreiche Studien haben eine erhöhte IMAT-Masse bei Personen mit Fettleibigkeit, Typ-2-Diabetes, metabolischem Syndrom und Insulinresistenz dokumentiert 6,7,8,9. Nichtsdestotrotz stehen die zellulären und biologischen Eigenschaften von IMAT erst am Anfang der Entschlüsselung. Die eingeschränkte Zugänglichkeit und die Variation der IMAT-Lokalisationen und des IMAT-Gehalts im gesamten Körper haben die Entnahme von Proben aus diesem einzigartigen Fettdepot2 erschwert. Darüber hinaus können die Proben bei der Entnahme leicht mit der Skelettmuskulatur (SM) "kontaminiert" werden, was die Trennung zwischen dem biologischen Beitrag der verschiedenen Gewebe schwer zu entschlüsseln macht (Abbildung 1C). Zu diesem Zweck dient die Einzelkern-RNA-Sequenzierung (snRNA-seq), die in den letzten zehn Jahren stark an Aufmerksamkeit gewonnen hat, als ideale Methode, um die Trennung von IMAT- und SM-abgeleiteten Genexpressionsmustern mit Einzelzellauflösung zu ermöglichen. Darüber hinaus ist die Isolierung von Zellkernen besonders nützlich für Fettgewebe, da es sich um große lipidbeladene Adipozyten handelt, die nicht in Einzelzellsuspension dissoziiert werden können, ohne die Integrität der Zellen zu beeinträchtigen. Schließlich birgt diese Technologie das Potenzial, neuartige Marker für IMAT-spezifische Adipozyten zu entdecken und die Zusammensetzung und das Vorhandensein verschiedener Vorläuferzellpopulationen aufzudecken sowie die Variation der Zellzusammensetzung unter pathologischen und normalen Bedingungen zu untersuchen.

Abbildung 1: Bilder von IMAT. Repräsentatives Magnetresonanzbild (MRT) der IMAT von (A) einer schlanken Frau mittleren Alters und (B) einem Mann mittleren Alters mit Adipositas. Rot: subkutanes Fettgewebe, gelb: intermuskuläres Fettgewebe, grün: Skelettmuskulatur, blau: Knochen. Bild mit freundlicher Genehmigung von Heather Cornnell, AdventHealth Translational Research Institute. (C) Frische Gewebeprobe mit IMAT (eingezehrt durch eine gestrichelte schwarze Linie). Bild mit freundlicher Genehmigung von Meghan Hopf, AdventHealth Translational Research Institute und Bryan Bergman, University of Colorado. Diese Abbildung wurde mit Genehmigung von Goodpaster et al.1 geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Aus der Viehwirtschaft wurde eine Reihe von Studien veröffentlicht, die die Marmorierung von Fleisch (insbesondere IMAT) bei Schweinen, Hühnern und Rindern mit Einzelzell- (sc) und snRNA-seq10 untersuchen. Diese Studien haben mehrere Subpopulationen von Adipozyten und Marker potenzieller Vorläuferzellen von IMATidentifiziert 11,12,13; ob sich diese zellulären Zusammensetzungen auf die humane IMAT übertragen lassen, ist jedoch nicht bekannt. Unseres Wissens hat nur eine Studie die zelluläre Heterogenität von menschlichen Muskeln mit Fettinfiltration untersucht, die von männlichen Patienten mit Hüftarthrose unter Verwendung von snRNA-seq14 gewonnen wurden. Die Forscher berichteten über eine kleine Adipozytenpopulation und mehrere fibro-adipogene Vorläuferpopulationen (FAP) innerhalb der großen Population von Myonuklei14. Unsere Studie ist die erste, die eine Methode entwickelt, um IMAT, das manuell aus menschlichen Muskeln seziert wurde, mittels snRNA-seq direkt auf die zelluläre Zusammensetzung zu untersuchen.
Wichtig ist, dass die Protokolle für snRNA-seq für das untersuchte Gewebe angepasst werden müssen, da die Menge des verfügbaren Gewebes und die physikalischen Eigenschaften des spezifischen Gewebes die optimalen Verarbeitungsschritte bestimmen. Die Gewebeausbeute für IMAT ist in der Regel gering und überschreitet oft nicht 50 mg, selbst wenn ultraschallgesteuerte Biopsien durchgeführt werden. Daher ist die richtige Verarbeitung dieses knappen Gewebes unerlässlich. Wir glauben, dass dieses Protokoll als wertvolle Ressource für Forscher dienen wird, die die IMAT beim Menschen untersuchen.
Access restricted. Please log in or start a trial to view this content.
Protokoll
Die für dieses Protokoll verwendete Stichprobe war Teil der Study of Muscle, Mobility, and Aging (SOMMA)15, die vom Institutional Review Board der Western IRB-Copernicus Group (WCG) genehmigt wurde und in Übereinstimmung mit der Deklaration von Helsinki durchgeführt wurde. Die Teilnehmer gaben eine schriftliche Einverständniserklärung für ihre Teilnahme an der Studie ab.
HINWEIS : Dieses Protokoll wurde von einem früheren Protokoll adaptiert, bei dem 100 mg subkutanes Fettgewebe des menschlichen Abdomens auf einer Nanowell-basierten Plattform16 verwendet wurden. Das aktuelle Protokoll ist für 50 mg humane IMAT und Bibliotheksvorbereitung unter Verwendung einer tröpfchenbasierten Plattform optimiert. Eine weitere Optimierung dieses Protokolls für die Zellkernisolierung aus nicht-humanen IMAT oder anderen Fettdepots könnte erforderlich sein.
1. Herstellung von Puffern und Reagenzien (Tabelle 1 und Tabelle 2)
HINWEIS: Bereiten Sie die Puffer am Tag des Versuchs frisch vor und verwenden Sie sie nicht wieder.
- Eine Zentrifuge auf 4 °C vorkühlen.
- Bereiten Sie den Homogenisierungspuffer und das Kernisolationsmedium vor.
- Besorgen Sie sich zwei Eimer Eis und kühlen Sie 2 x 15 mL konische Röhrchen vor.
- Mischen Sie alle Reagenzien für den Homogenisierungspuffer (HB) in einem konischen 15-ml-Röhrchen in der in Tabelle 1 aufgeführten Reihenfolge. Bleiben Sie auf Eis. Durch Vortexen mischen.
- Mischen Sie alle Reagenzien für das Kernisolationsmedium (NIM) in einem konischen 15-ml-Röhrchen in der geordneten Liste in Tabelle 2. Bleiben Sie auf Eis. Durch Vortexen mischen.
- Bereiten Sie 10% Triton-X vor, indem Sie 100 μl Triton X-100 auf 900 μl nukleasefreies Wasser hinzufügen. Vortex, um das richtige Mischen zu gewährleisten. Bei Raumtemperatur (RT) aufbewahren.
| Reagenz | Volumen (μL) | Endkonzentration (mM) | |
| 1x | 2x | ||
| 1 M MgCl2 | 10 | 20 | 5 |
| 1 M Tris-Puffer, pH 8,0 | 20 | 40 | 10 |
| 2 M KCl | 25 | 50 | 25 |
| 1,5 M Saccharose (-4°C) | 334 | 668 | 250 |
| 1 mM DVB-T | 2 | 4 | 0,001 (~1 μM) |
| 100x Protease-Hemmer | 20 | 40 | 1x |
| Superasin 20 U/μL | 40 | 80 | 0,4 U/μL |
| Nukleasefreies Wasser | 1549 | 3098 | - |
| Gesamtvolumen | 2000 | 4000 | - |
Tabelle 1: Homogenisierungspuffer (HB). Bleiben Sie auf Eis. Durch Vortexen mischen.
| Reagenz | Volumen (μL) | Endkonzentration (mM) | |
| 1x | 2x | ||
| EDTA | 0.4 | 0.8 | 0.1 |
| Ribolock-RNAse-Inhibitor (40U/μL) | 40 | 80 | 0,8 U/μL |
| 1% BSA-PBS (-/-) | 1959.6 | 3919.2 | - |
| Gesamtvolumen | 2000 | 4000 | - |
Tabelle 2: Kernisolationsmedium (NIM). Bleiben Sie auf Eis. Durch Vortexen mischen.
2. Pulverisierung von gefrorenem Gewebe (Abbildung 2A)
- Richten Sie den Arbeitsplatz für die Homogenisierung ein.
- Füllen Sie den Kanister mit flüssigem Stickstoff (LN2).
ACHTUNG: Tragen Sie bei der Arbeit mit LN2 immer eine Schutzbrille und Kryohandschuhe. - Besorge dir 2x Mörser, 1x Stößel, 1x Mikroschaufelspatel, 1x Glasspachtel und 1x Edelstahlstößel für den automatischen Douncer.
- Richten Sie den automatischen Douncer ein.
- Füllen Sie ein Becherglas mit Eis und kühlen Sie das Glas vor.
- Füllen Sie den Kanister mit flüssigem Stickstoff (LN2).
- Füllen Sie die 2 Mörser (mit Stößel und Spatel) mit LN2 , um die Instrumente abzukühlen. Lassen Sie den LN2 verdampfen und wiederholen Sie den Vorgang.
- Während die Instrumente abkühlen, geben Sie 1 mL HB in den Glasdounce.
- Füllen Sie beide Mörser ein letztes Mal mit LN2 und gießen Sie die 50 mg IMAT-Probe in einen der Mörser.
- Pulverisieren Sie den IMAT mit dem Stößel, indem Sie ihn vorsichtig auf das Stück Taschentuch drücken, um es in kleine Stücke zu brechen. Stellen Sie sicher, dass alle Teile pulverisiert sind.
- Der LN2 verdampft langsam, während das Gewebe pulverisiert wird. Wenn das Gewebe ordnungsgemäß pulverisiert ist und noch 1/4 - 1/2 Mörser LN2 übrig ist, kippen Sie den Mörser in Richtung der Lippe des Mörsers, um das pulverisierte Gewebe an der Lippe zu sammeln. Lassen Sie den LN2 vollständig verdampfen.
- Unmittelbar nachdem das letzte LN2 verdampft ist, wird das pulverisierte Gewebe in den Glasdounce mit 1 ml HB geschöpft.
3. Homogenisierung von pulverisiertem Gewebe
- Homogenisieren Sie das pulverisierte Gewebe mit dem automatischen Douncer. Bringen Sie das Glas auf dem Edelstahlstößel für 10 Hübe in Vorwärtsrichtung auf und ab, gefolgt von 10 Hüben in umgekehrter Richtung.
- Stellen Sie sicher, dass die Lösung nach der Homogenisierung trüb ist und keine sichtbaren Gewebestücke enthält. Oft wird eine hellrosa Farbe aufgrund der Kontamination mit Muskelgewebe erwartet.
- Übertragen Sie das Homogenat in ein vorgekühltes 1,7-ml-Röhrchen mit geringer Bindung auf Eis.
- Verwenden Sie 400 μl HB, um den Dounce zu spülen, um sicherzustellen, dass das gesamte Material übertragen wird, und geben Sie es in das Röhrchen.
HINWEIS: Es können zwei Proben gleichzeitig verarbeitet werden. Verdoppeln Sie dazu die Menge an HB und NIM. Pulverisieren und homogenisieren Sie eine Gewebeprobe und unmittelbar danach pulverisieren und homogenisieren Sie die zweite Gewebeprobe, um die Isolations- und Reinigungsschritte parallel durchführen zu können.
4. Isolierung und Reinigung von Zellkernen (Abbildung 2B)
- 14 μl Triton-X (10 %) zum Homogenat hinzufügen, um eine Konzentration von 0,1 % zu erhalten.
- Halten Sie die Tube 10-15 Minuten lang auf Eis und im Dunkeln, während Sie alle 3 Minuten vortexen.
- Befeuchten Sie ein 100 μm und ein 40 μm Zellsieb (pro Probe) mit jeweils 100 μl RT DPBS in einem konischen 50 mL Röhrchen.
- Filtrieren Sie das Homogenat durch das 100 μm Zellsieb.
- Spülen Sie das 1,7 mL Röhrchen mit 400 μl HB und filtrieren Sie es durch das 100 μm Zellsieb.
- Filtrieren Sie anschließend die Lösung durch das 40 μm Zellsieb.
- Übertragen Sie die gleiche Menge Lösung in zwei vorgekühlte 1,7 mL Low-Bind-Röhrchen, was ~900 μl in jedem Röhrchen entspricht.
- Die Röhrchen 10 min bei 2700 x g bei 4 °C zentrifugieren. Nach der Zentrifugation sollte ein kleines Pellet sichtbar sein.
- Entfernen und verwerfen Sie die oberste Lipidschicht und den verbleibenden Überstand, wobei ~50 μl Lösung aus dem ersten Röhrchen übrig bleiben.
- Wiederholen Sie den Vorgang für die zweite Tube.
- Resuspendieren Sie das Pellet im ersten Röhrchen gründlich, indem Sie es 20x vorsichtig auf und ab pipettieren und in ein neues 1,7-ml-Röhrchen mit geringer Bindung umfüllen. Vermeiden Sie die Bildung von Blasen.
- Wiederholen Sie diesen Vorgang für das zweite Röhrchen und übertragen Sie die resuspendierte Lösung in dasselbe Röhrchen.
- 500 μl NIM zugeben und durch Pipettieren mischen.
- Das Röhrchen mit einer Waage bei 1000 x g für 10 min bei 4 °C zentrifugieren.
- Entfernen Sie den Überstand, lassen Sie ~50 μl übrig und pipettieren Sie vorsichtig auf und ab, bis das Pellet wieder suspendiert ist. Optional können Sie das resuspendierte Pellet in ein neues, sauberes Röhrchen umfüllen, wenn etwas Lipidrest an der Seite des Röhrchens verbleibt.
- 200 μl NIM zugeben und durch Pipettieren mischen.
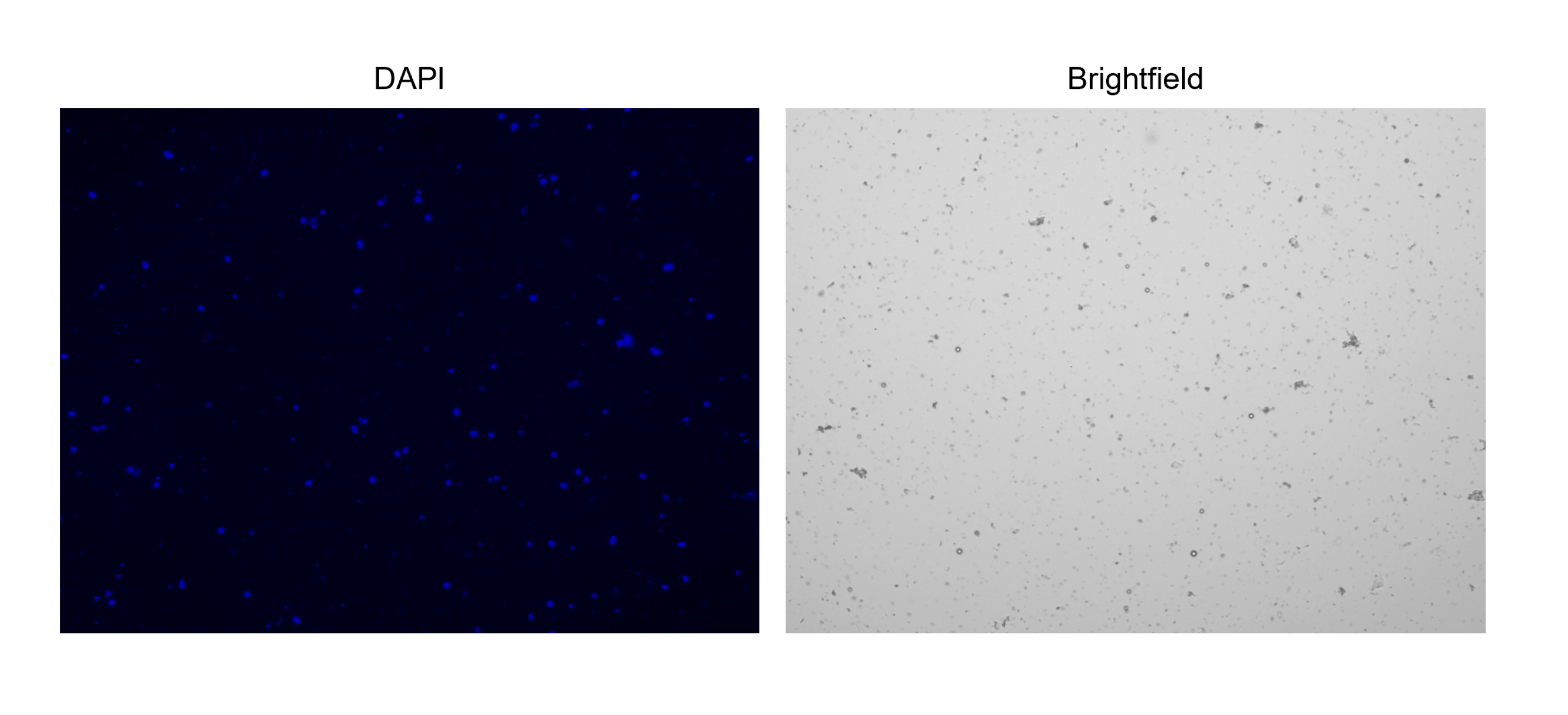
5. Färbung und Zählung von Zellkernen (Abbildung 2C und Abbildung 3)
HINWEIS : Um die Zählung zu erleichtern, richten Sie ein Protokoll zur "Kernzählung" auf einem automatisierten Zellzähler ein, da die Anpassung des Hellfelds und der DAPI-Kanäle die Zählung stark beeinflussen kann. Passen Sie die Kanäle so an, dass nur Kerne und keine Trümmer erfasst werden. Stellen Sie sicher, dass der Hellfeldkanal nur "Objekte" markiert, die auch einen DAPI-Fleck aufweisen.
- Fügen Sie 1 Tropfen der Färbelösung für lebende Zellen hinzu und lassen Sie sie 15 Minuten lang im Dunkeln auf Eis.
- Die Lösung durch ein 30 μm Zellsieb filtrieren.
- Mischen Sie die Zellkernlösung durch Pipettieren und geben Sie 10 μl der Lösung in einen Objektträger der Zellzählkammer.
- Zählen Sie die Zellkerne mit einem automatisierten Zellzähler.
HINWEIS: Die optimale Konzentration beträgt 1000 Kerne/μL, was 1,0 x 106/ml entspricht.- Stellen Sie sicher, dass keine Klumpen von Kernen vorhanden sind, da dies den Chip zur Erzeugung von Einzelkerntröpfchen verstopfen könnte (Abbildung 3).
- Wenn die Kernkonzentration nicht hoch genug ist, schleudern Sie die Lösung bei 1000 x g für 10 min bei 4 °C herunter, um ein Pellet zu erhalten, entfernen Sie den Überstand und resuspendieren Sie sie in einem kleineren Volumen.
- Wenn der Schmutzgrad in der Lösung hoch ist, resuspendieren Sie die Kernlösung in einem größeren Volumen NIM (d. h. 1 mL) und filtrieren Sie erneut durch ein 30-μm-Zellsieb. Dann bei 1000 x g für 10 min bei 4 °C herunterschleudern und in einem angemessenen Volumen im Verhältnis zur Kernkonzentration resuspendieren.
- Nachdem Sie die Kernkonzentration erhalten haben, fahren Sie direkt mit dem ersten Schritt der Bibliotheksvorbereitung fort.

Abbildung 3: Färbung von isolierten Zellkernen. Bild vom Zellzähler von Zellkernen, die mit NucBlue/DAPI gefärbt wurden (linkes Bild) und das entsprechende Hellfeldbild (rechtes Bild). Das Vorhandensein geringer Mengen an Schmutz ist im Hellfeldbild offensichtlich. Der hier verwendete automatische Zellenzähler verfügt nicht über eine Option zum Einschließen von Maßstabsleisten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
6. Parameter für die Bibliotheksvorbereitung und -sequenzierung
- Es wird auf ein gründliches Protokoll für die Bibliotheksvorbereitung unter Verwendung des tröpfchenbasierten Einzelkernansatzes verwiesen, das auf der Webseite17 des Anbieters verfügbar ist.
- Streben Sie eine angestrebte Zellkernrückgewinnung von 10.000 an. Bei Proben mit einem hohen Gehalt an Trümmern oder zerbrechlichen Kernen wird jedoch eine geringere Anzahl der gewonnenen Kerne erwartet.
- Die Proben werden nach Schritt 2.3 bis zu 72 h bei 4 °C gelagert. im Protokoll zur Bibliotheksvorbereitung, um die parallele Verarbeitung mehrerer Proben zu kombinieren. Verarbeiten Sie dazu zwei Proben bis Schritt 2.3 an zwei aufeinanderfolgenden Tagen und verarbeiten Sie am dritten Tag die 4 Proben zusammen aus Schritt 3 und weiter im Protokoll zur Bibliotheksvorbereitung.
- Sequenzierungsparameter: Sequenzierung auf einer Sequenzierungsplattform mit dem Ziel von 50.000 Paired-End-Reads pro Kern.
HINWEIS: Die in diesem Protokoll präsentierten Daten wurden auf der NovaSeq 6000-Plattform sequenziert, mit dem Ziel, 50.000 Paired-End-Reads pro Kern zu erreichen.

Abbildung 2: Protokoll-Workflow. Schematische Darstellung des Arbeitsablaufs in (A) Schritten 2 und 3, (B) Schritt 4 und (C) Schritt 5 des Protokolls. Die Figur ist mit BioRender.com entstanden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
7. Datenverarbeitung und -analyse
HINWEIS : In diesem Protokoll werden einige der empfohlenen Software- und R-Pakete, die zur Verarbeitung der resultierenden Sequenzierungsdaten verwendet werden, kurz vorgestellt, wobei der Schwerpunkt auf den Schritten nach der anfänglichen Vorverarbeitung liegt (Tabelle 3). Diese Studie enthält allgemeine Metriken zur Qualitätskontrolle (QC) und ein Beispiel für eine einheitliche Mannigfaltigkeitsapproximation und -projektion (UMAP) in Abbildung 4. Eine detaillierte Beschreibung der bioinformatischen Analyse würde jedoch den Rahmen dieses Protokolls sprengen. Daher können sich die Leser auf die kürzlich erschienene Übersichtsarbeit zu Best Practices für die Einzelzellanalyse von Heumos et al.18 beziehen.
- Vorverarbeitung von Sequenzierungsdaten
- Ordnen Sie die einzelnen Kernlesevorgänge dem humanen Referenzgenom GRCh38 zu.
- Beziehen Sie die Intron-Lesevorgänge in die Zählung ein.
- Führen Sie die Qualitätskontrolle und Filterung der Daten mit dem Seurat R-Paket 19 durch.
- Berechnen Sie einen Zellkomplexitätswert, indem Sie die log(10)-Anzahl der nachgewiesenen Gene durch die log(10)-Anzahl der erkannten Lesevorgänge dividieren.
- Stellen Sie die wichtigsten QC-Metriken mithilfe eines Histogramms oder Violin-Diagramms dar, einschließlich der Anzahl der pro Zellkern nachgewiesenen Gene, des prozentualen Anteils der mitochondrialen Lesevorgänge und der Zellkomplexitätsbewertung.
- Filtern Sie Zellkerne mit weniger als 200 oder mehr als 10.000 Genen pro Zellkern, mehr als 10 % mitochondrialen Lesevorgängen und einem Komplexitätswert von unter 0,8 heraus.
- Normalisieren Sie Daten und führen Sie eine Dimensionalitätsreduzierung durch.
- Verwenden Sie die SCTransform-Funktion von Seurat, um die Daten anhand von 2000 variablen Features zu normalisieren.
- Cluster-Daten mit den folgenden Funktionen aus dem Seurat R-Paket: RunPCA, FindNeighbors, FindClusters und RunUMAP.
- Plotten Sie eine UMAP, um das Clustering der Daten zu visualisieren.
- Filtern Sie vorhergesagte Dubletten mit dem DoubletFinder R-Paket 20 heraus und gruppieren Sie die Daten neu.
- Annotation von Clustern unter Verwendung bekannter Genmarker der Zelltypen, von denen erwartet wird, dass sie im Gewebe vorhanden sind (überwachter Ansatz) oder basierend auf den Top 5 unterschiedlich exprimierten Genen zwischen den Clustern (unüberwachter Ansatz).
- Verwenden Sie decontX21, um den Grad der Kontamination von Ambient-RNA zu bestimmen und die Genexpressionsmatrix für Ambient-RNA anzupassen.
- Fügen Sie die rohe Genmatrix als Hintergrund hinzu.
- Speichern Sie das Seurat-Objekt für die zukünftige Erkundung der Daten.
HINWEIS: Der Code für die QC- und Clustering-Analyse ist in der Zusatzdatei 1 verfügbar.
| Software/R-Pakete, die im Datenworkflow verwendet werden | Alternative Software/Pakete | Verarbeitungsschritt |
| CellRanger | STARsolo, kallisto | Trimmen, Ausrichten, Mapping |
| Seurat | SingleCellExperiment, Cellranger | QC, Analyse und Datenexploration |
| DoubletFinder | scds, scdblFinder, Scrublet | Dublett-Erkennung |
| DecontX | SoupX, CellBender | Anpassung der Umgebungs-RNA |
Tabelle 3: Software/Tools für den Daten-Workflow.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Dieser Arbeitsablauf wurde entwickelt, um die Verarbeitung von gefrorenen humanen IMAT-Proben zu steuern, um Genexpressionsprofile mit Einzelkernauflösung zu erhalten und so die Identifizierung von Zelltypen zu ermöglichen. Hier wird eine repräsentative IMAT-Stichprobe eines Teilnehmers der SOMMA-Studie vorgestellt.
Der erste Schritt jeder Analyse von snRNA-seq-Daten besteht darin, die Qualität der Daten zu bewerten, um Zellkerne von schlechter Qualität zu identifizieren, die möglicherwe...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Die Arbeit mit IMAT ist mit mehreren Herausforderungen verbunden. Neben der eingeschränkten Zugänglichkeit ist die Ausbeute an Probenmaterial oft sehr gering, und eine "Kontamination" der Skelettmuskulatur ist fast unmöglich zu vermeiden. Um die beste Probenqualität zu erhalten, sollte man beim Einführen der Biopsienadel in die Muskelfaszie eindringen (um sicherzustellen, dass kein subkutanes Fettgewebe entnommen wird) und so viel Muskelgewebe wie möglich entfernen, indem man die Probe unmittelbar nach der Entnahme...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Die Autoren danken Bryan Bergman, PhD an der University of Colorado, für die Bereitstellung des Bildes der IMAT-Biopsie in Abbildung 1C aus der MoTrIMAT-Studie (R01AG077956). Wir sind dankbar für die Study of Muscle, Mobility and Aging, die die IMAT-Stichprobe zur Verfügung gestellt hat, deren Daten im Abschnitt "Repräsentative Ergebnisse" gezeigt werden. Das National Institute on Aging (NIA) finanzierte die Study of Muscle, Mobility and Aging (SOMMA; R01AG059416) und deren Nebenstudien SOMMA AT (R01AG066474) und SOMMA Knee OA (R01AG070647). Die Unterstützung der Studieninfrastruktur wurde teilweise von NIA Claude D. Pepper, den Older American Independence Centers an der University of Pittsburgh (P30AG024827) und der Wake Forest University (P30AG021332) sowie den Clinical and Translational Science Institutes, finanziert vom National Center for Advancing Translational Science, an der Wake Forest University (UL1 0TR001420) finanziert.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.2 µm corning syringe filters | Millipore Sigma | CLS431229 | |
| 1.7 mL DNA LoBind tubes | Eppendorf | 22431021 | low-bind tubes |
| 10% Tween 20 | Bio-Rad | 1662404 | |
| 100x protease inhibitor | Thermo Fisher Scientific | 78437 | |
| 10X Magnetic Separator | 10X Genomics | 230003 | |
| 10X Vortex Adapter | 10X Genomics | 330002 | |
| 15 mL canonical tubes | Sarstedt | 6,25,54,502 | |
| 2100 Bioanalyzer | Agilent | G2939BA | |
| 50 mL conical tubes | Sarstedt | 6,25,47,254 | |
| CellRanger | Genomics | N/A | |
| Chromium iX accesory kit | 10X Genomics | PN1000323 | |
| Chromium iX Controller | 10X Genomics | PN1000326 | |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | PN1000127 | |
| Chromium Next GEM Single Cell 3' Gel Bead Kit v3.1 | 10X Genomics | PN1000129 | |
| Chromium Next GEM Single Cell GEM Kit v3.1 | 10X Genomics | PN1000130 | |
| Countess 3 Automated Cell Counter | Thermo Fisher Scientific | AMQAX2000 | Automated cell counter |
| Countess cell counting chamber slides | Thermo Fisher Scientific | C10228 | |
| DoubletFinder | R | N/A | |
| DPBS (no calcium, no magnesium) | Thermo Fisher Scientific | 14190144 | |
| DTT | Thermo Fisher Scientific | R0861 | |
| Dual Index Kit TT Set A, 96 rxns | 10X Genomics | PN1000215 | |
| Dynabeads MyOne SILANE | 10X Genomics | PN2000048 | |
| Falcon 100 µm Cell strainer | Corning Life Science | 352360 | |
| Falcon 40 µm Cell strainer | Corning Life Science | 352340 | |
| Glycerin (glycerol), 50% (v/v) Aqueous Solution | Ricca Chemical Company | 3290-32 | |
| KCL | Thermo Fisher Scientific | AM9640G | |
| Library Construction Kit v3.1 | 10X Genomics | PN1000196 | |
| MACS SmartStrainers (30µm) | Miltenyi Biotec | 130-098-458 | |
| Mastercycler Nexus Gradient Thermal cycler | Eppendorf | 6331000017 | |
| MgCl2 | Ambion | AM9530G | |
| Mortar and pestel | Health care logistics | 14075 | |
| NucBlue Live Ready Probes Reagent | Thermo Fisher Scientific | R37605 | |
| Nuclease Free Water (not DEPC treated) | Thermo Fisher Scientific | AM9930 | |
| Probumin Bovine Serum Albumin Fatty Acid Free, Powder | Sigma-Aldrich | 820024 | |
| Qiagen Buffer EB | Qiagen | 19086 | |
| Ribolock RNAse inhibitor | Thermo Fisher Scientific | EO0382 | |
| Seurat | R | N/A | |
| Sucrose | Sigma-Aldrich | S0389 | |
| SUPERasin 20 U/µL | Thermo Fisher Scientific | AM2695 | |
| ThermoMixer C | Eppendorf | 5382000015 | |
| Tissue homogenizer | Glass-Col | 099C K54 | |
| Tris buffer pH 8.0 | Thermo Fisher Scientific | AM9855G | |
| Triton X-100 | Thermo Fisher Scientific | AC327372500 | |
| UltraPure 0.5M EDTA pH 8.0 | Gibco | 15575020 |
Referenzen
- Goodpaster, B. H., Bergman, B. C., Brennan, A. M., Sparks, L. M. Intermuscular adipose tissue in metabolic disease. Nat Rev Endocrinol. 19 (5), 285-298 (2023).
- Sparks, L. M., Goodpaster, B. H., Bergman, B. C. The metabolic significance of intermuscular adipose tissue: Is IMAT a friend or a foe to metabolic health. Diabetes. 70 (11), 2457-2467 (2021).
- Gallagher, D., et al. Adipose tissue in muscle: A novel depot similar in size to visceral adipose tissue. Am J Clin Nutr. 81 (4), 903-910 (2005).
- Manini, T. M., et al. Reduced physical activity increases intermuscular adipose tissue in healthy young adults. Am J Clin Nutr. 85 (2), 377-384 (2007).
- Addison, O., Marcus, R. L., LaStayo, P. C., Ryan, A. S. Intermuscular fat: A review of the consequences and causes. Int J Endocrinol. 2014, 309570(2014).
- Goodpaster, B. H., et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch Intern Med. 165 (7), 777-783 (2005).
- Goodpaster, B. H., Thaete, F. L., Kelley, D. E. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr. 71 (4), 885-892 (2000).
- Goodpaster, B. H., et al. Association between regional adipose tissue distribution and both type 2 diabetes and impaired glucose tolerance in elderly men. Diabetes Care. 26 (2), 372-379 (2003).
- Sachs, S., et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am J Physiol Endocrinol Metab. 316 (5), E866-E879 (2019).
- Ford, H., Liu, Q., Fu, X., Strieder-Barboza, C. White adipose tissue heterogeneity in the single-cell era: From mice and humans to cattle. Biology (Basel). 12 (10), 1289(2023).
- Wang, L., et al. Single-nucleus and bulk RNA sequencing reveal cellular and transcriptional mechanisms underlying lipid dynamics in high marbled pork. NPJ Sci Food. 7 (1), 23(2023).
- Li, J., et al. Identification of diverse cell populations in skeletal muscles and biomarkers for intramuscular fat of chicken by single-cell RNA sequencing. BMC Genomics. 21 (1), 752(2020).
- Lyu, P., Qi, Y., Tu, Z. J., Jiang, H. Single-cell RNA sequencing reveals heterogeneity of cultured bovine satellite cells. Front Genet. 12, 742077(2021).
- Fitzgerald, G., et al. MME+ fibro-adipogenic progenitors are the dominant adipogenic population during fatty infiltration in human skeletal muscle. Commun Biol. 6 (1), 111(2023).
- Cummings, S. R., et al. The study of muscle, mobility and aging (SOMMA): A unique cohort study about the cellular biology of aging and age-related loss of mobility. J Gerontol A Biol Sci Med Sci. 78 (11), 2083-2093 (2023).
- Whytock, K. L., et al. Isolation of nuclei from frozen human subcutaneous adipose tissue for full-length single-nuclei transcriptional profiling. STAR Protoc. 4 (1), 102054(2023).
- 10x Genomics. Chromium Single Cell 3' Reagent Kits User Guide (v3.1 Chemistry Dual Index), Document Number CG000315 RevE. , Available from: https://cdn.10xgenomics.com/image/upload/v1668017706/support-documents/CG000315_ChromiumNextGEMSingleCell3-_GeneExpression_v3.1_DualIndex__RevE.pdf (2022).
- Heumos, L., et al. Best practices for single-cell analysis across modalities. Nat Rev Genet. 24 (1), 550-572 (2023).
- Hao, Y., et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. 42 (2), 293-304 (2023).
- McGinnis, C. S., Murrow, L. M., Gartner, Z. J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8 (4), 329-337 (2019).
- Yang, S., et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 21 (2), 57(2020).
- Common considerations for quality control filters for single cell RNA-seq data. 10X Genomics. , Available from: https://www.10xgenomics.com/analysis-guides/common-considerations-for-quality-control-filters-for-single-cell-rna-seq-data (2022).
- Luecken, M. D., Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 15 (6), e8746(2019).
- Emont, M. P., et al. A single-cell atlas of human and mouse white adipose tissue. Nature. 603 (7903), 926-933 (2022).
- Hildreth, A. D., et al. Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat Immunol. 22 (5), 639-653 (2021).
- Whytock, K. L., et al. Single cell full-length transcriptome of human subcutaneous adipose tissue reveals unique and heterogeneous cell populations. iScience. 25 (8), 104772(2022).
- Probst, V., et al. Benchmarking full-length transcript single cell mRNA sequencing protocols. BMC Genomics. 23 (1), 860(2022).
- CG000148 Rev A Technical Note - Resolving cell types as a function of read depth and cell number. Technical note. 10X Genomics. , Available from: https://assets.ctfassets.net/an68im79xiti/6gDArDPBTOg4IIkYEO2Sis/803be2286bb a5ca67f353e6baf68d276/CG000148_10x_Technical _Note_Resolving_Cell_Types_as_Function_of_ Read_Depth_Cell_Number_RevA.pdf (2018).
- Gupta, A., et al. Characterization of transcript enrichment and detection bias in single-nucleus RNA-seq for mapping of distinct human adipocyte lineages. Genome Res. 32 (2), 242-257 (2022).
- Bakken, T. E., et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 13 (12), e0209648(2018).
- Wu, H., Kirita, Y., Donnelly, E. L., Humphreys, B. D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol. 30 (1), 23-32 (2019).
- Kim, N., Kang, H., Jo, A., Yoo, S. -A., Lee, H. -O. Perspectives on single-nucleus RNA sequencing in different cell types and tissues. J Pathol Transl Med. 57 (1), 52-59 (2023).
- Avila Cobos, F., Alquicira-Hernandez, J., Powell, J. E., Mestdagh, P., De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Commun. 11 (1), 5650(2020).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten