
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Primer para inmunohistoquímica en el tejido cryosectioned cerebro de rata: Ejemplo de tinción de microglía y neuronas
En este artículo
Resumen
This introductory level protocol describes the reagents, equipment, and techniques required to complete immunohistochemical staining of rodent brains, using markers for microglia and neuronal elements as an example.
Resumen
La inmunohistoquímica es una técnica ampliamente utilizada para detectar la presencia, localización, y la abundancia relativa de antígenos in situ. Este protocolo de nivel introductorio describe los reactivos, equipos y técnicas requeridas para completar la tinción inmunohistoquímica de tejido de cerebro de roedores, el uso de marcadores para microglia y elementos neuronales como un ejemplo. En concreto, este trabajo es un protocolo de paso a paso para la visualización fluorescente de la microglia y neuronas a través de inmunohistoquímica para Iba1 y Pan-neuronal, respectivamente. Fluorescencia doble etiquetado es particularmente útil para la localización de múltiples proteínas dentro de la misma muestra, proporcionando la oportunidad de observar con precisión las interacciones entre tipos de células, receptores, ligandos, y / o la matriz extracelular en relación el uno al otro, así como co-proteína localización dentro de una sola célula. A diferencia de otras técnicas de visualización, intensidad de la tinción inmunohistoquímica de fluorescencia puede disminuir enlas semanas a meses después de la tinción, si no se toman las precauciones adecuadas. A pesar de esta limitación, en muchas aplicaciones se prefiere la fluorescencia doble etiquetado sobre alternativas tales como tetrahidrocloruro de 3,3'-diaminobencidina (DAB) o fosfatasa alcalina (AP), como fluorescencia es más eficiente en el tiempo y permite la diferenciación más precisa entre dos o más marcadores. La discusión incluye sugerencias para solucionar problemas y consejos para promover el éxito.
Introducción
La inmunohistoquímica es un proceso para la detección de antígenos (es decir, proteínas) en secciones de tejido mediante el uso de anticuerpos primarios que se unen específicamente a los antígenos de interés. La inmunohistoquímica fue iniciado por JR Marrack en 1934 cuando se determinó que los anticuerpos podrían localizar antígenos con gran especificidad 1. A partir de 1942, algunos de los primeros estudios in vitro usando anticuerpos fluorescentes para visualizar inmunohistoquímica se publicaron 2,3, después de lo cual el primer estudio in vivo en histoquímica fue publicado 4. Durante la década de 1960, tres décadas después de la creación de métodos de inmunohistoquímica, anticuerpos conjugados a enzima comenzaron a ser utilizados como reactivos secundarios. Estos métodos se han desarrollado de forma simultánea e independientemente en Francia y los EE.UU. 5,6. Hoy en día, una amplia gama de anticuerpos ofrece un sinfín de posibilidades para los estudios de inmunohistoquímica 7.
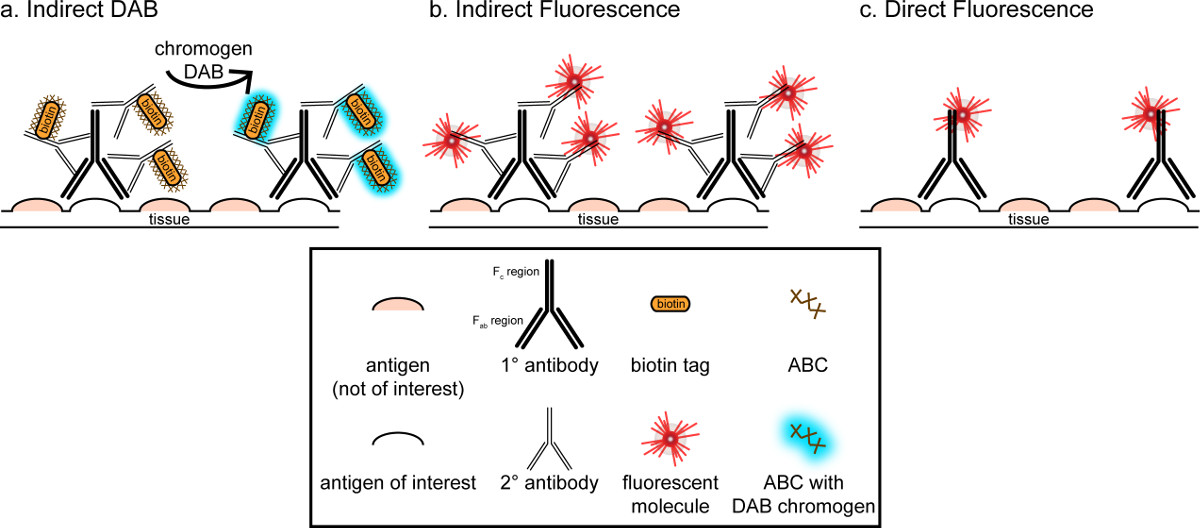
"> El objetivo general de esta correspondencia es proporcionar una breve introducción en la tinción inmunohistoquímica, no pretende ser una revisión completa y exhaustiva de esta técnica en el método descrito, técnicas inmunohistoquímicas para dos antígenos son presentados (marcadores de la microglía y. neuronas) para la tinción de paraformaldehído perfundido, sacarosa cryoprotected, cerebro de rata cryosectioned. La tinción inmunohistoquímica comienza con el bloqueo de la unión no específica de antígeno para reducir la tinción de fondo. A continuación, la incubación con el anticuerpo primario permite por la unión a un antígeno específico en el tejido. Siguiendo el anticuerpo primario, otro anticuerpo, denominado anticuerpo secundario, se aplica a enlazar el anticuerpo primario a una señal de visualización conjugado 8. El anticuerpo secundario se dirige a la inmunoglobulina G (IgG) de dominio específico a las especies en las que se planteó el anticuerpo primario. El anticuerpo secundario amplifica la señal del anticuerpo primario desde las regiones Fab de tque se unen anticuerpo secundario a múltiples sitios en el dominio IgG del anticuerpo primario. Cualquiera de enzimas o moléculas fluorescentes conjugados con las regiones C de F del anticuerpo secundario permitir la visualización. Por ejemplo, un conejo anti-Iba1 anticuerpo primario es una molécula de IgG de conejo específico para Iba1. Cuando se aplica burro anti-IgG de conejo como anticuerpo secundario, se reconocer y unirse a múltiples regiones del anti-IgG de conejo Iba1 (ver Figura 1). El anticuerpo de burro puede ser visualizado por diversos métodos. Esta correspondencia se centra en la detección de un fluoróforo conjugado con el anticuerpo secundario, que reconoce el anticuerpo primario, para la visualización por microscopía fluorescente. En inmunohistoquímica fluorescente, un tinte nuclear, tales como Hoechst o DAPI se puede utilizar para visualizar todos los núcleos.
Figura 1: Schrepresentación Ematic de vs. directa técnicas de marcaje de anticuerpos indirecto. Los anticuerpos se unen al antígeno de interés y puede ser amplificada por anticuerpos secundarios dirigidos contra las especies de los anticuerpos primarios. Esta técnica se puede realizar utilizando complejo avidina-biotina (ABC) para la amplificación y DAB para la visualización (A), o un anticuerpo secundario fluorescente directamente conjugado (B). Alternativamente, los anticuerpos primarios se pueden conjugar directamente con muchas etiquetas diferentes, incluyendo biotina o un fluoróforo (C). Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Un método alternativo para la visualización de la tinción inmunohistoquímica utiliza 3,3'-diaminobenzidina tetrahidrocloruro (DAB; véanse las Figuras 1 y 2). Esto difiere de fluorescencia mediante el uso de un biotinilado operoxidasa de rábano picante (HRP) anticuerpo secundario conjugado, que proporciona una enzima para convertir DAB a un precipitado que es visible bajo microscopía de campo brillante. En los casos en que un solo antígeno es de interés o se requiere tinción a ser de larga duración, DAB puede ser más apropiado que la tinción fluorescente. Sin embargo, la tinción DAB no está bien adaptado para la diferenciación entre múltiples marcadores, especialmente si dos antígenos nucleares son de interés. Para obtener información sobre materiales DAB y modificaciones del protocolo, consulte la Tabla 1. Alternativamente, cloruro de nitro azul tetrazolio / 5-Bromo-4-cloro-3-indolil fosfato (NBT / BCIP) se puede utilizar para visualizar una fosfatasa alcalina (AP) secundario conjugado anticuerpo.

Figura 2:. Imágenes representativas de secciones de tejido cerebral de níquel-DAB mejorada de marcaje único rata rata cerebro del SEcciones que se etiquetan con DAB-níquel mejorada para Iba1 (A) y Pan-neuronal (B) permitir el análisis de larga duración de la microglia o neuronas solas. Barra de escala 20 micras. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Hay que tener en cuenta la abundancia estimada del antígeno de interés dentro del tejido que se está analizando. Los métodos indirectos (como se describe más arriba) son útiles para los objetivos con baja abundancia. Cuando el antígeno de interés se encuentra en gran abundancia, los métodos directos se pueden aplicar. Los métodos directos implican un anticuerpo primario que se conjuga directamente a una señal de visualización, y por lo tanto no se requiere ningún anticuerpo secundario. Este método simplifica el proceso de tinción, pero elimina la amplificación logrado por métodos indirectos. El uso de un anticuerpo primario conjugado directamente también elimina la reactividad cruzada de anticuerpos secundariosal hacer doble etiquetado.
Esta comunicación se detalla el protocolo de doble etiquetado con Iba1 y Pan-neuronales (detalles en la Tabla 1). Iba1 manchas microglia en muchos estados de activación, incluyendo ramificada, hiper-ramificados, que se activa, ameboide, y la varilla. Manchas Pan-neuronales neuronal axones, dendritas, y soma. Desde Iba1 tiñe la mayoría de la microglía y metas Pan-neuronales la neurona, esta combinación de manchas es útil en la obtención de una amplia comprensión de las interacciones microglia neuronas.
En suma, la tinción inmunohistoquímica se basa en la cuidadosa selección de anticuerpos. Como la pregunta de investigación se hace más específica, los anticuerpos producidos contra antígenos alternativos pueden ser deseables. Para conseguir un estado de activación microglial específica, se puede optar por utilizar anticuerpos CD45 o CD68, en lugar de Iba1. Además, en el trabajo con ratones, F4 / 80 puede proporcionar los resultados necesarios. Del mismo modo, los elementos neuronales pueden ser dirigidos específicamente con anticuerpos razada contra el núcleo, sinapsis (pre o post), axón, y el cono de crecimiento. Además, hay otros marcadores que diferencian la edad de la neurona (doble Cortin, NeuN), y la regeneración neuronal (GAP-43).
Protocolo
NOTA: Todos los procedimientos se llevaron a cabo de conformidad con el Cuidado y Uso de Animales Comité Institucional (IACUC) de la Universidad de Arizona. Una lista de los materiales y equipos recomendados se puede encontrar en la Tabla 1.
1. Preparación del tejido
- Perfusión
- La eutanasia a los roedores con una sobredosis de pentobarbital sódico (25 mg / kg, IP), y perfundir transcardially con tampón fosfato salino (PBS) hasta que esté completamente desangrado (3-5 min) a una velocidad de flujo de 8 ml / min. Para obtener instrucciones de perfusión en profundidad, ver Gage et al 2012 9.
- Inmediatamente después de ras PBS, fijar el tejido por perfusión con paraformaldehído al 4% en PBS durante 15-20 min a una velocidad de flujo de 8 ml / min.
- Eliminar cerebro y el lugar en el 4% de paraformaldehído durante 24 h, seguido por soluciones de sacarosa graduadas (15%, 30%, 30%, en secuencia; preparado en solución salina tamponada con Tris) a 4 ° C. Transferir el cerebro para la posterior solución de sacarosa oólo después de que el cerebro se ha hundido en cada solución. Nota: Por lo general, 5 días en cada solución es tiempo suficiente para que el tejido se hunda.
- La congelación de tejidos y cryosectioning
- Coloque el cerebro en medio de inclusión, tal como el compuesto OCT y se sumerja en isopentano a una temperatura de -35 ° C. Deje que el cerebro se congele por un mínimo de 10 minutos, y luego almacenar a -80 ° C. Pueden surgir problemas si no se toma la diligencia a la temperatura; por favor, véase la discusión para la solución de problemas de información.
- Cortar secciones coronales en serie con un espesor de 20 micras y una temperatura de -20 ° C. Recoger el tejido en portaobjetos cargados positivamente. Secciones cerebrales pueden ser colocados en una caja portaobjetos envuelto en papel en una bolsa zip-top y almacenada a largo plazo a -80 ° C. Este método de almacenamiento crea una frontera doble para evitar la exposición al aire y las heladas.
Procesamiento 2. Tejido
NOTA: equipos y materiales Ejemplo rEQUERIDO para la tinción se muestran en la Figura 3. Las alternativas están disponibles, sin embargo, estas imágenes ayudarán a los nuevos en la tinción inmunohistoquímica para visualizar elementos apropiados antes de la compra.

Figura 3: Ejemplo de elementos necesarios para la tinción inmunohistoquímica El cuadro negro se muestra en (A) es una cámara de humedad ideal para inmunofluorescencia, como diapositivas están protegidos de la luz sin la necesidad de envolver la caja de papel de aluminio.. Después de seccionamiento, diapositivas se pueden almacenar en una caja como el cuadro amarillo se muestra en (B). Envolver la caja herméticamente en papel de aluminio y colocarlo en una bolsa zip-top antes del congelador ayuda a proteger las muestras de tejido de las quemaduras por congelación. Un ejemplo de diapositivas se da en (C), con diferentes platos de tinción representados en (D) y (E ). Cubreobjetos pueden variar en tamaño y espesor (F), sin embargo 1,2 cubreobjetos gruesas proporcionan buenos resultados de las imágenes en la mayoría de los microscopios verticales y confocal. Un lápiz tal como la mostrada en (G) se puede utilizar para etiquetar diapositivas. Marcadores permanentes deben ser evitados ya que la tinta puede correr, que afecta tanto a la tinción y la capacidad de determinar lo que la muestra es. Un mini pluma pap tal como la mostrada en (H) permite una frontera repelente que se puede sacar en las diapositivas.
- Preparación de diapositivas
- Eliminar diapositivas de congelador y descongelar a temperatura ambiente.
- Opcional: Si las secciones han flotado previamente de diapositivas, coloque las diapositivas descongeladas en un horno a 60 ° C durante no más de 4 horas para ayudar a prevenir las secciones de tejido de flotando diapositivas.
- Coloque los portaobjetos en un rack de diapositivas y el plato correspondiente.
- Lavar los portaobjetos tres veces en PBS durante 5 minutos cada uno, el cambio de solución entre lavados. A partir de este paso adelante, no tener sections sin líquido durante largos períodos de tiempo. Nota: Si las secciones se secan, la tinción de fondo se incrementa y datos significativos no se puede obtener de forma fiable.
- Eliminar diapositivas de congelador y descongelar a temperatura ambiente.
- Tinción de tejidos
- En una caja de tinción hermética a la luz, crear una "cámara de humedad", con tejidos sin pelusa humedecido con agua destilada.
- Seque los bordes de la diapositiva con un pañuelo que no suelte pelusa, use un mini pluma pap para hacer un borde repelente de líquidos en el mismo borde de la diapositiva, lejos de las secciones de tejido. Esta frontera debe garantizar un amplio espacio entre el menisco del líquido y el borde del tejido de modo que la tensión superficial no afecta a la tinción.
NOTA: El repelente frontera pluma pap se puede aplicar antes de la 2.1.3 si los anticuerpos de interés no requieren microondas recuperación de antígeno. Si la pluma de Papanicolaou se ha aplicado antes del lavado en PBS, la integridad de la frontera-líquido repelente debe comprobarse en este paso. Utilice una pluma mini-pap para llenar los vacíos en la frontera. - Con las diapositivas en posición horizontal, el antígeno inespecífico bloque de unión incubando en el 4% v / v de suero en PBS (solución de bloqueo). Pipetear 300 l de solución de bloqueo por portaobjetos durante 1 hora a temperatura ambiente. Asegúrese de que la solución de bloqueo se extiende a la pluma pap en el borde de la corredera y cubre completamente el tejido para evitar la tinción desigual causada por la tensión superficial cerca del tejido.
- Utilice el suero de la misma especie en la que se hace anticuerpo secundario. Nota: En este procedimiento, los anticuerpos secundarios se hacen en burro, y por lo tanto se utiliza suero de burro. Si se utilizan anticuerpos secundarios de dos o más especies diferentes, incluir el suero de cada especie.
- Pipetear anticuerpo primario en portaobjetos. Nota: Las concentraciones de anticuerpos para esta tinción se han optimizado a escala 1: 5.000 y 1: 500 para Iba1 y Pan-neuronal, respectivamente. Se han encontrado Estas concentraciones para mostrar tinción significativa con una ausencia de tinción de fondo.
- Diluir la solución de bloques deSuero 1% en PBS y añadir anticuerpos primarios. Pipetear 300 l de solución de anticuerpo primario en el 1% de suero por diapositiva. Una vez más, asegúrese de que el fluido es el borde de la pluma pap. Incubar durante la noche a 4 ° C.
- Incluya tres portaobjetos de control: uno que contiene anticuerpos ni Iba1 ni Pan-neuronales, una con Iba1 sin anticuerpo pan-neuronal, y uno con el anticuerpo pan-neuronal sin Iba1. Tinción de estas diapositivas en la misma carrera con las mismas soluciones, sin embargo omitir los anticuerpos primarios para probar la unión no específica de los anticuerpos secundarios.
- A la mañana siguiente, se desliza de lavado tres veces en PBS durante 5 minutos cada uno, el cambio de solución entre lavados.
- Anticuerpos fluorescentes son sensibles a la luz, por lo tanto, a partir de este paso adelante, reducir al mínimo la exposición de luz, garantizando contenedores de lavado se envuelven en papel de aluminio y cajas de hibridación son negro o se incubaron en la oscuridad. Pipetear los anticuerpos secundarios apropiados en todas las diapositivas y se incuba durante60 minutos a temperatura ambiente a una concentración de 1: 250 en solución de bloqueo (vea el paso 2.2.3) en una "cámara de humedad" hermética a la luz (véase el paso 2.2.1).
- Utilice anticuerpos secundarios de diferentes longitudes de onda. Aquí, para el anticuerpo primario de conejo anti-Iba1, utilice burro anti-conejo 594 como el anticuerpo secundario apropiado. Para el ratón anticuerpo primario anti-Pan-neuronal, utilice burro anti-ratón 488 como el anticuerpo secundario apropiado. Alternativamente, use anti-conejo 488 y anti-ratón 594.
- Lavar los portaobjetos tres veces en PBS durante 5 min cada uno.
- Opcional: realizar la tinción nuclear.
- Place en Hoechst (u otra mancha nuclear) a una concentración de 0,03 g / ml en doble H 2 0 destilada durante exactamente 60 segundos.
- Lavar los portaobjetos tres veces en PBS durante 5 min cada uno.
- Lave en ddH 2 0.
- Cubrición
- Diapositivas el cubreobjetos con un medio de montaje acuoso, tal como Fluoromount-G o ProlongGold. Tenga cuidado para eliminar todas las burbujas utilizando un aplicador con punta de algodón.
Nota: Otros agentes de montaje podrían utilizarse, sin embargo alta derrame entre tintes ha sido señalado por algunos pocos días de cubreobjetos. - Utilice esmalte de uñas transparente para sellar los bordes, evitando que las secciones se seque debido a la evaporación. Deje que se seque el esmalte de uñas en un recipiente hermético a la luz, mientras que las diapositivas permanecen planas y a temperatura ambiente, y luego almacenar en un recipiente hermético a la luz envuelto en papel de aluminio a 4 ° C.
- Diapositivas el cubreobjetos con un medio de montaje acuoso, tal como Fluoromount-G o ProlongGold. Tenga cuidado para eliminar todas las burbujas utilizando un aplicador con punta de algodón.
3. Proyección de imagen del Tejido manchado
- Microscopía
- Deje que el esmalte de uñas se seque durante al menos una hora antes de comenzar la microscopía, que debería tener lugar en una habitación oscura.
- Adquirir microfotografías utilizando un microscopio confocal o de investigación con una fuente de luz fluorescente y un accesorio de cámara digital. Utilizando el software que lo acompaña, ajustar la exposición para cada longitud de onda - 405, 488, y 594 - separado. Nota: en profundidad instrucciones de imágenes deben estar disponibles en línea desde el fabricante microscopio.
- Adquirir microfotografías en cada canal sin mover las secciones o ajustar el enfoque. Tome imágenes en color o en escala de grises, alternativamente y convertir a color después.
Nota: Las imágenes en escala de grises de cada canal de color o pueden ser cotejados en el post-procesamiento. - Asegúrese de que los cortes de tejido no se exponen a la luz ambiental o la luz microscópica durante largos períodos de tiempo, como foto-blanqueo de las secciones se producirá. Para evitar esto, aumentar el tiempo de exposición en lugar de intensidad de la luz / laser en aumento.
- No apague la fuente de luz fluorescente en los 30 minutos de estar encendido.
Nota: Cambio de la fuente de encendido y apagado en rápida sucesión puede disminuir la vida útil de la bombilla fluorescente.
Resultados
Este protocolo de tinción resultados en secciones de tejido de cerebro de rata que han microglia marcados con fluorescencia en el canal 594 (rojo) y neuronas marcadas en el canal 488 (verde; véase la Figura 4). Si una mancha nuclear se ha hecho, se mostrará en el canal 405 (azul). Las imágenes pueden ser tomadas en diferentes canales y superpuestos para la comparación directa de los tres canales, o entre dos canales. Muchos paquetes de software de adquisición digitales incluyen esta funcionalidad....
Discusión
El objetivo general de esta comunicación fue la introducción de procedimientos de inmunohistoquímica para el lector. Para ello, el ejemplo de doble etiquetado con Iba1 y antígenos Pan-neuronales para observar microglia y neuronas en paraformaldehído perfundido, se utilizó sacarosa cerebro cryoprotected, rata cryosectioned.
Esta técnica se puede adaptar para servir a los propósitos sin fin. Una serie de diferentes antígenos en una variedad de tipos de tejidos tales como, pero sin lim...
Divulgaciones
The authors have nothing to disclose.
Agradecimientos
The authors would like to thank Mr. Ryan Hart and Mr. Arriz Lucas for their invaluable feedback on this communication. This work was supported by NIH NINDS R01 NS065052 and Phoenix Children’s Hospital Mission Support Funds.
Materiales
| Name | Company | Catalog Number | Comments |
| Fisherbrand Superfrost Plus Glass Slides | Fisher Scientific | 22-034-979 | Used for tissue mounting (1.2.2) |
| Oven | Thermo Scientific | 51028112 | Used for tissue drying (2.1.1) |
| Mini Pap pen | Life Technologies | 00-8877 | Used in step 2.2.2 |
| Andwin Scientific Tissue-tek Slide Staining Dish | Fisher Scientific | 22-149-429 | Used for all washes during staining (2.2), as well as the Hoechst step (2.2.8) |
| Kimwipes | Fisher Scientific | 06-666-A | Used for drying slides (2.2) |
| Black Staining Box | Ted Pella | 21050 | Used for blocking and staining steps (2.2) |
| Normal Donkey Serum | Fisher Scientific | 50-413-253 | Used for block and antibody incubation (2.2) |
| Mouse α-Pan-neuronal | Millipore | MAB2300 | Used for primary antibody (2.2.4) |
| Rabbit α-Iba1 | Wako Chemical | 019-19741 | Used for primary antibody (2.2.4) |
| Donkey α-rabbit 594 | Jackson ImmunoResearch | 711-585-152 | Used for secondary antibody (2.2.6) |
| Donkey α-mouse 488 | Jackson ImmunoResearch | 715-545-150 | Used for secondary antibody (2.2.6) |
| Caterer's foil | Any | N/A | Used in steps 1.2.2 and 2.3.2 |
| Fluoromount-G | Southern Biotech | 0100-01 | Used for coverslipping (2.2.8) |
| Coverslips | Fisher Scientific | 12544E | Used for coverslipping (2.2.8) |
| Clear Nail Polish | Any | N/A | Used for coverslipping (2.2.8) |
| Axio Observer.Z1 and LSM 710 (laser scanning, confocal) | Carl Zeiss | N/A | Used for imaging (3) |
| Axioskop A2 | Carl Zeiss | N/A | Used for imaging (3) |
| CitriSolv | FisherScientific | For DAB protocol | |
| ABC | Vector Laboratories | PK-6100 | For DAB protocol |
| DAB Peroxidase kit | Vector Laboratories | SK-4100 | For DAB protocol |
| Biotinylated horse α-rabbit IgG | Vector Laboratories | BA-1100 | For DAB protocol |
| Biotinylated horse α-mouse IgG | Vector Laboratories | BA-2001 | For DAB protocol |
| 30% Hydrogen Peroxide | FisherScientific | H325-500 | For DAB protocol |
| Wheaton slide racks and staining dishes | TedPella | 21043 | For DAB protocol |
| Masterflex perfusion pump and tubing | Cole-Parmer | Used for perfusion (1.1.1 and 1.1.2) | |
| Andwin scientific tissue-tek CRYO-OCT compound (case of 12) | Fisher Scientific | 14-373-65 | Used for tissue freezing (1.2.1) |
| Thermometer (-50 to 50 C) | Fisher Scientific | 15-059-228 | Used for tissue freezing (1.2.1) |
| Cryostat | Leica | CM3500S | Used for tissue sectioning (1.2.2) |
| Staining Dish, Plastic with 2 Lids | Grale Scientific | 353 | For antigen retrival |
| 20 Place Staining Rack, Slides Horizontal | Grale Scientific | 354 | For antigen retrival |
| Microwave | Any | N/A | For antigen retrival |
Referencias
- Marrack, J. R. Chemistry of antigens and antibodies. Nature. 134, 468-469 (1934).
- Coons, A. H., Creech, H. J., Jones, R. N., Berliner, E. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 45, 159-170 (1942).
- Marshall, J. M. Localization of adrenocorticotropic hormone by histochemical and immunochemical methods. The Journal of experimental medicine. 94, 21-30 (1951).
- Mellors, R. C. Histochemical demonstration of the in vivo localization of antibodies: antigenic components of the kidney and the pathogenesis of glomerulonephritis. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 3, 284-289 (1955).
- Nakane, P. K., Pierce, G. B. Enzyme-labeled antibodies: preparation and application for the localization of antigens. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 14, 929-931 (1966).
- Avrameas, S., Uriel, J. Method of antigen and antibody labelling with enzymes and its immunodiffusion application. Comptes rendus hebdomadaires des seances de l'Academie des sciences. Serie D: Sciences naturelles. 262, 2543-2545 (1966).
- Cuello, A. C. . Immunohistochemistry. , (1983).
- Junqueira, L. C. U., Mescher, A. L. . Junqueira's basic histology : text and atlas. , (2013).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of visualized experiments : JoVE. , (2012).
- Christensen, N. K., Winther, L., Kumar, G. L., Rudbeck, L. . Education Guide: Immunohistochemical (IHC) staining methods. , 103-108 (2009).
- Wang, G., Achim, C. L., Hamilton, R. L., Wiley, C. A., Soontornniyomkij, V. Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy). Methods. 18, 459-464 (1999).
- Feldengut, S., Del Tredici, K., Braak, H. Paraffin sections of 70-100 mum: a novel technique and its benefits for studying the nervous system. Journal of neuroscience methods. 215, 241-244 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados