
Method Article
Plásmido derivado de ADN Strand Displacement Puertas de Redes de reacción de Ejecución Química
En este artículo
Resumen
This protocol describes a method for deriving DNA strand displacement gates from plasmids and testing them using fluorescence kinetics measurements. Gates can be modularly composed into multi-component systems to approximate the behavior of formal chemical reaction networks (CRN), demonstrating a new use for CRNs as a molecular programming language.
Resumen
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
Introducción
La previsibilidad de Watson-Crick ha permitido a la nanotecnología de ADN dinámico para emerger como una forma programable para diseñar dispositivos moleculares con propiedades dinámicas 1,2. En particular, cadena de ADN desplazamiento - una reacción de hibridación competitiva programable - ha demostrado ser un poderoso mecanismo para la ingeniería de sistemas dinámicos de ADN. En una reacción de desplazamiento de cadena de ADN, un oligonucleótido entrante desplaza una "salida" hebra previamente atado de una pareja de unión complementaria. Múltiples tales reacciones pueden ser encadenados juntos en cascadas de reacción de varios pasos con un alto grado de control sobre el orden y el tiempo de reacción individual 3 pasos. ADN cascadas de desplazamiento de cadena se han utilizado para crear circuitos digitales y analógicas moleculares 4-7, 8-10 nanoestructuras conmutables, motores moleculares autónomas 11-15, y amplificadores catalíticos no covalentes 13,16-21. Por otra parte, DDispositivos NA utilizando reacciones de desplazamiento de hebra se pueden simular y diseñados para diversas aplicaciones que usan el ordenador con ayuda de software de diseño 22-24.
Actualmente, el ADN sintetizado químicamente sirve como material principal para la nanotecnología de ADN. Sin embargo, los errores en el proceso de la síntesis de ADN, y los oligonucleótidos resultantes imperfectos, se cree que limitar el rendimiento de los dispositivos dinámicos de ADN al causar reacciones secundarias erróneos. Por ejemplo, las reacciones de "fugas" pueden resultar en la liberación de un oligonucleótido de salida incluso en la ausencia de un disparador de reacción. Estos efectos son más evidentes en las cascadas de reacción autocatalíticos donde incluso una cantidad mínima de fuga inicial con el tiempo como resultado la activación completa del 19,20 cascada. Por el contrario, las reacciones a menudo fallan en alcanzar el nivel esperado de activación debido a que algunos componentes no activan incluso en la presencia de la entrada prevista 7,25. Para hacer que el rendimiento de ADN a base denanodispositivos comparables a sus homólogos basados en proteínas biológicas, tales modos de error deben reducirse dramáticamente.
Plásmidos bacterianos u otro ADN biológica podrían servir como una fuente relativamente barata de ADN de alta pureza para aplicaciones de la nanotecnología. Grandes cantidades de ADN se pueden generar por la replicación en bacterias y las capacidades de corrección de pruebas intrínsecas de los sistemas vivos asegurar la pureza del ADN resultante. De hecho, varios estudios recientes han reconocido la utilidad potencial de ADN biológico para aplicaciones de la nanotecnología 21,26-28. Sin embargo, la naturaleza completamente de doble hebra de ADN del plásmido hasta ahora ha prohibido su uso como material para la fabricación de dispositivos dinámicos de ADN, que normalmente consisten en múltiples oligonucleótidos y contienen ambos dominios de doble cadena y de cadena sencilla. En un trabajo reciente 29 de este tema fue abordado y una nueva arquitectura de puerta de ADN que se compone principalmente de ADN de doble cadena mellado (ndsDNA) fue introducirre.
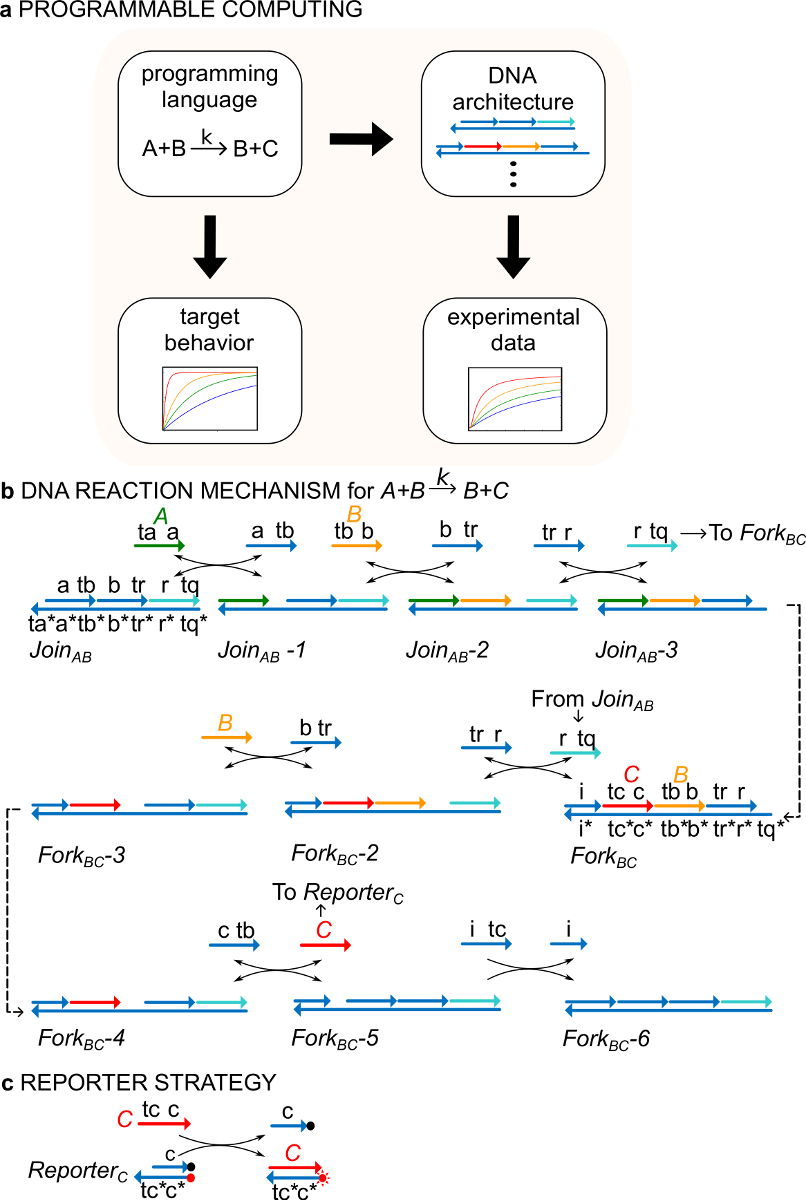
Es importante destacar que los sistemas de puertas ndsDNA pueden ser diseñados que dan cuenta de la dinámica especificados por cualquier red reacción química formales (CRN) 29. puertas ndsDNA tanto, se podrían utilizar, en principio, para crear sistemas dinámicos que presentan oscilaciones y caos, biestabilidad y la memoria, la lógica de Boole o comportamientos algorítmicos 30-38. Por ejemplo, Ref. 29 demostraron una CRN de tres reacción que proporciona una implementación molecular de un protocolo de "consenso", un tipo de algoritmo de computación distribuida 29,39,40. Este trabajo demuestra por primera vez un nuevo uso para el formalismo CRN como un "lenguaje de programación" para sintetizar rápidamente los sistemas moleculares funcionales (Figura 1).
Aquí, se proporciona un protocolo detallado para derivar puertas ndsDNA de ADN plásmido. Primero es una revisión del proceso de diseño de la secuencia. Luego sigue una explicación de cómo oligonucleótidos sintéticos que contienenlas secuencias de compuerta se clonaron en plásmidos y la secuencia verificados y amplificados a través de cultivo bacteriano. A continuación, se muestra cómo ndsDNA puertas se pueden derivar de los plásmidos mediante procesamiento enzimático (véase la Figura 2). Finalmente, un método para probar el comportamiento puerta utilizando ensayos de cinética de fluorescencia se describe.
Mecanismo de reacción
Como ejemplo, el protocolo se centra en el catalítica reacción química A + B-> B + C. La especie A, B, y C ("señales", Figura 1B) todos corresponden a una molécula de ADN de una sola hebra diferente. Las secuencias de estas moléculas son completamente independientes y las hebras no reaccionan entre sí directamente. Las secuencias de todas las señales tienen dos dominios funcionales diferentes, es decir, subsecuencias que actúan juntos en reacciones de desplazamiento de hebra: 1) un dominio de punto de apoyo corto (etiquetas ta, tb, tc) que se utiliza para la iniciación de desplazamiento de cadena reaction, y 2) un dominio de largo (etiquetas a, b, c) que determina la identidad de la señal.
Las interacciones entre los hilos de señal están mediadas por ADN (ndsDNA) complejos de puerta de doble cadena con muescas (llamados Únete AB y Tenedor aC) y las especies de cadena sencilla auxiliares (, , y ). La reacción A + B- formales> B + C se ejecuta a través de una serie de etapas de reacción de desplazamiento de cadena, donde cada paso de reacción expone un punto de apoyo para una reacción posterior (Figura 1B). En este ejemplo, las señales A y B son inicialmente libre en solución, mientras que la señal C está unido a la puerta de tenedor. Al final de la reacción B y C están en solución. De manera más general, las señales que están obligados a una puerta están inactivas mientras que las señales que están libres en solución son activos, es decir, que pueden participar en una reacción de desplazamiento de cadena comouna entrada. El curso temporal de la reacción se sigue utilizando una estrategia de indicador fluorescente (Figura 1C). En un trabajo anterior 29, se demostró que este mecanismo de reacción no sólo da cuenta de la estequiometría correcta, sino también la cinética de la reacción objetivo.
Protocolo
1. Secuencia de Diseño
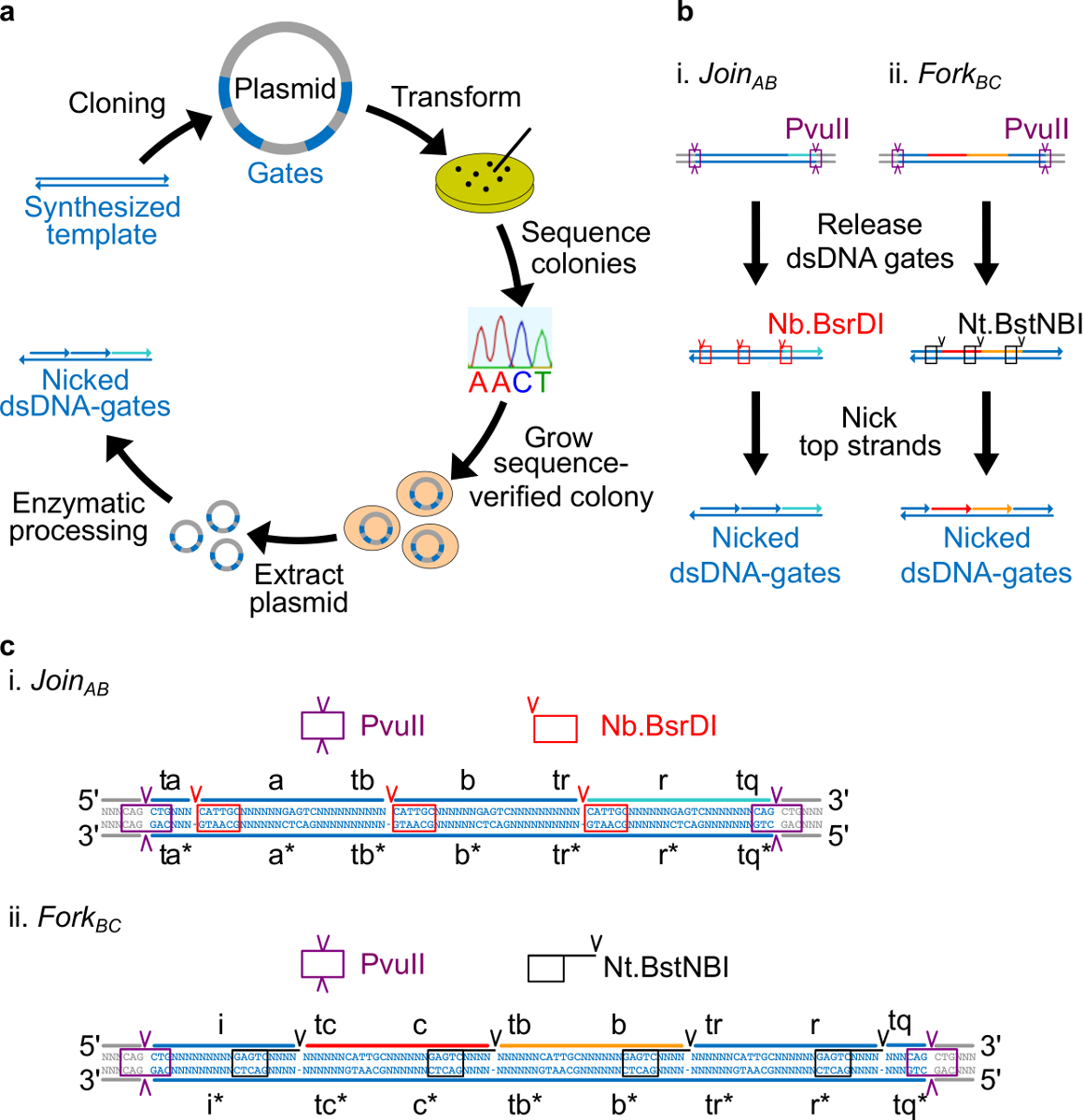
Nota: visión general de diseño de secuencia: En esta sección, la estrategia para el diseño de las puertas de ADN plásmido derivado se describe. Sitios de enzimas colocados en cada extremo de las puertas para permitir la liberación de puertas completamente de doble hebra después de la digestión. Sitios mellar se colocan entonces de tal manera que las enzimas crean mellas en la cadena superior para crear las puertas ndsDNA finales. Por último, las secuencias restantes se eligen de manera que los dominios independientes son ortogonales entre sí y no presentan estructura secundaria.
- Coloque el sitio nicking Nt.BstNBI cuatro nucleótidos lejos del extremo 3 'de cada dominio de largo (a, b, c, r, e i). Coloque el sitio nicking Nb.BsrDI en el extremo 5 'de cada dominio de largo (a, b, c, y r. Tenga en cuenta que el dominio i no tiene ningún sitio de mellar Nb.BsrDI). Figura 2C muestra la vista secuencia detallada de Únete a AB y BC Tenedor puertas.
- Coloque el sitio de restricción PvuII en ambos extremos de puertas ndsDNA de modo que la digestión PvuII puede liberar las puertas a partir de plásmidos (véase la Figura 2C).
- Diseño otras secuencias no restringidos siguiendo dos principios: (a) hebras no deben exhibir estructuras secundarias (estructuras de ADN pueden predecirse usando Nupack 41), y (b) todos los dominios deben ser ortogonales para minimizar la diafonía.
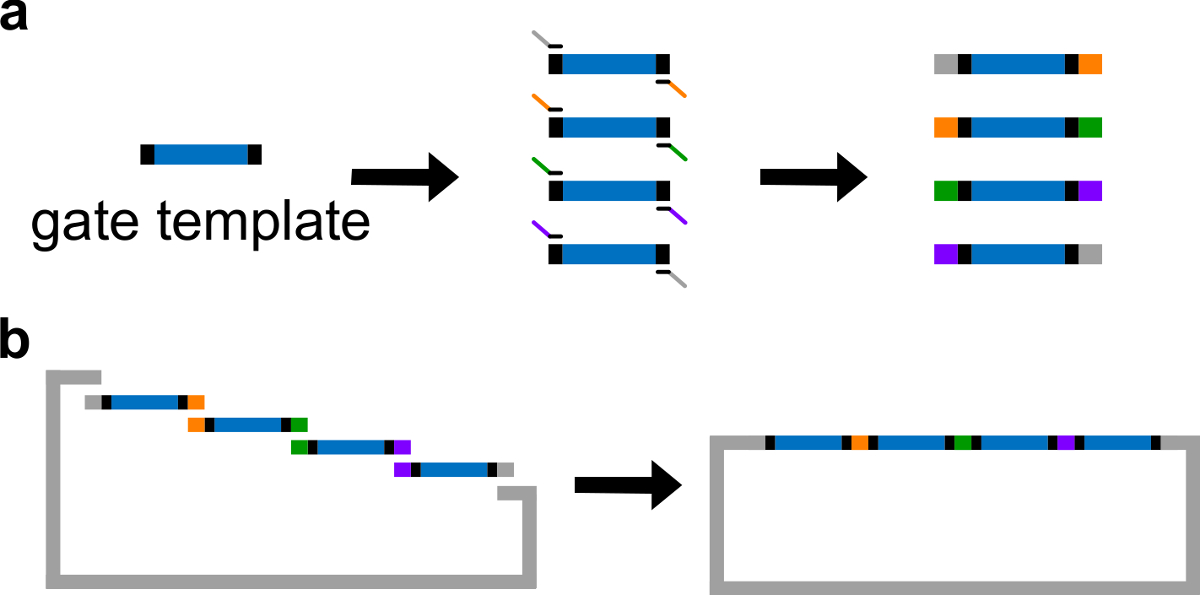
- Coloque secuencias ndsDNA en el centro de una plantilla de puerta. Coloca 30-40 secuencias espaciadoras pb al azar en ambos extremos de la plantilla de puerta, cada espaciador sirve como un sitio de unión única para la siguiente reacción en cadena de polimerasa (PCR).
2. Clonación de NdsDNA puertas en plásmidos
Nota: En esta sección se describe el método de clonación Gibson para la inserción de 4 copias de la puerta en un esqueleto del plásmido.
- Plantillas de puerta Solicitar ndsDNA como bloques genómicos de doble cadena de un fabricante de ADN (secuencias modelo de puerta se muestranen la Tabla 1; hebras ocurren en puertas ndsDNA se muestran en la Tabla 2; secuencias nivel de dominio se muestran en la Tabla 3).
- Después de recibir el ADN ordenada, girar los tubos que contienen bloques genómicos en 10,000-14,000 xg durante 1 min para asegurar que todo el ADN seco es en la parte inferior del tubo.
- Resuspender los bloques genómicos secos en agua libre de DNasa para lograr una concentración final de 10 ng / l.
Nota: Como alternativa, el ADN puede ser resuspendió utilizando ácido etilendiaminotetraacético (EDTA) 1 x tampón Tris (tampón TE: Tris 10 mM y EDTA 1 mM, pH 8,0). Sin embargo, EDTA es un agente quelante para cationes divalentes y podría inhibir la PCR. - Generar 4 fragmentos de puerta con diferentes regiones de solapamiento a través de una PCR estándar con una ADN polimerasa de alta fidelidad (ver Figura 3A). Las secuencias de los cebadores se detallan en la Tabla 4 (temperatura de fusión de estos cebadores es 62 ° C).
- Ejecutar un gel de agarosa al 2% a 140 V durante 30 minutos a temperatura ambiente (por un protocolo de electroforesis en gel de agarosa detallada ver 42) y cortar las bandas correspondientes a cada fragmento amplificado por PCR del gel. Entonces purificar los cortes de gel utilizando un kit de extracción de gel (consulte los Materiales) siguiendo las instrucciones del fabricante.
- Digerir un alto número de copias del plásmido columna vertebral (ver materiales) con PvuII-HF y PstI-HF a 37 ° C durante 1 hora (ver Tabla 5) de acuerdo con el protocolo del fabricante. PvuII-HF y PstI-HF son enzimas de restricción de alta fidelidad, que reducen drásticamente los recortes inespecíficos.
- Ejecutar un gel de agarosa al 1,5% y cortar la columna vertebral linealizado (típicamente correr el gel a 140 V durante 30-40 minutos a temperatura ambiente). A continuación, extraer el ADN de la porción de gel usando el kit de extracción de gel siguiendo las instrucciones del fabricante.
- Realizar el montaje Gibson 43 con el vector linealizado y fragmentos de PCR purificados (ver Tabla 6 y la Figura 3B ) a 50 ° C durante 1 hr.
- Transformar el producto de montaje Gibson desde el paso 2.8 en Escherichia coli (E. coli) y la placa sobre una placa de agar Lisogenia Broth (LB) que contiene los antibióticos ampicilina (a una concentración de 100 mg / ml). Realizar la transformación a través de electroporación o un método de choque térmico 44,45, y el uso de la E. apropiada coli cepa. Por ejemplo, utilice E. coli cepa JM109 para la transformación de choque térmico, y el uso de DH5a electrocompetentes E. células de E. coli para la electroporación.
Nota: El esqueleto del plásmido utilizado contiene un casete de resistencia a ampicilina. Si se utiliza un marcador de selección diferente, utilice los antibióticos apropiados en lugar de ampicilina.
3. bacteriana Cultura Amplificación y Control de Calidad
Nota: Esta sección describe la producción en masa y el aislamiento de plásmidos que contienen las puertas de ADN después de control de calidad.
- Elija una sola coloniade la placa selectiva Ampicilina desde el paso 2.9 y se incuba un cultivo de 3 ml de medio enriquecido que contiene los antibióticos ampicilina (a una concentración de 100 mg / ml). Marque la colonia tal que puede ser utilizado de nuevo en etapas experimentales posteriores. Mantener el cultivo a 37 ° CO / N con agitación vigorosa (200-300 rpm). Por lo general, se incuba durante 16 a 24 horas.
- Se extrae el ADN plasmídico a partir del cultivo bacteriano usando un kit Mini-prep siguiendo las instrucciones del fabricante.
- Mida el ADN de plásmido purificado usando un espectrofotómetro siguiendo las instrucciones del fabricante. Niveles de rendimiento típicos de 50-1,000 ng / l.
- Obtener el ADN plasmídico extraído secuenciado mediante el envío de la muestra a una empresa de secuenciación de ADN. Iniciadores de secuenciación deben estar situados aproximadamente 100 nucleótidos aguas arriba y aguas abajo de la región para ser secuenciados; el cebador de secuenciación para el plásmido (ver Materiales para el plásmido) tiene la siguiente secuencia: ATTACCGCCTTTGAGTGAGC.
Note: Si hay error de secuencia o recombinación en el insertada ndsDNA puertas, seleccione una colonia diferente de la placa desde el paso 2.9. Siga los pasos 3.1 a 3.4 para verificar que las secuencias de las puertas insertadas son correctos. - Después de verificar que las secuencias son correctos, elegir la colonia correspondiente de la placa selectiva Ampicilina (del paso 2.9), y se incuba una cultura de 800 ml Caldo Terrific (TB) que contiene antibióticos ampicilina (a una concentración de 100 mg / ml). Mantener el cultivo a 37 ° C durante 16-24 horas con agitación vigorosa (200-300 rpm). TB en particular es muy adecuado para la producción de plásmido de alto rendimiento.
Nota: Como alternativa, LB también podría ser utilizado para crecer bacterias aunque el rendimiento de plásmido puede ser un problema. - Se purifica el ADN utilizando un kit Maxi-prep siguiendo las instrucciones del fabricante.
- Siga el paso 3,3-3,4 para comprobar si las secuencias son correctos. Si se ha producido ninguna recombinación, consulte la siguiente nota. De lo contrario, vaya al paso 4.
Nota: Un posible problema aquí es que varias copias de puertas insertadas en el plásmido pueden recombinarse, debido a la reparación del ADN. Para abordar este problema, utilice un E. cepa de E. coli que carecen de la proteína recA (una proteína relacionada con la reparación del ADN), tales como JM109 o DH5 para transformar un plásmido previamente secuencia verificada (es decir, sin ningún error de secuencia y la recombinación). Después elige una colonia de esta placa y verificar la secuencia del plásmido mediante el envío de la muestra a una empresa de secuenciación de ADN.
4. enzimática Procesamiento
Nota: En esta sección se describe el proceso de digestión de los plásmidos de tal manera que se cortan y melladas en los lugares correctos y listos para ser utilizados para experimentos de cinética.
- Digerir el plásmido de ADN purificado de la etapa 3.7 con enzima de restricción PvuII-HF durante 1 hora a 37 ° C (ver Tabla 7). Típicamente digerir el plásmido con 4 unidades de PvuII-HF por 1 mg de plásmido. Alta fidenzimas de restricción elity se recomiendan para su uso, ya que reducen drásticamente los recortes inespecíficos.
- Efectuar precipitación con etanol en la muestra.
- Agregue 2 volúmenes equivalentes de etanol absoluto enfriado con hielo a la muestra.
- Incubar la mezcla a -80 ° C durante al menos 1 hora (esta mezcla también se puede sentar a -80 ° C para O / N).
- Centrifugar a 10,000-14,000 xg a 0 ° C durante 30 min.
- Eliminar el sobrenadante.
- Añadir 1.000 l de RT 95% de etanol a la muestra, y se invierte 10-15 veces.
- Centrifugar a 10,000-14,000 xga 4 ° C durante 10 min.
- Eliminar el sobrenadante y el aire seco en el banco durante 10-20 min.
- Resuspender los sedimentos de ADN en un volumen apropiado de nucleasa libre de H 2 O (típicamente de 100-200 l). La adición de más de 200 l por lo general hacer la muestra demasiado diluida para su uso en experimentos cinéticos.
- Mida el DNA se resuspendió utilizando un espectrofotómetro después de la mlas instrucciones del anufacturer.
- Digesto unirse puertas con mellar enzima Nb.BsrDI a 65 ° C durante 1 hora con 4 unidades de enzima por 1 g de plásmido (ver Tabla 8); digerir horquilla puertas con mellar Nt.BstNBI enzima a 55 ° C durante 1 hora usando 8 unidades de enzima por 1 g de plásmido (véase la Tabla 9).
Nota: El paso 4.2 elimina tampón de digestión de enzima y ayuda a concentrar las puertas para experimentos cinéticos. Paso 4.2 se puede omitir para unirse a puertas debido a que tanto la enzima de restricción PvuII-HF y la enzima mellar Nb.BsrDI comparten el mismo tampón de digestión. En el paso 4.2.8, nucleasa libre de H 2 O se utiliza en lugar de TE porque EDTA es un agente quelante para cationes divalentes y puede inhibir las enzimas de restricción que necesitan estos iones para funcionar.
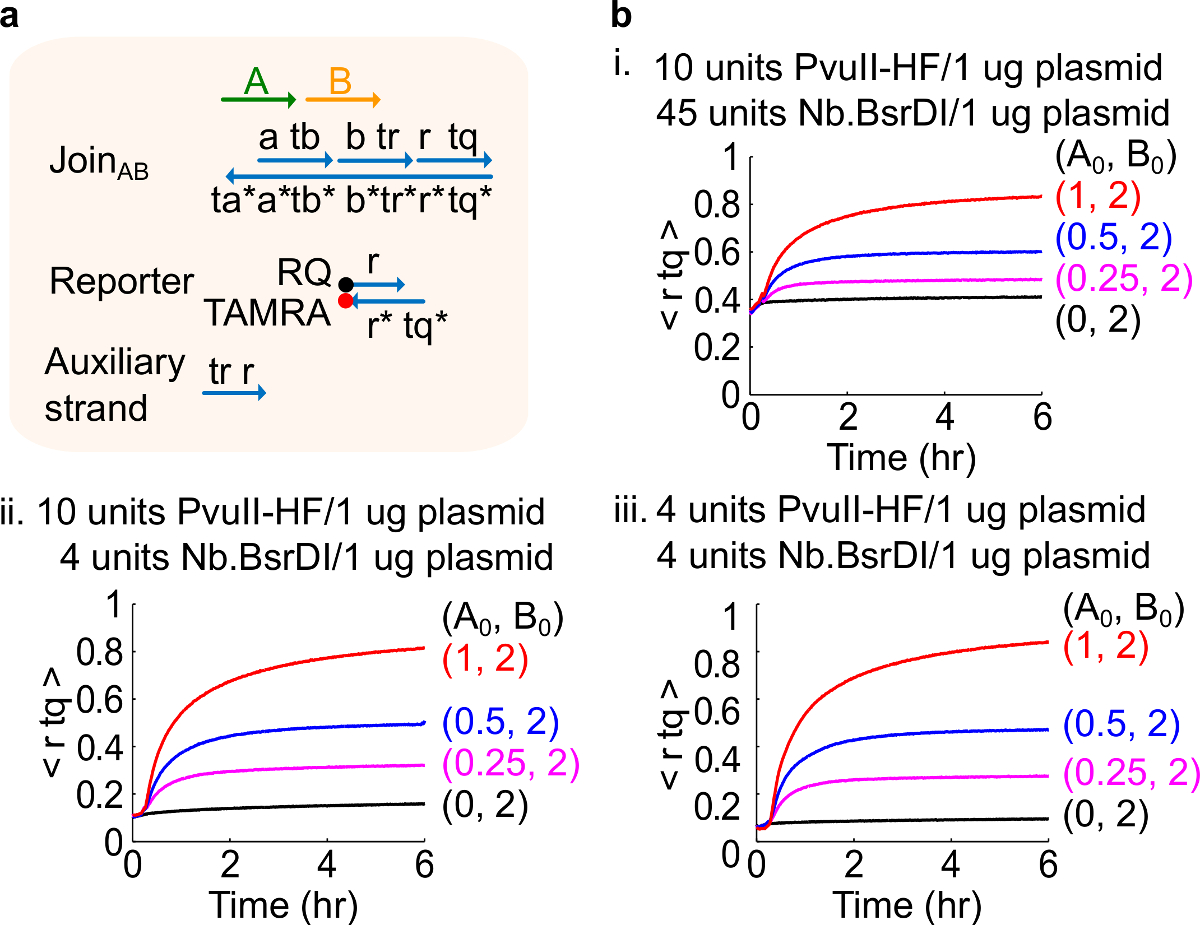
Nota: La adición de cantidades excesivas de enzimas puede conducir a altas cantidades de fugas del circuito inicial (Figura 4), que es muy probablemente causado por un exceso de digestión 46. Este problema can abordarse mediante la optimización de las cantidades de enzimas (véase la Figura 4). Intervalo típico de enzimas es de 1-10 unidades / 1 g de plásmido.
5. Preparación de oligonucleótidos de cadena simple
Nota: Esta sección describe el protocolo para resuspender y cuantificar el ADN de una sola cadena sintetizado químicamente (ssDNA) que se utilizará para hilos de señal y hebras auxiliares. Para hebra secuencias véase la Tabla 10. Tenga en cuenta que el protocolo siguiente es un ejemplo de preparación 10 M ssDNA. Otras concentraciones de ssDNA se pueden preparar de manera similar.
- Después de recibir oligos de ADN del fabricante, girar los tubos que contenían ADN en 10,000-14,000 xg durante 1 min para asegurar que todo el ADN seco es en la parte inferior del tubo.
- Resuspender el ADN usando 1x Tris ácido etilendiaminotetraacético (EDTA) tampón (tampón TE: Tris 10 mM y EDTA 1 mM, pH 8,0) para alcanzar una concentración final de 100 mM. porejemplo, resuspender 8 nmol de ADN en 80 l de tampón TE.
- Mezclar 10 l de la DNA a 100 m con 90 l de agua molecular en un tubo de microcentrífuga, que deben alcanzar una concentración final de 10 mM.
- Medir la concentración exacta de la muestra de ADN usando un espectrofotómetro siguiendo las instrucciones del fabricante. El siguiente protocolo da un ejemplo de cómo se puede medir la concentración de ADN.
- En blanco el espectrofotómetro con 2 l de agua molecular.
- Medir la absorbancia a 260 nm (A 260) de la muestra de ADN. Utilice la siguiente ecuación para calcular la concentración de valores.
Nota: La concentración de la muestra es M = Un coeficiente de 260 / extinción. El coeficiente de extinción se puede encontrar en la hoja de datos de especificación por el fabricante de ADN.
6. Preparación de Reporteros fluorescentes
Nota: En esta sección se describe elProtocolo para la preparación de Reportero C, Otros reporteros fluorescentes se puede montar de manera similar.
- Ordene cromatografía líquida de alta resolución (HPLC) oligonucleótidos purificados ROX- (la cadena superior del Reportero C) y -RQ (la cadena inferior de Reportero C) del fabricante de ADN (ver Tabla 10 para las secuencias ).
- Después de recibir el sintetizado oligonucleótidos, resuspender y cuantificar las muestras como se explica en el paso 5.
- Mezclar la parte superior reportero y hebras inferiores (es decir, ROX- y -RQ) en 1x Tris-acetato-EDTA (TAE) con 12,5 mM Mg 2 + (véase la Tabla 11 para la receta detallada ). Tenga en cuenta que aquí se añade el 30% de exceso de cadena extintor etiquetada -RQ para montar el reportero, que asegura que todos los capítulos marcados con fluoróforo se apagaron incluso con estequiometría imperfecto.
- Recocer el complejo reportero C usando un ciclador térmico, enfriamiento desde 95 ° C a 20 ° C a una velocidad de 1 ° C / min. Las muestras pueden ser almacenadas a 4 ° C.
7. Las medidas de fluorescencia
Nota: La sección describe un protocolo general para la cinética de fluorescencia mediciones (vea la Figura 5 para el procedimiento experimental), y este protocolo se utilizará en los pasos 8, 9 y 10. También tenga en cuenta que este protocolo es para el uso de un espectrofluorímetro. Alternativamente, estos experimentos también se podrían realizar en un lector de placas a pesar de las variaciones de sensibilidad, bien-a bien y la falta de control de la temperatura en los experimentos a largo plazo pueden ser un problema.
- Ajuste el regulador de temperatura a 25 ° C, y esperar a que la temperatura se estabilice. El uso de un controlador de temperatura puede reducir la variabilidad en la señal que puede resultar de la variación de temperatura.
- Establecer parámetros adecuados FOr cinética de las mediciones en el software de adquisición de datos de la espectrofluorímetro. Ajustes detallados ejemplo son los siguientes:
- Ajuste el ancho de la ranura de 2,73 nm para ambas monocromadores de excitación y emisión.
- Ajuste el tiempo de integración de 10 segundos por cada punto de tiempo de 60 seg. Ajuste el tiempo total de medición a 24 horas.
- Establecer las longitudes de onda de excitación / emisión para que coincida con los fluoróforos utilizados en el experimento. Ejemplo longitudes de onda son las siguientes: ROX (588 nm / 608 nm), y TAMRA (559 nm / 583 nm).
- Añadir nucleasa H 2 O y libre de 10x tampón Tris-acetato-EDTA que contiene 125 mM Mg 2 + (10x TAE / Mg 2 +) a una celda de cuarzo sintético. Ver Tablas 12, 13 y 14, por ejemplo, volúmenes de usar.
- Añadir hebras poliT para lograr una concentración final de 1 M ~ (véase el cuadro 12, 13, y 14 para los volúmenes), y luego el vórtice sintéticacélulas de cuarzo de 10 a 15 seg. En general, las puntas de pipeta se unirán de forma no específica de ADN. La adición de altas concentraciones de hebras poliT puede reducir este error unión no específica.
- Añadir a la prensa y cordones auxiliares. Véase la Tabla 12, 13 y 14, por ejemplo, los volúmenes a utilizar. Tenga en cuenta que para la calibración reportero, no se necesitan cadenas auxiliares.
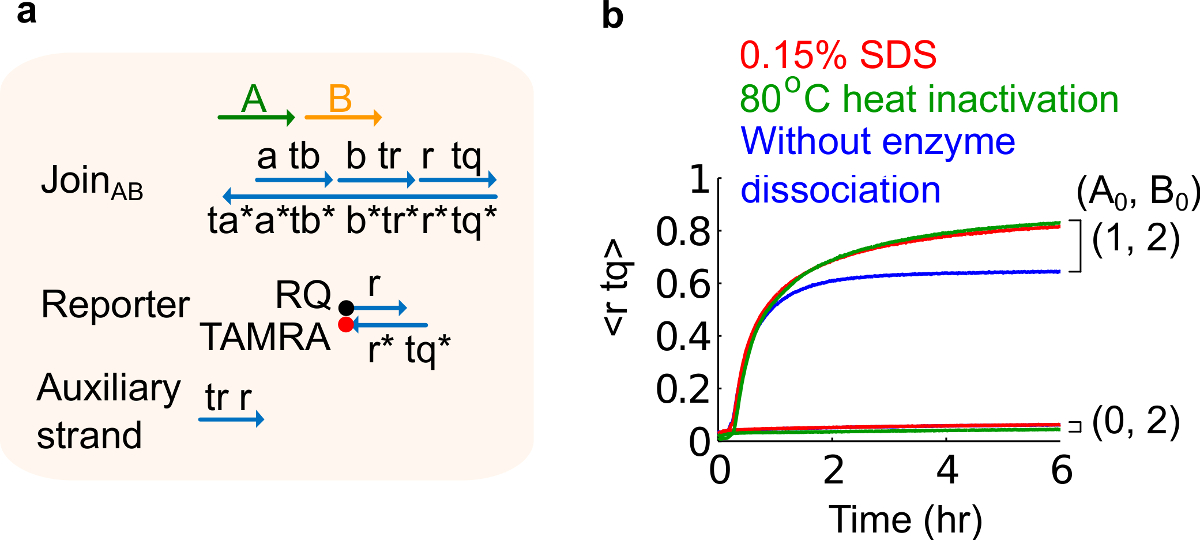
- Añadir dodecil sulfato de sodio al 10% (SDS) para lograr una concentración final de 0,15% de SDS. Nota: SDS se utiliza para disociar las enzimas de las puertas plásmido derivado porque las enzimas pueden interferir con la reacción de desplazamiento de cadena (véase la Figura 6). SDS se recomienda aquí en lugar de la desnaturalización térmica de las enzimas para evitar la disociación y la recombinación incorrecta de hebras de compuerta, que pueden afectar negativamente a la función del circuito.
- [Saltar este paso para la calibración reportero.]
- Añadir unirse y puertas de horquilla (véase la Tabla 13, y 14 para los volúmenes)a la celda de cuarzo sintético y mezclar la solución con la pipeta hacia arriba y hacia abajo durante al menos 20 veces (no vórtice la cubeta porque las soluciones de vórtice con SDS pueden dar lugar a burbujas que afectarán cinética de fluorescencia mediciones).
- Además, pasar a las siguientes etapas de medición tan pronto como sea posible porque la reacción de fugas inicia inmediatamente después de la adición de unirse y horquilla puertas a la celda de cuarzo sintético.
- Coloque las celdas de cuarzo sintético en la cámara de un espectrofluorímetro.
- Inicie la medición cinética.
- Después de 5 minutos de la medición, añadir hebras de entrada (véase la Tabla 12, 13, y 14 para los volúmenes) a la celda de cuarzo sintético y mezclar la reacción pipeteando arriba y abajo por lo menos 20 veces. Tenga en cuenta que la muestra se debe mezclar suavemente para evitar burbujas. Realice este paso mientras que el programa de adquisición de datos está en pausa para evitar la medición de señales desencadenada por externaligero.
- Registre la cinética de reacción hasta que alcance el estado estacionario. La cinética de la reacción se muestran en el ordenador.
8. Calibrar fluorescentes Reporteros
Nota: En esta sección se describe el protocolo para la toma de curvas de calibración de los reporteros fluorescentes. Las curvas de calibración se utilizan para convertir unidades arbitrarias de fluorescencia a la concentración molar de la señal.
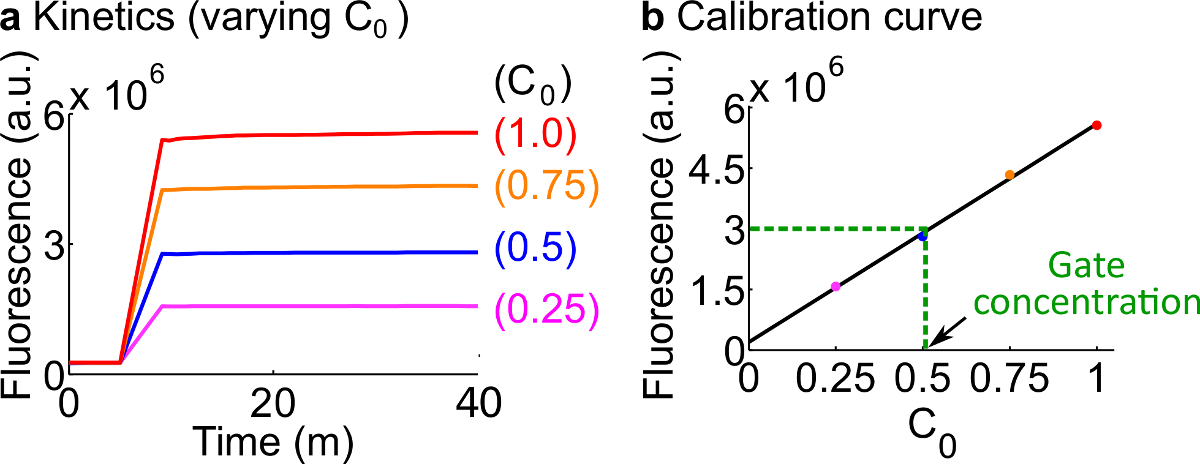
- Calibrar reporteros fluorescentes siguiendo el protocolo descrito en el Paso 7. Uso de los volúmenes de reactivos y tampones tal como se resume en la Tabla 12 La concentración estándar para este ejemplo es 50 nM (1x).; los periodistas están en 3x; entrada es 1x. Para los casos en que la entrada son en 0.25x, 0.5x, 0.75x, ajustar el volumen de nucleasa H 2 O gratis correspondientemente para mantener el volumen final de cada reacción que ser 600 l. Un dato de ejemplo se muestran en la Figura 7A.
- Hacer una curva de calibración delReportero C por un ajuste lineal de los valores de fluorescencia finales contra la concentración inicial de la señal C (Una curva de calibración se muestra ejemplo en la Figura 7B). Esta curva de calibración se puede utilizar para convertir las unidades arbitrarias de fluorescencia a su concentración señal correspondiente.
9. cuantificar la concentración de derivados de plásmido ndsDNA Puertas
Nota: Cada lote procesado independientemente de puertas ndsDNA plásmido derivado de los resultados con un rendimiento diferente de puertas funcionales, y esta sección describe un protocolo para la cuantificación de la concentración de puertas ndsDNA plásmido derivado.
- Cuantificar la concentración de puertas ndsDNA plásmido derivado siguiendo el protocolo descrito en el Paso 7. Uso de los volúmenes de reactivos como se resume en la Tabla 13 Nota:. Tabla 13 describe un ejemplo receta para Tenedor BC cuantificación Join. AB y otras puertas se pueden realizar de manera similar pero usando diferentes líneas de entrada, hilos auxiliares y periodistas.
- Convertir el valor de fluorescencia final medida en este experimento a una concentración de C de la señal usando la curva de calibración desde el paso 8.2. Luego de vuelta a calcular la concentración puerta ndsDNA. Por ejemplo, un valor de fluorescencia final para la puerta de cuantificación experimento corresponde a 25 nM de la señal C (0,5x) en base a la curva de calibración en la Figura 7B. Desde el stock de Tenedor aC se diluye 40 veces en esta reacción, la concentración de un balance de la puerta Tenedor BC es 1 M.
10. Cinética Medidas para la reacción A + B> B + C
Nota: Esta sección describe un protocolo para probar la realización de ADN de una reacción química formal de la cinética de fluorescencia usando mediciones.
- Pormedición de forma cinética siguiendo el protocolo descrito en el Paso 7. Use volúmenes de reactivos y tampones tal como se resume en la Tabla 14.
Resultados
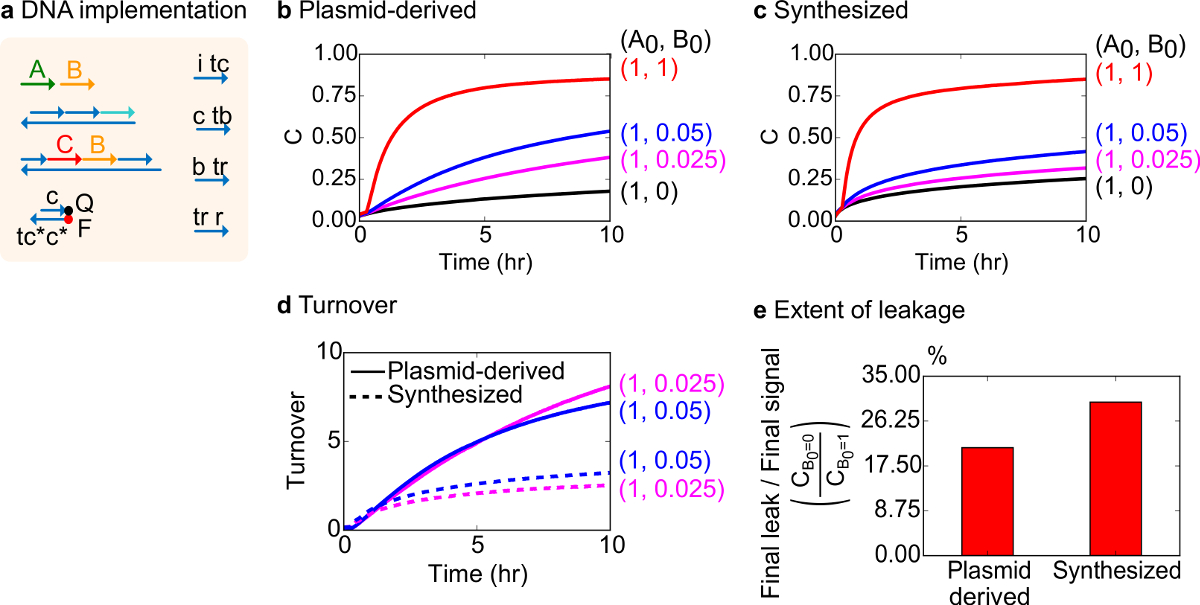
Para una prueba funcional, se ha creado una implementación de ADN de la reacción catalítica bimolecular (es decir, A + B-> B + C). El rendimiento de puertas plásmido derivado se comparó con puertas ensambladas a partir de ADN sintético. Reacciones catalíticas son una buena prueba de la pureza puerta porque una puerta defectuosa puede irreversiblemente trampa de un catalizador, provocando un efecto desproporcionado sobre la cantidad de producto producido 18,19. Al mismo tiempo, una reacción de fuga pequeña resulta en la liberación sin disparo de la señal catalítico se amplifica linealmente, lo que lleva a una señal de error desproporcionada. Los datos experimentales para las puertas de plásmido derivado y sintetizados se muestran en la Figura 8B y 8C, respectivamente. En los experimentos, la concentración de la señal de cadena A está fijo mientras se varía la cantidad de la señal catalítica B. Señal C se utiliza para leer el progreso de la reacción sin interrumpir el catalíticaciclo. Catálisis se puede observar en los datos ya que las reacciones se aproximan a la terminación incluso con cantidades de catalizador B mucho más pequeñas que la cantidad de A. Desde SDS no se agregó a experimentos realizados con el sistema sintetizado, la velocidad de reacción (que podrían verse afectados por la adición de SDS) no se compara y el enfoque analítico es en cambio en el volumen de negocios catalítica (se detalla como sigue).
Se llevó a cabo análisis más detallado de la facturación catalítica de esta reacción. Facturación se define como la cantidad de señal producida C para cada catalizador B en un momento dado. En concreto, el volumen de negocios se calcula a partir de los datos experimentales dividiendo la señal de fuga restado C por la cantidad inicial de catalizador B agregó. Para un sistema catalítico ideales, este número de recambio debería aumentar linealmente con el tiempo y ser independiente de la cantidad de catalizador siempre y cuando el sustrato no es limitante. En un sistema real, puertas defectuosas pueden desactivar gato alysts, y la facturación alcanzará un valor máximo, incluso si no todos sustrato disponible se convierte en producto. El valor máximo rotación indica cuántos sustratos (señal A) un catalizador (señal B) se pueden convertir antes de ser inactivado. Aquí, se observa que el sistema sintetizado se desvía de la incremento lineal ideal de rotación mucho antes que el sistema de plásmido derivado hace, lo que indica el secuestro del catalizador a través de una reacción colateral indeseable (Figura 8D). La comparación rotación sólo se muestra para concentraciones bajas debido a altas concentraciones de catalizadores, todas las puertas se activan y liberan señal C. La fuga de circuito también se compara, y se observa que la proporción de señal de fuga utilizando puertas de plásmidos derivados es de aproximadamente 8% menos que los que utilizan puertas sintetizados después de 10 h de reacción (Figura 8E).
iles / ftp_upload / 53087 / 53087fig1.jpg "/>
Figura 1. CRN (A) sirven como un lenguaje de programación prescriptivo. Redes de reacción de ADN pueden ser diseñados para aproximar la dinámica de un CRN formal de aplicación (B) el ADN de una instrucción química ejemplo:. A + B-> B + C. Hebras de ADN se dibujan como líneas con flechas en el extremo 3 'y * indica la complementariedad. Toda señal de hebras A ( a>, verde), B (, naranja), y C (, rojo) se consistieron en un dominio punto de apoyo (etiquetado como ta, tb y tc) y un dominio de identidad (etiquetados como a, b, y c). La reacción bimolecular A + B-> B + C requiere dos complejos multi-hebra Únete a AB y Tenedor AC, y cuatro hilos auxiliares , , y . La reacción tiene lugar a través de siete etapas de desplazamiento de la cadena, donde cada paso de inicios con la unión. (C) Estrategia Reportero punto de apoyo. La reacción se siguió usando un reportero en el que la cadena inferior está marcada con un fluoróforo (punto rojo) y la cadena superior está unido a un extintor (punto negro). Debido a la co-localización del fluoróforo y el extintor, la fluorescencia reportero se apaga en el Boletín intacto. La señal C puede sustituir a la cadena superior del reportero, lo que lleva a un aumento de la fluorescencia. (Esta cifra se ha modificado de la referencia 29). Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. puertas (A) NdsDNA elaborados a partir de ADN del plásmido bacteriano. Varias copias de la hebra plantilla puerta ndsDNA doble se clonan en un plásmido. Los plásmidos clonados son lan transformado en E. coli células y colonias en la placa son secuencia verificada. Una vez que se confirmó la secuencia, el ADN plásmido se amplifica y se extrajo. Por último, el plásmido de doble cadena se procesa en las puertas ndsDNA deseados a través de procesamiento enzimático. (B) el tratamiento enzimático de puertas ndsDNA. La enzima de restricción PvuII se utiliza para liberar la puerta desde el plásmido. Las puertas liberados se procesan adicionalmente utilizando enzimas mellar: Nb.BsrDI se utiliza para generar nicks para Únete AB (Grupo I); Nt.BstNBI se utiliza para generar nicks para Tenedor BC (Panel ii). Los sitios de restricción y mellar se indican como cajas con códigos de colores. (C) Vista de secuencia de la plantilla puerta del Únete AB (Panel i) y Tenedor aC (Grupo II). El sitio de restricción PvuII (resaltado en cuadro morado) es en ambos extremos de las puertas ndsDNA. El Nb.BsrDI y Nt.BstNBI Sitios mellar se destacan en cajas de color rojo y negro, respectivamente. Las ubicaciones de corte están marcados con puntas de flecha. Secuencia N es cualquier nucleótido. (Esta cifra se ha modificado con el permiso de 29 Ref.) Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. (A) PCR de una plantilla de ADN puerta. Una plantilla de puerta de ADN contiene las secuencias ndsDNA puerta en el centro (una región azul), y secuencias espaciadoras en ambos extremos (regiones negras; estas dos secuencias finales son ortogonales). Los cebadores pueden unirse a las secuencias del espaciador de la plantilla de la puerta, y generar cuatro fragmentos de ADN superpuestas a través de PCR (secuencias superpuestas están codificados por color en la figura). Montaje (B) Gibson. Los cuatro Fragmen de ADN amplificadosts se ensamblan en un plásmido linealizado espina dorsal a través del método de montaje Gibson 43. (Esta cifra se ha modificado con el permiso de 29 Ref.) Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4. Circuito rendimiento con diferentes cantidades de enzimas. (A) Una representación simplificada de la puerta, reportero, filamentos auxiliares, y de señal hilos utilizados para los experimentos correspondientes. (B) experimentos cinéticos con plásmido derivado Únete AB procesa con diferentes cantidades de enzimas . yo. 10 unidades de PvuII-HF y 45 unidades de Nb.BsrDI por 1 g de plásmido; ii. 10 unidades de PvuII-HF y 4 unidades de Nb.BsrDI por 1 g de plásmido; iii. 4 unidades de PvuII-HF y 4 unidades de Nb.BsrDI por 1 g de plásmido. Todos los hilos auxiliares estaban en 2x (1x = 10 nM). El complejo puerta era 1,5 veces, y los experimentos se realizaron a 35 ° C en 1x TAE / Mg 2+. (Esta cifra se ha modificado con el permiso de 29 Ref.) Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

. Figura 5 Diagrama de flujo de cinética experimentos Blue:. Materiales a añadir a la cubeta (celda de cuarzo 0,875 ml sintético). La tabla de referencia 14 para volúmenes específicos para agregar para el experimento cinética de A + B -> B + C. Verde: Instrucciones de un espectrofluorímetro (etiquetado como SPEX). Rojo: Mezcla instrucciones.53087fig5large.jpg "target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 6. Enzima comportamiento de disociación y el circuito. (A) Una representación simplificada de la puerta, reportero, filamentos auxiliares, y de señal hilos utilizados para los experimentos correspondientes. (B) experimentos cinética del calor plásmido derivado Únete AB utilizando 80 ° C inactivación (trazas verdes), sulfato de 0,15% dodecil de sodio (SDS) (rojo), y un control sin inactivación por calor o adición de SDS (azul). La concentración estándar era 1x = 10 nM, y todos los hilos auxiliares y la entrada B estaban en 2x. El complejo puerta era 1,5 veces, y los experimentos se realizaron a 35 ° C en tampón 1x Tris-acetato-EDTA que contiene 12,5 mM Mg 2+ (1x TAE / Mg 2+ ). (Esta cifra se ha modificado de la referencia 29). Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura calibración 7. Reporter. (A) Reportero C cinética. La concentración reportero estaba en 3x (1x = 50 nM), y la concentración inicial de la señal C está indicado en la figura. (B) Los niveles de fluorescencia de la señal C en el punto (40 min) extremo de medición muestra una relación lineal con el concentración inicial de la señal C. En un ejemplo cuantificación de Tenedor BC puerta (verde línea discontinua), el valor de fluorescencia de Tenedor aC fue medidad como 3 x 10 6 (au), que corresponde a 25 nM (0.5x), basado en la curva de calibración. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

La Figura 8. Cinética de reacción catalíticos bimoleculares (A + B-> B + C). (A) una representación de la puerta, reportero, hebras auxiliares, y hebras de señal utilizado para los experimentos correspondientes simplifica. Los experimentos se llevaron a cabo en tampón 1x Tris-acetato-EDTA que contiene 12,5 mM Mg 2+ (1x TAE / Mg 2+). Todos los complejos de compuerta estaban en la concentración de 75 nM (1,5x), y hebras auxiliares estaban en la concentración de 100 nM (2x). Cinética de datos para las puertas y los datos de plásmidos derivados de puertas sintetizados se muestran en (B) y (C) , respectivamente. Señal estaba en 50 nM (1x). Diferentes cantidades de señal (catalizador) se introdujeron en el sistema, y la reacción se ensayó a 35 ° C. Puertas derivados de plásmido (D) mostraron una mayor rotación de puertas de ADN sintetizados cuando se añadieron pequeñas cantidades de entrada. (E) Alcance de fuga. El gráfico de barras muestra la relación de la fuga final para la señal de final (C B0 = 0 / C B0 = 1) en los puntos extremos (10 h). (Esta cifra se ha modificado de la referencia 29). Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Plantillas Gate | Secuencias | Longitud (nt) |
| JoinAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
Cuadro 1. Las secuencias de las plantillas de compuerta ndsDNA.
| puerta | Hebra | Longitud de cadena inferior (nt) |
| JoinAB | JoinAB-Inferior, , , | 87 |
| ForkBC | ForkBC-Inferior, , , , | |
| 108 |
Tabla 2. Hebras comprenden Únete AB y Tenedor BC. (Esta tabla se ha modificado de la referencia 29).
| Dominio | Secuencia | Longitud (nt) |
| ejército de reserva | CTGCTA | 6 |
| tuberculosis | TTCCAC | 6 |
| tc | TACCCA | 6 |
| tr | TCCTAC | 6 |
| tq | AACCAG | 6 |
| la | CATTGCTTCTACGAGTCATCC | 21 |
| segundo | CATTGCACCTTAGAGTCCGAA | 21 |
| do | CATTGCCACATCGAGTCCCTT | 21 |
| r | CATTGCTTAACCGAGTCTCAC | 21 |
| yo | CTGCCATCATAAGAGTCACCA | 21 |
| Primer capítulo | Secuencias | Longitud (nt) |
| Cebador directo 1- | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| Imprimación-1 Reverse | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Cebador 2- | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Imprimación-2 Reverse | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Cebador 3- | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| Cebador-3 inversa | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Cebador 4- | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Cebador-4 Reverse | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
Tabla 4. Las secuencias de cebador para las plantillas de compuerta PCR de ndsDNA.
| Reactivo | El volumen para la reacción 1x (l) |
| Alta de copias del plásmido columna vertebral (~ 300 ng / l) | 10 |
| PvuII-HF (20.000 unidades / ml) | 2 |
| PstI-HF (20.000 unidades / ml) | 2 |
| Búfer inteligente 10x Cut | 2 |
| H2O | 4 |
| Volumen total | 20 d> |
Tabla 5. Protocolo para el plásmido columna vertebral resumen.
| Reactivo | El volumen para la reacción 1x (l) |
| Vector de ADN (~ 50 ng / l) | 1 |
| Fragmento amplificado por PCR-1 (~ 50 ng / l) | 1 |
| Fragmento amplificado por PCR-2 (~ 50 ng / l) | 1 |
| Fragmento amplificado por PCR-3 (~ 50 ng / l) | 1 |
| Fragmento amplificado por PCR-4 (~ 50 ng / l) | 1 |
| 2x Asamblea Gibson Mix Master | 5 |
| Volumen total | 10 |
| Reactivo | El volumen para la reacción 1x (l) |
| El plásmido de ADN (~ 1 g / l de concentración) | 1000 |
| PvuII-HF (20.000 unidades / ml) | 200 |
| Búfer inteligente 10x Cut | 133.3 |
| Volumen total | 1333.3 |
Tabla 7. Protocolo para puertas ndsDNA insertados plásmido digerir con la enzima de restricción PvuII-HF.
| Reactivo | Volumen (l) |
| Únete puertas (~ 5 g / l de concentración) | 150 |
| Nb.BsrDI (10.000 unidades / ml) | 300 |
| Búfer inteligente 10x Cut | 50 |
| Volumen total | 500 |
Tabla 8. Protocolo para unirse puertas digieren con mellar enzima Nb.BsrDI.
| Reactivo | Volumen (l) |
| Fork puertas (~ 5 g de concentración / l) | 150 |
| Nt.BstNBI (10.000 unidades / ml) | 600 |
| 10x tampón NEB 3.1 | 83.3 |
| Volumen total | 833.3 |
Tabla 9 Protocolo para horquilla puertas a digerir con mellar enzima Nt.BstNBI.
. Tabla 10 secuencias Strand para la implementación de la reacción química A + B -.> B + C (. Esta tabla se ha modificado de la referencia 29)
| Reactivo | Volumen (l) | La concentración final |
| ROX- a 100 M | 10 | 10 M (1x) |
| -RQ a 100 μ;METRO | 13 | 13 M (1.3x) |
| 10x TAE con 125 mM Mg 2+ | 10 | 1x TAE con 12,5 mM Mg 2+ |
| H2O | 67 | - |
| Volumen total | 100 | 10 M (1x) |
Tabla 11. Protocolo para el montaje de Reportero C.
| Reactivo | Volumen (l) | La concentración final |
| H2O | 514 | - |
| 10x TAE con 125 mM Mg 2+ | 60 | 1x TAE con 12,5 mM Mg 2+ |
| PoliT en 300 μ;METRO | 2 | 1 M |
| Reportero C a 10 micras | 9 | 150 nM (3x) |
| 10% de SDS | 9 | 0,15% |
| a 5 micras | 6 | 50 nM (1x) |
| Volumen total | 600 | - |
Tabla 12. Protocolo para la calibración de Reportero C. Los volúmenes proporcionados aquí es para un volumen de reacción total de 600 l (correspondientes a la utilización de una celda de cuarzo sintético 0,875 ml), pero se puede ajustar para trabajar con diferentes células de tamaño.
| Reactivo | Volumen (l) | Con finalcentración |
| H2O | 493 | - |
| 10x TAE con 125 mM Mg 2+ | 60 | 1x TAE con 12,5 mM Mg 2+ |
| poliT en 300 M | 2 | 1 M |
| Reportero C a 10 micras | 9 | 150 nM (3x) |
| en 100 M | 3 | 10x |
| en 100 M | 3 | 10x |
| en 100 M | 3 | 10x |
| 10% de SDS | 9 | 0,15% |
| Tenedor BC en ~ 1 mM (concentración desconocida) | 15 | ~ 0.5x |
| en 100 M | 3 | 10x |
| Volumen total | 600 | - |
Tabla 13. Protocolo para la calibración de Tenedor BC. Los volúmenes proporcionada aquí es para un volumen total de reacción de 600 l, pero se puede ajustar para trabajar con diferentes células de tamaño.
| Reactivo | Volumen (l) | La concentración final | |
| H2O | 407.2 | - | |
| 10x TAE con 125 mM Mg 2+ | 52.8 | 12,5 mM Mg 2 + | |
| poliT en 300 M | 2 | 1 M | |
| Reportero C a 10 micras | 9 | 150 nM (3x) | |
| a los 10 M | 6 | 100 nM (2x) | |
| a los 10 M | 6 | 100 nM (2x) | |
| a los 10 M | 6 | 100 nM (2x) | |
| 6 | 100 nM (2x) | ||
| 10% de SDS | 9 | 0,15% | |
| Únete a AB en 1 M | 45 | 75 nM (1.5x) | |
| Tenedor BC a 1 M | 45 | 75 nM (1.5x) | |
| a los 10 M | 3 | 50 nM (1x) | |
| a los 10 M | 3 | 50 nM (1x) | |
| Volumen total | 600 | - | |
Tabla 14. Protocolo para una reacción química A + B-> B + C. Los volúmenes proporcionada aquí es para un volumen total de reacción de 600 l, pero se puede ajustar para trabajar con diferentes células de tamaño.
| Puertas sintetizados | Puertas plásmido derivado de | ||||
| Descripción | Costo | Únete puertas | Puertas Fork | ||
| PÁGINA cadena larga purificada (100 nt; se desempeñó como los hilos inferiores de una puerta) | ~ $ 75 | Descripción </ strong> | Costo | Descripción | Costo |
| PÁGINA cadena corta purificada (~ 30 nt, sirvieron como principales cadenas de una puerta) | ~ $ 185 | Plantilla de Puerta | ~ $ 100 | Plantilla de Puerta | ~ $ 100 |
| Total | ~ $ 260 | Kit de extracción de plásmido | ~ $ 26 | Kit de extracción de plásmido | ~ $ 26 |
| La enzima de restricción (PvuII-HF) | ~ $ 11 | La enzima de restricción (PvuII-HF) | ~ $ 11 | ||
| Mellar enzima (Nt.BsrDI, Ingreso puertas) | ~ $ 29 | Enzima mellar (Nt.BstNBI, puertas Fork) | ~ $ 62 | ||
| Total | ~ $ 166 | Total | ~ $ 199 |
fo:.. keep-con-previous.within-page = "always"> Cuadro 15 Comparación de costos entre las puertas de plásmidos derivados y portones sintetizados (. Esta tabla se ha modificado de la referencia 29)
| Puertas sintetizados | Puertas plásmido derivado de | ||
| Tratamiento | Tiempo de procesamiento | Tratamiento | Tiempo de procesamiento |
| Recocido | 1 hr | Clonación | 5 horas |
| Purificación PÁGINA | 2 horas | La extracción del plásmido | 2 horas |
| Total | 3 horas | Dos pasos de digestión con enzimas | 0,5 horas |
| Precipitación con etanol | 1 hora | ||
| Total | 8.5 hr | ||
Tabla 16. Comparación de Procesamiento de tiempo entre las puertas de plásmidos derivados y portones sintéticos. (Esta tabla se ha modificado de la referencia 29).
Discusión
Este documento describe un método para derivar puertas ndsDNA a partir de ADN plásmido de alta pureza. Por otra parte, un protocolo se presenta para caracterizar el rendimiento de puerta usando un ensayo de la cinética de fluorescencia. Los datos experimentales muestran que el sistema de plásmido derivado supera a su contraparte sintética incluso si el sistema sintético se ensambla a partir de hebras purificó usando electroforesis en gel de poliacrilamida (PAGE). Probablemente, el mejor desempeño de puertas plásmido derivado se debe principalmente a la muy alta pureza del ADN biológica. ADN sintético contiene una variedad de errores, en supresiones particulares que resultan en oligonucleótidos de longitud n-1, y dichos productos secundarios son normalmente no se elimina por completo en PAGE o cromatografía líquida de alta resolución (HPLC) procedimientos de purificación. También se observaron mejoras similares a los reportados aquí en un estudio previo de un amplificador de horquilla catalizada que utiliza ADN derivado de fuentes biológicas 21.
Sin embargo, incluso el uso de puertas de plásmido derivado no puede eliminar por completo los errores en el desempeño de puerta, para los que hay al menos dos razones: en primer lugar el exceso de la digestión o la falta de corte de precisión puede conducir a puertas con demasiados muescas o hendiduras en las posiciones equivocadas. En cualquier caso, las puertas son más propensos a participar en reacciones no deseadas. Tales problemas pueden aliviarse mediante la optimización de la cantidad de enzima utilizada (véase la Figura 4). En segundo lugar, en estos experimentos, la mayoría de las entradas y las hebras eran auxiliares ADN sintético y de este modo contenían deleciones y mutaciones. En principio, todas las entradas de una sola hebra y hebras auxiliares también se pueden obtener a partir de ADN fagémido a través de una digestión con enzimas de nicking del genoma viral m13 pre-codificada 26. Tal vez el funcionamiento del circuito se puede mejorar adicionalmente usando ssDNA derivado del genoma bacteriano.
Mientras que se encontró el uso de puertas de plásmidos derivados para mejorar el rendimiento del circuito, un análisisde los tiempos de procesamiento de coste y reveló que mientras la producción de puertas de plásmido derivado es ligeramente más barato (Tabla 15), se tarda 2-3 veces más largo el tiempo de procesamiento en comparación con el montaje y la purificación de puertas a partir de oligos sintetizados comercialmente (Tabla 16). Los costes primarios de puertas plásmido derivado son la síntesis de genes y el uso de enzimas de restricción. Por 300 pmoles de puertas (suficientes para 15 reacciones en 30 nM), el costo estimado para Únete puertas es de aproximadamente $ 170 y $ 200 para Fork puertas, la diferencia de costos se debe a la utilización de diferentes enzimas mellar. Por el contrario, la síntesis química de los hilos para los mismos costes de compuerta alrededor de $ 260 incluyendo una cuota de purificación PÁGINA. El costo de tiempo principal para puertas plásmido derivado está en el procedimiento de clonación, que, al igual que la síntesis de ADN, puede ser subcontratado a una empresa de síntesis de genes. Sin embargo, una vez ensamblados, puertas plásmido derivado tienen la ventaja de que los plásmidos de acogida fácilmente se pueden replicar unand se puede almacenar en forma de stocks de glicerol bacterianas. Esto hace posible la reutilización de las puertas muchas veces.
De cara al futuro, la mejora del rendimiento de las puertas de plásmidos derivados podría permitir a una gama mucho más amplia de la dinámica de los comportamientos que han sido experimentalmente demostrado hasta ahora con ADN CRN. Por ejemplo, la reciente 47,48 trabajo teórico sugiere que los patrones espaciales auto-organizados en la escala macro se pueden realizar con el ADN CRNs a través de un mecanismo de difusión de reacción. El método que aquí se presenta proporciona una ruta viable para la construcción de los componentes moleculares subyacentes para tales materiales de ADN auto-patrones. Aunque difícil, el desarrollo de morfologías escala macro de una manera programable tendría implicaciones significativas en áreas que van desde la investigación de biomateriales para la medicina regenerativa.
Divulgaciones
The authors declare no competing financial interests.
Agradecimientos
Figuras 1, 2, 3, 4, 6, 8 y Tablas 2, 3, 10, 15, 16 se modifican de la referencia 29. Este trabajo fue apoyado por la Fundación Nacional de Ciencia (NSF conceder-CCF 1.117.143 y NSF-CCF 1.162.141 de GS). Y.-JC fue apoyado por becas del Gobierno de Taiwán. SDR fue apoyada por el Programa Nacional Science Foundation Graduate Research Fellowship (GRFP).
Materiales
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S | |
| PvuII-HF | NEB | R3151L | |
| PstI-HF | NEB | R3140S | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Terrific Broth, Modified | SIGMA-ALDRICH | T0918-250G | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAGEN Hispeed Maxi-prep Kit | QIAGEN | 12662 | |
| Nb.BsrDI | NEB | R0648L | |
| Nt.BstNBI | NEB | R0607L | |
| NanoDrop 2000c | Thermo Scientific | ||
| Double-stranded Genomic Blocks | IDT | ||
| Horiba Jobin-Yvon Spex Fluorolog-3 Fluorimeter | Horiba/Jobin Yvon | ||
| Synthetic Quartz Cells | Starna | 23-5.45-S0G-5 | |
| QIAGEN Gel Extraction Kit | QIAGEN | 28706 | |
| Plasmid Backbones | BioBrick | E0240-pSB1A2 | High copy number plasmid with Ampicillin resistance. Sequence can be found from http://parts.igem.org |
Referencias
- Zhang, D. Y., Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103-113 (2011).
- Krishnan, Y., Simmel, F. C. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 50, 3124-3156 (2011).
- Zhang, D. Y., Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303-17314 (2009).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475, 368-372 (2011).
- Qian, L., Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 332, 1196-1201 (2011).
- Zadegan, R. M., Jepsen, M. D., Hildebrandt, L. L., Birkedal, V., Kjems, J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 11, 1811-1817 (2015).
- Seelig, G., Soloveichik, D., Zhang, D. Y., Winfree, E. Enzyme-free nucleic acid logic circuits. Science. 314, 1585-1588 (2006).
- Zadegan, R. M., et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 6, 10050-10053 (2012).
- Andersen, E. S., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 459, 73-76 (2009).
- Zhang, D. Y., Hariadi, R. F., Choi, H. M., Winfree, E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 4, (1965).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406, 605-608 (2000).
- Green, S. J., Lubrich, D., Turberfield, A. J. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 91, 2966-2975 (2006).
- Venkataraman, S., Dirks, R. M., Rothemund, P. W., Winfree, E., Pierce, N. A. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2, 490-494 (2007).
- Green, S. J., Bath, J., Turberfield, A. J. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 101, 238101 (2008).
- Omabegho, T., Sha, R., Seeman, N. C. A bipedal DNA Brownian motor with coordinated legs. Science. 324, 67-71 (2009).
- Turberfield, A. J., et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 90, 118102 (2003).
- Dirks, R. M., Pierce, N. A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 101, 15275-15278 (2004).
- Seelig, G., Yurke, B., Winfree, E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 128, 12211-12220 (2006).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318, 1121-1125 (2007).
- Yin, P., Choi, H. M., Calvert, C. R., Pierce, N. A. Programming biomolecular self-assembly pathways. Nature. 451, 318-322 (2008).
- Chen, X., Briggs, N., McLain, J. R., Ellington, A. D. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 110, 5386-5391 (2013).
- Phillips, A., Cardelli, L. A programming language for composable DNA circuits. J. R. Soc. Interface. 6, S419-S436 (2009).
- Lakin, M. R., Youssef, S., Polo, F., Emmott, S., Phillips, A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 27, 3211-3213 (2011).
- Lakin, M. R., Youssef, S., Cardelli, L., Phillips, A. Abstractions for DNA circuit design. J. R. Soc. Interface. 9, 470-486 (2012).
- Zhang, D. Y., Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182-4197 (2010).
- Ducani, C., Kaul, C., Moche, M., Shih, W. M., Hogberg, B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 10, 647-652 (2013).
- Lin, C., et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 105, 17626-17631 (2008).
- Bhatia, D., et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 48, 4134-4137 (2009).
- Chen, Y. J., et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 8, 755-762 (2013).
- Arkin, A., Ross, J. Computational functions in biochemical reaction networks. Biophys. J. 67, 560-578 (1994).
- Érdi, P., Tóth, J. . Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. , (1989).
- Magnasco, M. O. Chemical kinetics is Turing universal. Phys. Rev. Lett. 78, 1190 (1997).
- Oishi, K., Klavins, E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 5, 252-260 (2011).
- Senum, P., Riedel, M. Rate-independent constructs for chemical computation. PLoS One. 6, (2011).
- Soloveichik, D., Cook, M., Winfree, E., Bruck, J. Computation with finite stochastic chemical reaction networks. Natural Computing. 7, 615-633 (2008).
- Soloveichik, D., Seelig, G., Winfree, E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 107, 5393-5398 (2010).
- Tyson, J. J., Chen, K. C., Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 15, 221-231 (2003).
- Cardelli, L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 23, 247-271 (2013).
- Angluin, D., Aspnes, J., Eisenstat, D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 21, 87-102 (2008).
- Cardelli, L., Csikasz-Nagy, A. The cell cycle switch computes approximate majority. Sci. Rep. 2, 656 (2012).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170-173 (2011).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. , (2012).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. , e253 (2007).
- Lessard, J. C. Transformation of E. coli via electroporation. Methods Enzymol. 529, 321-327 (2013).
- Nasri, M., Thomas, D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 15, 7677-7687 (1987).
- Dalchau, N., Seelig, G., Phillips, A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. , 84-99 (2014).
- Scalise, D., Schulman, R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2, 55-66 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados