
Method Article
Genoma toda la Proteína-proteína interacción Screening por Complementación de ensayo de proteínas-fragmento (PCA) en las células vivas
En este artículo
Resumen
Proteins interact with each other and these interactions determine in a large part their functions. Protein interaction partners can be identified at high-throughput in vivo using a yeast fitness assay based on the dihydrofolate reductase protein-fragment complementation assay (DHFR-PCA).
Resumen
Proteins are the building blocks, effectors and signal mediators of cellular processes. A protein’s function, regulation and localization often depend on its interactions with other proteins. Here, we describe a protocol for the yeast protein-fragment complementation assay (PCA), a powerful method to detect direct and proximal associations between proteins in living cells. The interaction between two proteins, each fused to a dihydrofolate reductase (DHFR) protein fragment, translates into growth of yeast strains in presence of the drug methotrexate (MTX). Differential fitness, resulting from different amounts of reconstituted DHFR enzyme, can be quantified on high-density colony arrays, allowing to differentiate interacting from non-interacting bait-prey pairs. The high-throughput protocol presented here is performed using a robotic platform that parallelizes mating of bait and prey strains carrying complementary DHFR-fragment fusion proteins and the survival assay on MTX. This protocol allows to systematically test for thousands of protein-protein interactions (PPIs) involving bait proteins of interest and offers several advantages over other PPI detection assays, including the study of proteins expressed from their endogenous promoters without the need for modifying protein localization and for the assembly of complex reporter constructs.
Introducción
Las redes de interacción de proteínas (PIN) ofrecen un mapa de baja resolución de cómo las proteínas se organizan funcionalmente en la celda 1. Cada conexión física entre dos proteínas, o la interacción proteína-proteína (PPI), pueden representar una asociación que es estable en el tiempo, tales como las que se encuentran dentro de los complejos de proteínas y que contribuyen a la organización estructural de la célula. Estas conexiones también pueden representar asociaciones transitorias que regulan la actividad, la estabilidad, la localización y la interacción de los dos socios. La identificación de los socios de la interacción física de una determinada proteína, por tanto, ofrece amplia información sobre la función y regulación de esa proteína 2,3. Por estas razones, grandes esfuerzos se han puesto hacia el mapeo de PINs en organismos modelo, incluyendo Escherichia coli 4-6, Arabidopsis thaliana 7, Saccharomyces cerevisiae 8-12, Drosophila melanogaster </ Em> 13, Caenorhabditis elegans 14 y 15 Homo Sapiens. Estos estudios han proporcionado información importante sobre cómo se organizan las proteínas en la célula y por lo tanto la información clave sobre las proteínas con funciones anteriormente desconocidas.
Varias estrategias se han desarrollado a lo largo de los años para estudiar los PIN. Estas tecnologías se pueden agrupar en términos generales en tres categorías basadas en el tipo de información que proporcionan sobre los IBP (revisado en 16 a 18). El primero de ellos se basa en dos híbridos de levadura y sus derivados 19. Estas tecnologías proporcionan información sobre la asociación directa entre pares de proteínas, lo que permite la construcción de redes binarias. La segunda familia se basa en la purificación por afinidad de proteínas cebo y la identificación de sus socios asociados, tales como la purificación por afinidad seguido de espectrometría de masas 20. Estos enfoques identifican grupos de proteínas que son directamenteo indirectamente asociado, en general, de una manera estable, y son extremadamente poderosa para identificar los complejos de proteínas. El tercer enfoque se basa en ensayos de complementación de proteínas fragmento (ACC) 11,21. Este enfoque proporciona un nivel intermedio de resolución entre los dos enfoques anteriores, ya que permite la detección de asociaciones directas y proximal entre proteínas. Cada técnica tiene sus propias fortalezas y debilidades, como recientemente revisado 18.
El PIN eucariota mejor descrito es de lejos la una de la levadura Saccharomyces cerevisiae en ciernes, en parte debido a su proteoma es relativamente menos complejas que las de otros eucariotas modelo y porque ensayos de alto rendimiento para detectar los IBP primero se han ensayado y es más eficiente implementado en este organismo modelo 9-12. Un método particularmente potente para el sistema de levadura es la dihidrofolato reductasa ensayo de complementación fragmento de proteína (DHFR-PCA), un ensayo que ha sidoutilizado en diferentes contextos para estudiar el PIN levadura en condiciones estándar y perturbados 11,22-26. Este método se basa en un ensayo de supervivencia que permite la detección de los IBP directos y cerca-directos para una proteína cebo dado en ambos niveles de expresión endógenos y localizaciones subcelulares nativos de las parejas de interacción 11,21 de una manera cuantitativa 27. La señal obtenida utilizando este ensayo (es decir, tamaño de la colonia en matrices de colonias de alta densidad) por lo tanto refleja la cantidad de complejos de proteínas formadas entre el cebo y presa en un entorno celular casi equivalente a la de las células de tipo salvaje. El ensayo se basa en la reconstitución de una enzima informadora involucrado en el metabolismo del folato, la dihidrofolato reductasa (DHFR), por el que dos fragmentos complementarios de la DHFR que se fusionan a las dos proteínas de interés se ponen en proximidad cuando las dos proteínas interactúan, que a su vez conduce a la reconstitución reversible de la actividad de la enzima 11 y el crecimiento de la cepa en un medio que contiene metotrexato (MTX; Figura 1). Este compuesto inhibe la enzima DHFR endógena, pero no el mutado utilizado en el ensayo 28. Dos colecciones de cepas de PCA, uno que contiene ~ 4300 MATa cepas con un ORF fusionado al DHFR F [1,2] fragmento y una que contiene ~ 4800 MAT α cepas con un ORF fusionado al [3] fragmento DHFR, se pueden comprar a implementar DHFR-PCA en pequeña o gran escala en cualquier laboratorio. Aquí se describe un protocolo general pero detallada para la detección de los IBP entre una proteína cebo y ~ 4.800 presa proteínas utilizando este ensayo.
Protocolo
1. Construcción / verificación de cebo Cepas
- Si la cepa cebo de interés está disponible en el [1,2] colección MATa DHFR F, recuperarlo de la colección como se describe en el paso 1.1.1, de lo contrario la construcción de la cepa como se describe en el paso 1.1.2.
NOTA: El protocolo descrito aquí utiliza un DHFR F [1,2] cepa como un cebo y el DHFR F [3] colección como presas, ya que esta colección contiene más cepas que la DHFR F [1,2] colección. Sin embargo, es posible realizar la pantalla al revés si la cepa cebo sólo está disponible en el [3] colección de DHFR F o en ambas orientaciones si se requiere una mayor cobertura de la interactome.- Descongelar la placa de stock de glicerol que contiene la cepa cebo en hielo durante una hora. Esterilizar la lámina de aluminio que cubre la placa con etanol al 95%. Pierce la lámina con una punta estéril, una pipeta hacia arriba y abajo para volver a suspender las células y la racha de 2-3 l de glicerol en selectivo peptona extracto de levaduradextrosa (YPD) + 100 mg / ml nourseotricina (Nat) a fin de aislar colonias individuales. Incubar durante dos días a 30 ° C.
- Construcción de una cepa PCA cebo (Mata DHFR F [1,2]).
- Uso de alta fidelidad de la polimerasa y un protocolo estándar de PCR, amplificar la DHFR F [1,2] casete a partir del plásmido pAG25-enlazador-DHFR F [1,2] -ADHterm utilizando oligonucleótidos con extremos salientes homóloga a la última 40 pb de la ORF de extremo 3 'excluyendo el codón de parada (cebador hacia delante) y a la primera 40 pb del gen de la 3'-UTR (cebador inverso) (Figura 1A).
- Transformar el producto de PCR en células de levadura competentes (por lo general en una cepa BY4741) utilizando el protocolo estándar de transformación de levadura LiOAc / PEG como en 29 (Figura 1A).
- Placa en YPD selectiva + Nat medio para aislar transformantes positivos.
- Realizar un PCR colonia de diagnóstico en colonias aisladas para confirmar el correcto DHFR F [1,2] de fusión. Utilice prIMers recocido 1) en la secuencia de codificación de gen (oligo Forward) aproximadamente 100 pb aguas arriba de la fusión DHFR y 2) en el terminador de ADH de la casete (oligo inverso) (Figura 1B).
- Secuenciar el producto de PCR mediante secuenciación Sanger confirmar fusión génica adecuada.
- Archivar la cepa cebo confirmado en el 25% de glicerol a -80 ° C.
NOTA: El protocolo puede ser pausado en este paso.
2. Pin-herramienta de esterilización y prensa Procedimientos
NOTA: El procedimiento de esterilización se describe a continuación se ha optimizado para las herramientas pin manipulados por el BM3-BC (S & P Robótica) plataforma robótica, pero se puede adaptar a otras plataformas también. Esta sección describe los procedimientos de esterilización e impresión Pin-herramientas que se utilizan para transferir las células de un medio a otro para el resto del protocolo. En casa de los scripts utilizados para realizar estas rutinas pueden obtenerse a petición. Nota del that todos los pasos se pueden realizar sin la necesidad de una plataforma robótica usando un pin-herramienta manual de 30.
- Montar el pasador-herramienta adecuada en la plataforma robótica.
- Preparar la limpieza y estaciones húmedas como sigue:
- Añadir 500 ml de agua estéril en la estación de baño de agua.
- Añadir 320 ml de agua estéril en la estación de cepillo.
- Añadir 380 ml de etanol al 70% en el baño de ultrasonidos al replicar de la placa de agar (86 x 128 mm omnitray, que contiene 35 ml de medio solidificado) a la placa de agar, o 400 ml cuando se replica a partir de una placa de microtitulación que contiene cultivos líquidos a una placa de agar.
- Añadir 35 ml de agua estéril en la estación húmeda (que consiste en un omnitray vacío estéril).
- Al comienzo de cada día que requiere la plataforma robótica, esterilizar los pin-herramientas cinco veces durante un minuto en el baño de ultrasonidos. Mientras tanto, encender la lámpara UV durante cinco minutos para esterilizar el recinto robot.
NOTA: Esto no es necesario si la robanot se encuentra bajo una campana estéril. - Entre cada pin-herramienta ronda de replicación, esterilizar la herramienta pin de la siguiente manera:
- Remoje la herramienta pin cinco veces durante 10 segundos en la estación de baño de agua para eliminar los agregados celulares.
- Remojar la herramienta pin dos veces atrás y adelante en la estación de cepillo.
NOTA: El cepillo giratorio elimina las células residuales. - Remoje la herramienta pin dos veces durante 20 segundos en la estación de ultrasonidos.
NOTA: Lo que queda células en los pines se eliminará mediante ultrasonidos o asesinado por etanol. - Asegúrese de que la profundidad de inmersión de los pines aumenta en cada baño sucesivas para asegurar una correcta esterilización.
- Seque la herramienta pin en la estación aérea de pelo durante 25 s.
- Antes de tomar las células en las placas de origen, mojar los pasadores en la estación húmeda, que contiene 35 ml de agua estéril en un omnitray.
- Sumergir las clavijas en dos ocasiones en las colonias de la placa de origen.
- Imprima en una placa de destino recién vertido tocando la agsuperficie ar dos veces (en adelante, como la acción de "imprimir" una matriz).
3. La condensación de la DHFR F [3] Colección en 1536 matrices usando una plataforma automatizada robótica
- Descongelar en hielo la DHFR F [3] de recogida (60 placas de 96 pocillos) y una placa de 96 pocillos adicional lleno de la DHFR L-F [3] [3] fragmento cepa de control (Figura 1C), que contiene el DHFR F y el enlazador aguas arriba expresa solo como control negativo.
NOTA: En principio, este fragmento no debe interactuar con cualquier proteína de fusión de DHFR fragmento (véase la discusión para más detalles). - Placas de centrífuga (vuelta rápida) antes de retirar el papel de aluminio para evitar los riesgos de contaminación cruzada entre los pozos.
- Condensar la colección en 16 conjuntos de 384 cepas (Figura 2a). Para ello, para cada matriz 384, cuatro placas de impresión de glicerol en los cuatro cuadrantes de un YPD selectiva + 250 mg / ml de higromicina B (hygB) omnitray usando ésimoe 96 pin-herramienta (aquí, una matriz 384 se puede subdividir en cuatro cuadrantes igualmente Interspaced, cada uno que consta de 96 posiciones en un diseño de matriz de 2 x 2). Inserte cuatro placas de 96 pocillos que contienen el [3] control negativo L-DHFR F entre las otras 60 placas con el fin de tener un conjunto final de 64 placas que llenan exactamente cuatro 1536 arrays. Esterilizar la herramienta pin entre cada ciclo de replicación, como se describe en el paso 2.
NOTA: Inserte la L-DHFR F [3] placas con el fin de tener uno de tales placa en cada uno de los cuatro finales 1536 arrays. - Incubar las placas durante dos días a 30 ° C. NOTA: En esta etapa, la colección de DHFR puede ser almacenada en un formato de 384 a 4ºC durante hasta un mes en placas de agar.
- Condensar la colección en cuatro matrices de 1.536 cepas (Figura 2a). Para ello, para cada matriz 1536, imprimir cuatro matrices de 384 cepas en los cuatro cuadrantes de una placa del mismo medio como en el paso 3.2 utilizando el pin-herramienta 384 (aquí, una matriz de 1536 se puede subdividir en cuatro equally Interspaced cuadrantes cada uno consistente en 384 posiciones en un diseño de matriz de 2 x 2).
- Incubar las placas durante dos días a 30 ° C. NOTA: En esta etapa, la colección de DHFR puede ser almacenada en un formato de 1536 a 4ºC durante hasta un mes en placas de agar.
- Replicar los cuatro arrays en el mismo medio para estandarizar tamaño de la colonia utilizando un pin-herramienta 1536.
- Incubar las placas durante dos días a 30 ° C.
4. Procedimiento DHFR-PCA de alto rendimiento
- Inocular un cultivo de la cepa cebo (DHFR F [1,2] fusión) obtenida de la etapa 1.1.1 o 1.1.2 en 20 ml de YPD líquido + Nat en un tubo de 50 ml (Figura 2B).
- Incubar durante dos días a 30 ° C con agitación a 250 rpm para permitir la cultura para alcanzar la saturación.
- Después de dos días de incubación, la placa 5 ml de la cultura en un YPD + Nat omnitray. Deje que las células absorben en la superficie durante 5-10 minutos y retirar el exceso de líquido (Figura 2B). Repita twice hacer tres repeticiones.
- Incubar durante dos días a 30 ° C.
- Imprimir la cepa cebo de los pasos 4.3 y 4.4 por 12 placas de YPD (suficiente para aparearse cuatro placas de la DHFR F [3] de recogida × tres repeticiones) con un pin-herramienta 1536 usando cada césped celular no más de cuatro veces.
- Imprimir el arreglo adecuado de la [3] colección DHFR F en la parte superior de las células de cebo utilizando el pin-herramienta 1536 (Figura 2C).
- Deje que las cepas se aparean incubando durante dos días a 30 ° C.
- Seleccione células diploides imprimiendo colonias en omnitrays contienen YPD + hygB + Nat (Figura 2C).
- Incubar durante dos días a 30 ° C.
- Selección diploide Repita como se describe en los pasos 4.8 y 4.9 (Figura 2B).
- Preparar placas con medios que contienen MTX (mediano MTX) un día antes de su uso siguiendo estos pasos 21 (PRECAUCIÓN:. Tener cuidado cuando se trabaja con MTX, ya que es un compuesto tóxico Use siempre guantes, protegafas cción y una bata de laboratorio cuando se manipulen) (Figura 2 C):
- Prepare los 10x -Lys / met / ade aminoácido deserción en agua desionizada. Filter-esterilizar el abandono en una botella estéril utilizando un filtro de jeringa estéril de 0,2 micras o, si es necesario en grandes cantidades, una tapa del embudo de filtración de 0,2 m botella (Tabla 1).
- Preparar una solución madre de 10 mg / ml MTX en sulfóxido de dimetilo (DMSO). Utilizar inmediatamente después de la preparación de la solución y congelar el resto a -20 ° C. Proteger de la luz, ya que es fotosensible. No congele MTX nuevo después de descongelarla.
- Preparar los medios de comunicación como sigue (ingredientes del medio y cantidades son los mismos que los utilizados en Tarassov et al. 11):
- Para un litro de medio, se mezcla en dos matraces separados: 1) 6,69 g de base nitrogenada de levadura sin aminoácidos y sin sulfato de amonio y 330 ml de agua desionizada; 2) 25 g de agar noble ml y 500 de agua desionizada.
- Frascos autoclave a 121 ° C durante 20 min.
- Equilibrar la temperatura en un baño de agua a 55 ° C durante al menos una hora.
- Mezclar los dos matraces juntos y agregar 50 ml de 40% de glucosa estéril, 100 ml de 10x de deserción estéril, y 20 ml de MTX 10 mg / ml.
- Verter 35 ml (ver nota abajo) de medio en omnitrays. Deje solidificar durante al menos una hora y media. Placas protectoras de la luz.
NOTA: Aquí, verter 35 ml de medio por omnitray es fundamental para garantizar grosor igual, que es importante para todos los pasos intermedios.
- Las placas radiográficas de la segunda ronda de selección diploide con la plataforma robótica o con una cámara digital normal con iluminación placa uniforme. Use estas imágenes para identificar posiciones vacías en las matrices al realizar el análisis de aguas abajo. Asegúrese de que los parámetros de la cámara son siempre los mismos y que la luz robot está encendido.
- Imprimir células diploides en MTX usi mediosón del pin-herramienta 1536.
- Incubar durante cuatro días a 30 ° C en bolsas de plástico para evitar el secado.
- Preparar un segundo lote de omnitrays contenían medio MTX como se describe en el paso 4.11.
- Después de cuatro días de incubación, las placas de imagen mediante la plataforma robótica o una cámara digital normal. Asegúrese de que los parámetros de la cámara son siempre los mismos y que la luz robot está encendido.
- Realizar una segunda ronda de selección MTX mediante la replicación de las células en el segundo lote de medios MTX.
NOTA: Esto disminuirá el crecimiento de fondo de las cepas de PCA y aumentar la resolución cuantitativa. - Incubar durante cuatro días a 30 ° C en bolsas de plástico para evitar el secado.
- Las placas radiográficas como se describe en el paso 4.16.
Análisis 5. Imagen
- Analizar imágenes de matrices de colonias con ImageJ encargo 31 secuencias de comandos o utilizando softwares publicados como colonizador, Ht analizador rejilla colonia, perfilador celular, Colonia imagenr, ScreenMill, YeastXtract y gitter (compilado en el 32). Análisis de la imagen de salida debe una o varias hojas de cálculo que contienen tamaños de colonia para cada posición de cada matriz, utilice estas colonias de tamaños para todos los análisis.
NOTA: En este estudio, hemos utilizado un script ImageJ encargo descrito en Leducq et al 33 (ver sección de discusión para más detalles)..
Análisis 6. Datos
NOTA: Los resultados de los análisis de las imágenes se pueden procesar en un tabulador como Excel o el uso de un lenguaje de programación, tales como R 34. Los siguientes pasos describen el procedimiento utilizando una costumbre ImageJ 31 script.
- El uso de un script personalizado, concatenar los archivos de salida a partir de análisis de imágenes y anotaciones en cada fila con la placa y la tensión de la información como en el complementario el cuadro 1.
- Entrar 2 transformar los valores de tamaño colonia (densidad integrada o área de la colonia; aquí, la columna "IntDenBackSub & #8221; desde el complementario el cuadro 1 se utilizó).
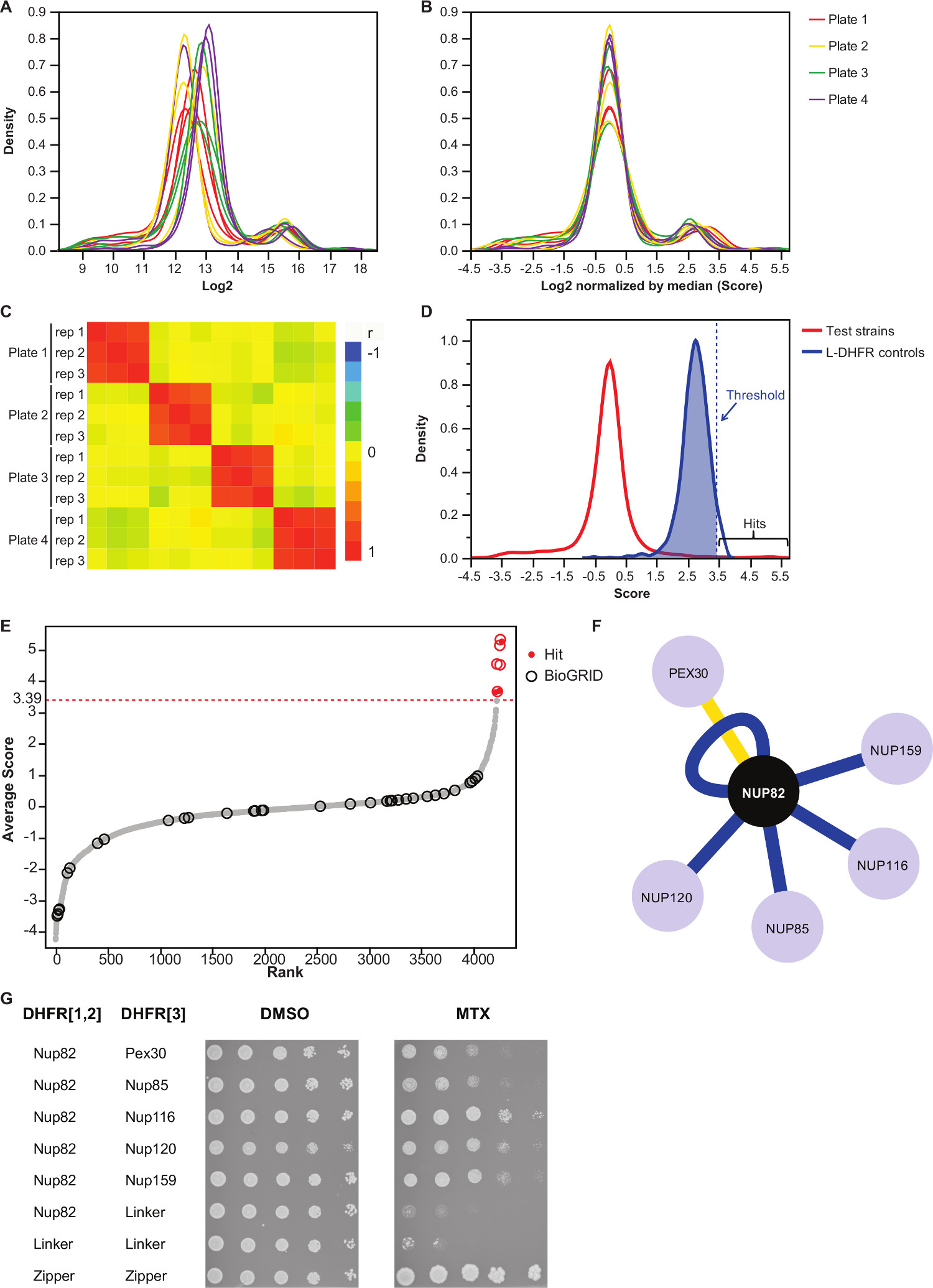
NOTA: Distribución de Valor se verá como en la Figura 3A. - Normalizar estos valores restando el valor de la mediana de cada placa.
NOTA: Este paso controles para sesgo placa que puedan deberse a la cantidad de medios desigual o variación en la adquisición automática de la imagen y reduce la varianza inter-replicarse (Figura 3B). - Verifique que las réplicas se correlacionan entre sí (Figura 3C) para evaluar la reproducibilidad de los experimentos.
- Para diferenciar la interacción de pares que no interactúan cebo-presa, establecer un umbral alto de confianza correspondiente al percentil 95 de la distribución de la L-DHFR F [3] controles.
NOTA: En este experimento, esto corresponde a 3,39 (Figura 3D). Alternativamente, se puede utilizar un umbral basado en la superposición con interactores físicos conocidos, tales como los reportados en la BIOGRID 35.Véase la discusión para más detalles. - Para cualquier cebo, filtro presas identificó como implicados en las interacciones positivas falsas en las pantallas de DHFR-PCA (véase la discusión para más detalles) y que figuran en la tabla complementaria 2 (identificado como "1" en la columna "filtrada").
- La media de los Log2 tamaños de colonia normalizada de las tres réplicas de cada interacción (columna "Calificación media" en el complementario el cuadro 2).
7. Validación de Interactianos física utilizando experimentos a pequeña escala
NOTA: Cualquier PPI de interés particular que tiene una puntuación por encima o cerca del umbral aplicado puede ser validado utilizando el ensayo de DHFR-PCA en un diseño experimental a pequeña escala usando un ensayo de crecimiento en medio MTX sólido o líquido. Los pasos siguientes muestran el procedimiento para construir manualmente cepas PCA diploides y realizar ensayos sobre el terreno de medio MTX. El experimentador debe realizar estos pasos para todos necessarcontroles Y (Bait-DHFR F [1,2] x L-DHFR F [3], la cremallera-enlazador-DHFR cepas diploides y enlazador-DHFR cepa diploide).
- Placa 2.3 l de la madre de glicerol de la cepa cebo generado en 1.1.2.6), la L-DHFR F [3] cepa de control, la cremallera-enlazador-DHFR y enlazador-DHFR cepas diploides en YPD + Nat, YPD + medios hygB y dos veces YPD + Nat + hygB, respectivamente.
- Recuperar la presa de interés en el [3] colección DHFR F y siga las instrucciones en el paso 1.1.1, pero la racha de la cepa en el medio en vez de medio YPD + Nat YPD + hygB.
- Realizar un diagnóstico de PCR como en 1.1.2.4 para confirmar la DHFR F [3] de fusión en el locus presa y la secuencia del producto.
- Inocular 1 ml de medio YPD líquido con las cepas haploides para aparearse (Bait x presa, [3] Control Bait x L-DHFR F) y crecer por lo menos dos días a 30 ° C para permitir que los diploides a la forma.
- Seleccione diploides estriando 4.5 l de la cultura en 7.4 en medio YPD + hygB + Nat sólido. Crecer dos días en 30 ° C.
- Select una colonia aislada y crecer durante la noche en cultivo líquido (1 ml) para llevar a cabo el ensayo de crecimiento.
- Preparar placas de MTX y DMSO (mismos ingredientes como medio MTX, pero sin MTX) un día antes de su uso. Vea el paso 4.11 para más detalles.
- Realizar ensayo de crecimiento mediante la detección de diluciones seriadas de las diferentes culturas en el control (DMSO) y placas de selección (MTX).
- Diluir precultivos a OD = 1.
- Realizar diluciones de cinco veces (hasta un factor de dilución de 625) en una placa de 96 pocillos estéril.
- Punto 4 l de cada dilución en los medios de comunicación PCA (DMSO y MTX).
- Incubar a 30 ° C en bolsas de plástico para evitar el secado.
- Las placas radiográficas de los días 1 a 7 de incubación utilizando la plataforma robótica o una cámara digital normal.
Resultados
Tabla complementaria 2 es un ejemplo de los resultados representativos obtenidos utilizando la proteína de levadura Nup82 fusionado a la DHFR F [1,2] fragmento como un cebo. El umbral definido con la L-DHFR F [3] controles se puede utilizar como un umbral empírico para determinar los accesos de alta confianza (Figuras 3D y 3E). Alternativamente, el ranking puntuación puede ser utilizado para realizar enriquecimientos de ontología de genes u otro funcional analiza 36 basado en estándares de oro 37. Los interactores físicas conocidas de la carnada se pueden recuperar de bases de datos como BIOGRID 35 y superpuestos en los datos (Figuras 3E y 3F). En este ejemplo, cinco de los ocho imparables alta confianza han sido señalados como interactores Nup82 y dos son parte de la subcomplejo Nup82, Nup116 y Nup159 (Figura 3F y 3G). El otro miembro del complejo, NSP1, no muestra ninguna interacciónción en nuestro experimento. Dos presas, Ade17 y TIF2 (no se muestra en la Figura 3F), tenían puntuaciones por encima del umbral duro aplicado, pero estos son propensos a ser falsos positivos, ya que interactúan con casi cualquier proteína cebo en pantallas de PCA que hemos realizado (resultados no publicados). Por otro lado, Pex30 puede representar una novela interactor física de Nup82 y pudimos confirmar esta interacción utilizando DHFR-PCA a bajo rendimiento (Figura 3G). Pex30 es una proteína de membrana peroxisomal y algunas interacciones directas se han reportado entre el complejo del poro nuclear (CPN) y este orgánulo. Una pantalla de dos híbridos identificó otras dos proteínas de la APN, Nup53 y Asm4 (Nup59), como interactores físicas de Pex30 38, y una interacción genética entre Pex30 y Nup170 ha informado 39. Otros dos compañeros de interacción detectada, Nup120 y Nup85 (Figura 3F y 3G), no forman parte de la sub-complejo Nup82, que ilustra la capacidadde la DHFR-PCA para detectar interacciones dentro y entre Subcomplejos en los complejos más grandes 11.

Figura 1:. Cepas de levadura de ingeniería de alto rendimiento para DHFR-PCA (. Figura adaptada de Leducq et al 2012 33) (A) Construcción de cepas Mata y MAT α haploides para fundir gene1 (G1) y Gene2 (G2) con el DHFR F [1,2] -NatMX y la DHFR F [3] -HPH casetes, respectivamente. Cassettes se amplifican a partir de plásmidos pAG25-DHFR F [1,2] y pAG32-DHFR F [3] con cebadores directos G1-5 "y G2-5", y atrás primers G1-3 "y G2-3", y luego insertado en el genoma en el extremo 3 'del gen diana mediante recombinación homóloga. Las proteínas resultantes, P1 y P2, respectivamente, se fusionan a la DHFR F [1,2] fragmento (Mata) y el DHFR F [3] fragmento (MATα) a través de un enlazador flexible. (B) Verificación de la construcción en (A) se lleva a cabo mediante la secuenciación de las uniones entre gene1 de ORFs y de Gene2 y los casetes de DHFR. Las cepas construidas PCA de oponerse a los tipos de apareamiento son entonces acopladas para formar un diploide. Cepas diploides crecen en medio MTX si los dos fragmentos complementarios de DHFR se ponen en proximidad por una interacción entre P1 y P2, que reconstituye la actividad de la enzima DHFR. (C) Construcción de las cepas de control diploide PCA para pantallas DHFR-PCA. Los controles negativos (L-DHFR) se construyen mediante la transformación de cepas Mata y MATα separado haploides con plásmidos p41-enlazador-DHFR F [1,2] y p41-enlazador-DHFR F [3] 11, respectivamente. Las dos cepas se acoplan resultando en una cepa diploide de control negativo en el que los fragmentos de DHFR son incapacespara complementarse entre sí (arriba). Los controles positivos se construyen utilizando el mismo planteamiento que para los controles negativos, pero los plásmidos transformados en cepas haploides (p41-cremallera-enlazador-DHFR F [1,2] (p41-ZL-DHFR F [1,2]) y p41 -zipper-enlazador-DHFR F [3] (p41-ZL-DHFR F [3])) contienen fragmentos de cremallera de leucina GCN4 paralelo dos fusionadas a los fragmentos complementarios de DHFR, lo que conduce a una interacción fuerte y constitutivo que reconstituye la actividad DHFR (parte inferior ). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Procedimiento de DHFR-PCA de alto rendimiento (figura adaptada de Leducq et al 33.) (A) El MAT α DHFR F [3] colección. se condensa en un formato de 1536 a través de dos rondas sucesivas de condensación. En primer lugar, las placas de glicerol se combinan por grupos de cuatro en selectivo medio YPD + hygB en un formato de 384 utilizando el pin-herramienta 96. En segundo lugar, 384 matrices son combinados por grupos de cuatro en selectivo medio YPD + hygB en un formato 1536 con el pin-herramienta 384. (B) céspedes celulares de la cepa cebo MATa PCA se preparan cultivando una cultura saturada de la cepa cebo en YPD selectiva + medio Nat y chapado a la cultura en un YPD + Nat omnitray. (C) Estos jardines se utilizan para acoplar la cepa cebo con la DHFR F [3] recogida en medio YPD. Las células se transfieren sucesivamente dos veces en medio YPD + hygB + Nat seleccionar para diploides y dos veces en medio MTX para realizar PCA. El crecimiento sólo se observó en el medio MTX si los fragmentos de DHFR se complementan entre sí después de una interacción entre el cebo y las proteínas presa.e.jpg "target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 3:.. Análisis de datos a través de etapas de normalización, la determinación de un umbral de significación y la identificación de las interacciones positivas (A) distribución de la densidad de tamaño de las colonias en cada placa (log 2) (B) La normalización por la mediana de cada imagen corrige los sesgos asociado con efectos de placa a placa. (C) Mapa de calor que muestra el coeficiente de correlación de Spearman entre las placas, lo que confirma la reproducibilidad del procedimiento. (D) Distribución de las puntuaciones de los IBP probados y el [3] controles L-DHFR F. Un umbral de fuerza se puede ajustar a los 95 percentil de la L-DHFR F [3] de distribución para identificar los IBP alta confianza (representado por una línea vertical de puntosdistribución de líneas). (E) orden de rango de la puntuación media de cada presa. Anteriormente reportados socios de interacción física de Nup82p en el BIOGRID 35 se identifican mediante círculos y los reportados en este estudio se identifican con puntos rojos. El umbral definido en (D) se muestra como una línea de puntos. (F) de red que muestra los IBP alta confianza identificadas en este estudio. Interactores bordes azules muestran previamente reportados físicas (BIOGRID 35) y bordes amarillos muestran una interacción no declarada previamente con Pex30. (G) ensayo de Spot-dilución de cepas diploides PCA implica Nup82-DHFR F [1,2] y la presa-DHFR F [3 ] pares identificados como interactores físicas de Nup82p en el presente estudio. Ensayo de crecimiento se realizó en medio DMSO (MTX disolvente, panel izquierdo) y medio MTX (panel derecho). Los controles negativos consisten en Nup82-DHFR F [1,2] - Enlazador-DHFR F [3] y el enlazador-DHFR F [1,2] - Enlazador-DHFR F [3] y un control positivo que consiste en tque fuerte interacción entre dos restos de cremallera de leucina (cremallera-DHFR F [1,2] - cremallera-DHFR F [3]) fueron incluidos. El crecimiento celular superior a los controles negativos en el medio MTX debe interpretarse como una interacción física. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Amino ácido | Cantidad (g) |
| Sulfato de adenina * | 0.2 |
| L-Triptófano | 0.4 |
| L-tirosina | 0.3 |
| L-fenilalanina | 0.5 |
| Ácido L-glutámico (sal monosódica) | 1.0 |
| L-asparagina | 1.0 |
| L-Valina | 1.5 |
| L-Treonina | 2.0 |
| L-Serina | 3.75 |
| Uracile | 0.2 |
| L-Histidina HCl | 0.2 |
| L-Arginina HCl | 0.2 |
| L-Metionina * | 0.2 |
| L-Lisina * | 0.2 |
| L-leucina | 0.6 |
| * Retirada al realizar norma PCA (como en este protocolo), pero se pueden añadir para otros fines. | |
Tabla 1:. Composición de la 10X -Lys / met / ade deserción en el medio MTX Las cantidades son para un litro de 10x deserción. Los asteriscos indican los compuestos que deben ser retirados por el medio estándar MTX, pero que se pueden agregar para otros fines.
Que complementa el cuadro 1:. Los datos combinados de las placas de 12 pruebas Tabla 1 contiene la da concatenadosta del análisis ImageJ realizó usando un script ImageJ personalizado con cada fila corresponde a una única posición en cada matriz 1536. Además, cada fila se ha anotado con la información sobre el archivo de imagen, la placa de colección DHFR, replicar, ORF y nombre de la proteína.
Tabla complementaria 2:. Crecimiento normalizada media por cepa Tabla 2 contiene el promedio Log 2 crecimiento normalizado para cada cepa de la colección junto con la desviación estándar. Presas filtradas, golpes y interactores físicas conocidas se identifican en columnas separadas.
Discusión
Se describe un protocolo basado en el ensayo de DHFR-PCA permite la identificación sistemática de interactores físicas para cualquier cebo de proteína dada en alto rendimiento. Este protocolo se puede adaptar mediante el cribado para más cebos, y esto en cualquier nivel deseado de replicación. Se demuestra la fiabilidad de este protocolo por la identificación de los socios de la interacción física para una proteína cebo involucrados en el complejo del poro nuclear: Nup82. Nuestro análisis permitió encontrar cinco interactores reportados previamente y uno interactor no notificados anteriormente (Figuras 3F y 3G), destacando la capacidad del método para estudiar la interactome proteína de levadura.
El protocolo descrito aquí incluye varios pasos críticos a los que el experimentador debe prestar atención. Recomendamos a 1) Asegúrese de que el DHFR cebo F [1,2] de fusión es correcta (Figura 1B); esto se puede lograr mediante la secuenciación de la construcción y measuring expresión de la proteína adecuada usando un anticuerpo anti-DHFR F [1,2] o anti-DHFR F [3] anticuerpo; 2) Antes de comenzar la pantalla, se recomienda verificar si cualquier cebo de interés exhibe interacciones promiscuos en pantallas de PCA. Esto se puede hacer mediante la realización de pantallas de control con cebos cruzados con el control L-DHFR apropiada o mediante apareamiento manualmente el cebo con el control L-DHFR apropiado y realizar un ensayo de crecimiento en medio MTX. 3) Las placas se deben verter el día antes de que se utilizan para que la humedad es óptimo para la adherencia de las células sobre la superficie del agar durante el proceso de impresión; 4) Fuente placas no deben utilizarse más de cuatro veces para transferir suficientes células en la placa de destino. Aumentar el número de copias de la placa de destino se puede hacer mediante etapas sucesivas de expansión (por ejemplo, 4 copias -> 16 copias -> 64 copias). Alternativamente, las células pueden ser recogidos en diferentes posiciones en el césped o en la colonia entre las diferentes rondas de replicación; 5) Si varios positions faltan después de la selección (s) diploide, asegúrese de que las placas de base no se usaron demasiadas veces en la etapa de apareamiento (pasos 4.5 a 4.7); 6) Asegúrese de que el medio MTX contiene todos los ingredientes esenciales en las concentraciones adecuadas. De hecho, si no hay crecimiento en absoluto se observa en el medio MTX, puede ser ya sea porque no hay interacción es detectable por PCA para las proteínas de interés o porque el medio MTX no estaba preparado adecuadamente. Para asegurarse de que el medio permite el crecimiento de las cepas que muestran fragmentos de DHFR complementación, una interacción constitutiva se puede añadir en las posiciones vacías de la colección y se utiliza como un control positivo, tal como DHFR-fragmentos fusionados a restos de cremallera de leucina 33 (Figura 1C). Pruebas paralelas utilizando los fragmentos enlazador-DHFR o los fragmentos de la cremallera-enlazador-DHFR permitirá discriminar entre condiciones que permiten que todas las células crezcan (baja concentración de MTX o cebo de proteína que tienden a hacer que las interacciones positivas falsas, como described a continuación) y las condiciones que impiden el crecimiento de todas las cepas (ingrediente concentración de MTX demasiado alto o esencial que falta en el medio); 7) Dado que la PCA se lleva a cabo a través de rondas sucesivas de repeticiones de un medio a otro, la contaminación cruzada entre las cepas entre diferentes placas puede ocurrir si, por ejemplo, la herramienta pin no se esteriliza adecuadamente entre las rondas de replicación y / o el agua del último baño (es decir, la estación húmeda) en el procedimiento de esterilización está contaminada por colonias de rondas de replicación anteriores. Varias posiciones en las matrices están vacíos y por lo tanto se pueden utilizar como posiciones de control donde se debe observó crecimiento para detectar contaminaciones cruzadas.
Análisis de imágenes se puede realizar utilizando varios softwares publicados (véase la sección 5 del protocolo) o cualquier script personalizado. En este estudio, la secuencia de comandos personalizada ejecuta los siguientes pasos: 1) El guión resta valores de píxel de un plato vacío al píxel valores de cada placa con el fin de correct los sesgos de iluminación. 2) La secuencia de comandos convierte cada imagen de fondo con corrección a binario utilizando un umbral de valor de píxel de 10. 3) Por cada 1.536 posiciones de cada placa, determinados mediante la superposición de un rectángulo en las colonias de borde, el script se ejecuta ImageJ "Analizar partículas ... "en función de una selección circular. La selección circular está configurado con un radio igual al intervalo entre dos posiciones menos 10 píxeles. 4) La secuencia de comandos selecciona la partícula más cercano del centro de la selección y la confirma como una colonia si su ubicación es no más de la mitad del intervalo entre dos colonias de distancia del centro de la selección. 5) El guión mide valores de píxeles de la partícula seleccionada en la imagen de fondo con corrección. 6) Para corregir además cualquier resto de sesgos de iluminación de fondo, el guión resta el valor medio de todos los píxeles de la selección circular que no formaban parte de una partícula a valores de píxeles de la colonia. La suma de estos valores de píxel corregidos, almacenados en la columna "IntDenBackSub" de la Tabla 1 complementario, se utilizan como medida del tamaño de la colonia.
Un paso crítico dentro de la parte de análisis es la elección del umbral de significación. Aquí, hemos elegido un umbral basado en la distribución de la negativa L-DHFR F [3] controles, pero dependiendo del objetivo de la pantalla, tal umbral puede ser demasiado estrictas. De hecho, L-DHFR F [3] controles se sobreexpresa (promotor fuerte TEF) de tal manera que los fragmentos complementarios pueden espontáneamente se complementan entre sí y por lo tanto éstos no son representativas de la expresión de la mayoría de las proteínas. Esto se pone de relieve por el hecho de que la distribución de la L-DHFR F [3] controles es más alto que el promedio de crecimiento de fondo (Figura 3D). Por lo tanto, algunas interacciones que tienen puntuaciones por debajo de este umbral estricto sino que están claramente fuera de la distribución del crecimiento fondo pueden ser considerados como éxitos supuestos que pueden representar, por ejemplo, entre transitoria o débilacciones. Estos se pueden estudiar más y validación cruzada si, por ejemplo, las dos proteínas no se expresan en niveles que pueden permitir la complementación espontánea de los fragmentos de DHFR como los controles de L-DHFR. Como alternativa, se podría establecer un umbral de significación basado en la proporción de superposición con interactores físicas reportadas en bases de datos como BIOGRID 35 a fin de maximizar la proporción de verdaderos positivos sobre falsos positivos. Sin embargo, a diferencia de la utilización de la distribución de L-DHFR, esta alternativa no siempre puede ser factible si, por ejemplo, el número de interactores físicos conocidos no es suficientemente alta. Además, la elección del umbral de significación tiene un impacto en la proporción de falsos positivos y falsos negativos en el conjunto de datos final. En efecto, como cualquier otro ensayo de detección de PPI, los falsos positivos pueden resultar de la interacción no específica de una proteína con la proteína DHFR-fusion si, por ejemplo, la proteína es muy abundante como se mencionó anteriormente. Estese ejemplifica por el hecho de que algunas presas interactúan sistemáticamente con todas las proteínas de cebo en las pantallas de PCA y, por tanto, necesitan ser retirados del análisis 11 (por ejemplo TIF2 y Ade17 y la Tabla complementaria 2). Para evitar este problema, una pantalla de PCA de control de las dos colecciones contra el control L-DHFR apropiado (F [1,2] o F [3]) para identificar cebos y presas que exhiben fragmentos de DHFR espontánea complementación se puede realizar en las condiciones específicas de cada pantalla. Por otra parte, la realización de un análisis de enriquecimiento de ontología de genes puede aumentar la confianza en los datos si se conoce la función de un cebo dado. Por otro lado, DHFR-PCA puede dar lugar a falsos negativos por varias razones: 1) no todas las proteínas se pueden fusionar a los fragmentos de DHFR ya que pueden desestabilizar las proteínas o modificar su localización si, por ejemplo, la fusión a la DHFR C-terminal interfiere con una señal de localización; 2) DHFR reconstitución en algunos compartimentos celulares may no producen folato si, por ejemplo, un precursor esencial para la síntesis de folato no está disponible; 3) C-termini necesidad de estar dentro de una distancia de 8 nm para DHFR complementación que se produzca 11. Por lo tanto, una interacción muy conocido no se puede detectar si su C-terminales no están lo suficientemente cerca en el espacio. Esto se ejemplifica aquí por el hecho de que una gran fracción de las interacciones físicas Nup82 reportadas en bases de datos, la mayoría de los cuales son indirecta, no se detectaron en nuestro ensayo. Del mismo modo, las interacciones entre proteínas de membrana para las que los extremos C-terminales están en trans con respecto a la membrana no darán lugar a fragmentos de DHFR complementación y no se detecta 11. Limitaciones 1) y 3) puede ser eludido de forma relativamente simple mediante la fusión del fragmento de DHFR a la N-terminales de la proteína. Hacerlo puede evitar interferir con una señal de localización, cerca de la C-terminal y puede permitir detectar una interacción entre las proteínas de membrana cuyo N y C-terminal son en relación cisa la membrana.
Varios desafíos permanecen en el estudio de PINs (revisado en 2,3). Los mapas de PINs producidos hasta ahora en gran parte se han descrito en una sola condición experimental para cada especie y así ofrecer una sola instantánea de cómo se podrían organizar redes de proteínas. Existe por tanto una necesidad para la exploración de las demás condiciones experimentales para ver cómo PINs pueden ser reorganizadas en respuesta a los cambios ambientales, estímulos específicos, a través de desarrollo o siguientes mutaciones. Estos retos se pueden superar mediante el desarrollo de nuevas tecnologías para interrogar a los IBP en tiempo real, en las células vivas y mediante la adaptación de las técnicas actuales para que puedan ser utilizados por una comunidad más amplia de laboratorios. Como técnica cuantitativa que puede detectar cambios en la cantidad de DHFR complementación complejos 27, DHFR-PCA se puede adaptar a superar estos retos y se ha utilizado para estudiar cómo los IBP se ven afectados por un ADN agente 22 dañar , agentes químicos 25, deleciones de genes 23,26 o en otras especies de levaduras y sus híbridos 33. Explorando estas nuevas dimensiones se hará más y más importante para revelar la dinámica del PIN.
Divulgaciones
Parte de los gastos de publicación de acceso abierto para este artículo fueron pagados por S & P Robótica.
Agradecimientos
Este trabajo fue apoyado por el Instituto Canadiense de Investigación en Salud (CIHR) Otorga 191.597, 299.432 y 324.265, una de Ciencias Naturales e Ingeniería de Investigación de subvención Canadá Descubrimiento y una subvención Human Frontier Science Program de CRL. CRL es un CIHR Nueva Investigador. Guillaume Diss es apoyado por una beca PROTEO. Samuel Rochette es apoyado por NSERC y FRQNT becas.
Materiales
| Name | Company | Catalog Number | Comments |
| BioMatrix Robot, Bench-top Configuration | S&P Robotics Inc. | BM5-BC | |
| 96-format Pin-tool | S&P Robotics Inc. | PH-96-10 | Standard 96-format Pin-tool with 96 high-precision floating pins |
| 384-format Pin-tool | S&P Robotics Inc. | PH-384-10 | Standard 384-format Pin-tool with 384 high-precision floating pins |
| 1536-format Pin-tool | S&P Robotics Inc. | PH-1536-05 | Custom 1536-format Pin-tool with 0.5mm high-precision floating pins |
| Automated imaging module | S&P Robotics Inc. | IMG-02 | |
| Methotrexate | Bioshop Canada Inc. | MTX440 | CAUTION: toxic compound |
| Hygromycin B | Bioshop Canada Inc. | HYG003 | |
| Nourseothricin dihydrogen sulfate | Werner BioAgents | 5010000 | |
| Yeast-Interactome Collection | Thermo Scientific | YSC5849 | |
| Omni Tray w/lid sterile | Thermo Scientific | 242811 | |
| Anti-DHFR F[1,2] antibody | Sigma-Aldrich | D1067 | |
| Anti-DHFR F[3] antibody | Sigma-Aldrich | D0942 |
Referencias
- Alberts, B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 92, 291-294 (1998).
- Diss, G., et al. Integrative avenues for exploring the dynamics and evolution of protein interaction networks. Curr Opin Biotechnol. 24, 775-783 (2013).
- Vidal, M., Cusick, M. E., Barabasi, A. L. Interactome networks and human disease. Cell. 144, 986-998 (2011).
- Hu, P., et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS biology. 7, e96 (2009).
- Arifuzzaman, M., et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16, 686-691 (2006).
- Rajagopala, S. V., et al. The binary protein-protein interaction landscape of Escherichia coli. Nature biotechnology. 32, 285-290 (2014).
- Arabidopsis-Interactome-Mapping-Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science. 333, 601-607 (2011).
- Babu, M., et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature. 489, 585-589 (2012).
- Krogan, N. J., et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 440, 637-643 (2006).
- Gavin, A. C., et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 415, 141-147 (2002).
- Tarassov, K., et al. An in vivo map of the yeast protein interactome. Science. 320, 1465-1470 (2008).
- Uetz, P., et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403, 623-627 (2000).
- Guruharsha, K. G., et al. A protein complex network of Drosophila melanogaster. Cell. 147, 690-703 (2011).
- Li, S., et al. A map of the interactome network of the metazoan C. elegans. Science. 303, 540-543 (2004).
- Rual, J. F., et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 437, 1173-1178 (2005).
- Landry, C. R., Levy, E. D., Abd Rabbo, D., Tarassov, K., Michnick, S. W. Extracting insight from noisy cellular networks. Cell. 155, 983-989 (2013).
- Berggard, T., Linse, S., James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 7, 2833-2842 (2007).
- Wodak, S. J., Vlasblom, J., Turinsky, A. L., Pu, S. Protein-protein interaction networks: the puzzling riches. Current opinion in structural biology. 23, 941-953 (2013).
- Fields, S., Song, O. A novel genetic system to detect protein-protein interactions. Nature. 340, 245-246 (1989).
- Dunham, W. H., Mullin, M., Gingras, A. C. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 12, 1576-1590 (2012).
- Michnick, S. W., Ear, P. H., Landry, C., Malleshaiah, M. K., Messier, V. A toolkit of protein-fragment complementation assays for studying and dissecting large-scale and dynamic protein-protein interactions in living cells. Methods Enzymol. 470, 335-368 (2010).
- Rochette, S., Gagnon-Arsenault, I., Diss, G., Landry, C. R. Modulation of the yeast protein interactome in response to DNA damage. Journal of proteomics. 100, 25-36 (2014).
- Diss, G., Dube, A. K., Boutin, J., Gagnon-Arsenault, I., Landry, C. R. A systematic approach for the genetic dissection of protein complexes in living cells. Cell Rep. 3, 2155-2167 (2013).
- Gagnon-Arsenault, I., et al. Transcriptional divergence plays a role in the rewiring of protein interaction networks after gene duplication. Journal of proteomics. 81, 112-125 (2013).
- Schlecht, U., Miranda, M., Suresh, S., Davis, R. W., St Onge, R. P. Multiplex assay for condition-dependent changes in protein-protein interactions. Proceedings of the National Academy of Sciences of the United States of America. 109, 9213-9218 (2012).
- Lev, I., et al. Reverse PCA, a systematic approach for identifying genes important for the physical interaction between protein pairs. PLoS Genet. 9, e1003838 (2013).
- Freschi, L., Torres-Quiroz, F., Dube, A. K., Landry, C. R. qPCA: a scalable assay to measure the perturbation of protein-protein interactions in living cells. Mol Biosyst. 9, 36-43 (2013).
- Pelletier, J. N., Campbell-Valois, F. X., Michnick, S. W. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proceedings of the National Academy of Sciences of the United States of America. 95, 12141-12146 (1998).
- Gietz, R. D., Woods, R. A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87-96 (2002).
- Schuldiner, M., Collins, S. R., Weissman, J. S., Krogan, N. J. Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions. Methods. 40, 344-352 (2006).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 9, 671-675 (2012).
- Wagih, O., Parts, L. gitter: A Robust and Accurate Method for Quantification of Colony Sizes From Plate Images. G3 (Bethesda). 4 (3), 547-552 (2014).
- Leducq, J. B., et al. Evidence for the robustness of protein complexes to inter-species hybridization. PLoS Genet. 8, e1003161 (2012).
- . . Development Core Team: A language and environment for statistical computing. , (2008).
- Stark, C., et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535-D539 (2006).
- Vinayagam, A., et al. Protein complex-based analysis framework for high-throughput data sets. Science signaling. 6, rs5 (2013).
- Jansen, R., Gerstein, M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Current opinion in microbiology. 7, 535-545 (2004).
- Ito, T., et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences of the United States of America. 98, 4569-4574 (2001).
- Costanzo, M., et al. The genetic landscape of a cell. Science. 327, 425-431 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados