
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
La detección de funcional no codificante-variantes genéticas Uso de movilidad electroforética Shift ensayo (EMSA) y el ADN afinidad Precipitación Ensayo (DAPA)
* Estos autores han contribuido por igual
En este artículo
Resumen
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
Resumen
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
Introducción
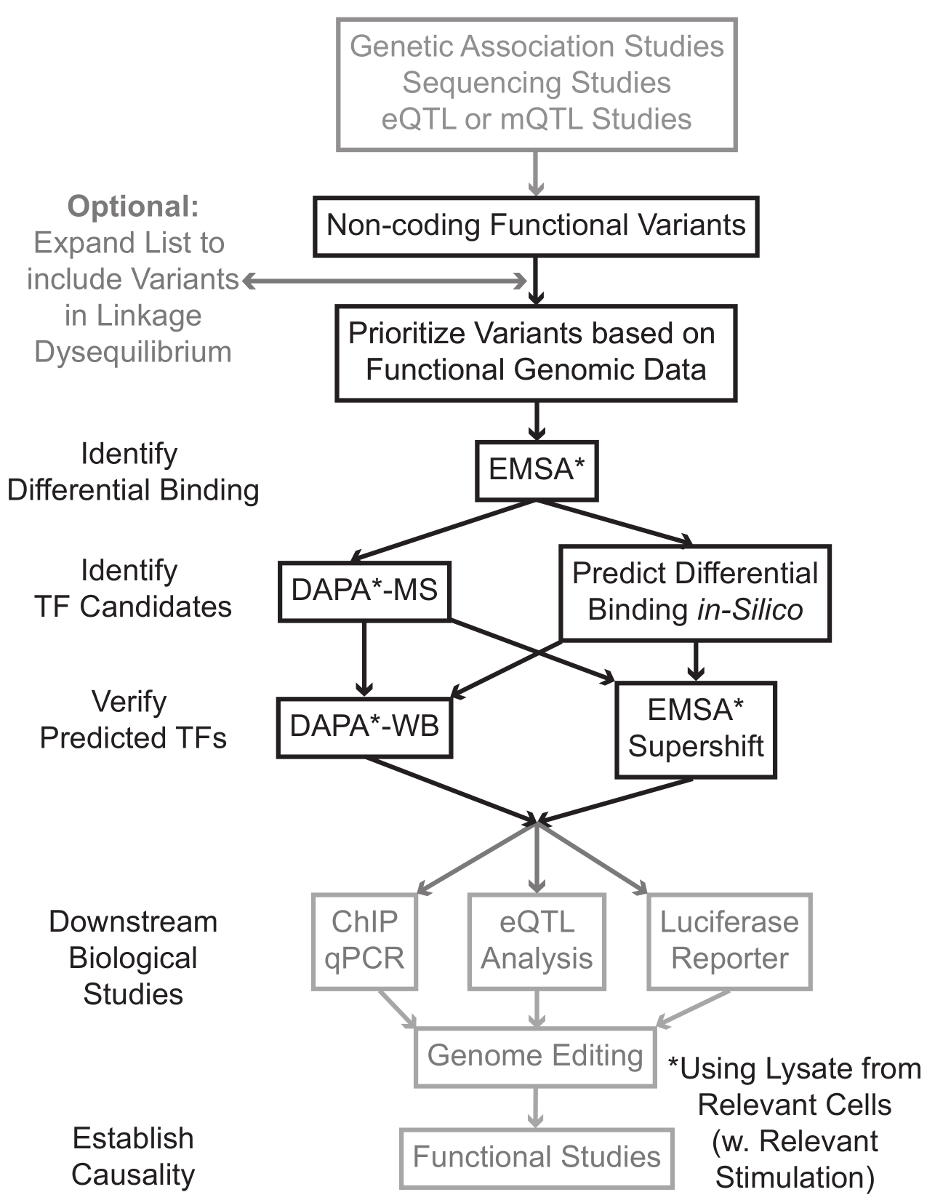
Secuenciación y genotipado basado en estudios, incluyendo estudios de genoma completo (GWAS Association), estudios candidato locus, y profundo de secuenciación de los estudios, han identificado muchas variantes genéticas que están estadísticamente asociados con una enfermedad, rasgo o fenotipo. Contrariamente a las predicciones iniciales, la mayoría de estas variantes (85-93%) se localizan en regiones no codificantes y no cambian la secuencia de aminoácidos de las proteínas de 1,2. Interpretación de la función de estas variantes no codificantes y la determinación de los mecanismos biológicos que los conectan a la enfermedad asociada, rasgo, o fenotipo ha demostrado ser un desafío 3-6. Hemos desarrollado una estrategia general para identificar los mecanismos moleculares que enlazan variantes a un fenotipo intermedio importante - la expresión génica. Esta tubería está específicamente diseñado para identificar la modulación de la unión por variantes genéticas TF. Esta estrategia combina los enfoques computacionales y técnicas de biología molecular destinadas a predecirefectos biológicos de las variantes candidatos in silico, y verificar estas predicciones empíricamente (Figura 1).

Figura 1:.. Enfoque estratégico para el análisis de los pasos no codificantes de variantes genéticas que no están incluidos en el protocolo detallado asociado a este manuscrito están sombreados en gris Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
En muchos casos, es importante comenzar mediante la ampliación de la lista de variantes para incluir a todos los de alta desequilibrio de unión (LD) con cada variante estadísticamente asociado. LD es una medida de asociación no aleatoria de los alelos en dos posiciones cromosómicas diferentes, que se puede medir por el r 2 Estadística 7. r 2 es una medida de la linKage desequilibrio entre dos variantes, con una unión perfecta r 2 = 1 denota entre dos variantes. Alelos en alto LD se encuentran a co-segregar en el cromosoma a través de poblaciones ancestrales. matrices de genotipos actuales no incluyen todas las variantes conocidas en el genoma humano. En su lugar, se aprovechan de la LD dentro del genoma humano e incluyen un subconjunto de las variantes conocidas que actúan como representantes de otras variantes dentro de una región particular del LD 8. Por lo tanto, una variante sin ninguna consecuencia biológica puede estar asociado con una enfermedad en particular, ya que es en LD con la variante de la causal variante con un efecto biológico significativo. Del procedimiento, se recomienda para convertir la última versión de los 1.000 genomas Proyecto 9 archivos variante llamada (VCF) en archivos binarios compatibles con PLINK 10,11, una herramienta de código abierto para el análisis de asociación de genoma completo. Posteriormente, todas las demás variantes genéticas con LD r 2> 0,8 con cada entrada va genéticaRiant puede ser identificado como candidatos. Es importante la utilización de la población de referencia apropiada para este paso- por ejemplo, si una variante fue identificada en sujetos de ascendencia europea, los datos de los sujetos de ascendencia debe emplearse la misma para la expansión LD.
expansión LD a menudo resulta en variantes de candidatos docenas, y es probable que sólo una pequeña fracción de estos contribuyen a mecanismo de la enfermedad. A menudo, no es factible para examinar experimentalmente cada una de estas variantes de forma individual. Es por lo tanto útil para aprovechar los miles de conjuntos de datos de genómica funcional públicamente disponibles como un filtro para priorizar las variantes. Por ejemplo, el consorcio ENCODE 12 ha realizado miles de experimentos chip-ss que describen la unión de TFS y co-factores, y las marcas de las histonas en una amplia gama de contextos, junto con los datos de la cromatina accesibilidad de las tecnologías tales como DNasa-ss 13, ATAC -seq 14 y ss Faire-15. DatabASES y servidores web, tales como la UCSC Genome Browser 16, Hoja de Ruta Epigenómica 17, 18 Epigenoma Blueprint, Cistrome 19 y 20 de RECONFIGURACIÓN proporcionan acceso libre a los datos producidos por estas y otras técnicas experimentales a través de una amplia gama de tipos y condiciones de las celdas. Cuando hay demasiadas variantes para examinar experimentalmente, estos datos se pueden utilizar para dar prioridad a los situados dentro de las regiones reguladoras probables en los tipos de células y tejidos pertinentes. Además, en los casos en que es una variante dentro de un pico de chip-ss para una proteína específica, estos datos pueden proporcionar pistas potenciales en cuanto a la TF (s) específico o co-factores cuya unión podría estar afectando.
A continuación, las variantes resultantes prioridad son examinados experimentalmente para validar la unión usando EMSA 21,22 proteína genotipo dependiente predicho. EMSA mide el cambio en la migración de la oligo en un gel de TBE no reductor. oligo marcado con fluorescencia se incuba con ellisado nuclear, y la unión de factores nucleares retardará el movimiento del oligo en el gel. De esta manera, oligo que se ha unido factores más nucleares presentará como una señal fluorescente más fuerte sobre la exploración. En particular, la EMSA no requiere predicciones acerca de las proteínas específicas cuya unión se verán afectados.
Una vez que se identifican las variantes que se encuentran dentro de las regiones reguladoras predichos y son capaces de factores nucleares diferencialmente de unión, se emplean métodos computacionales para predecir el TF (s) específico cuya unión podrían afectar. Nosotros preferimos utilizar CIS-BP 23,24, RegulomeDB 25, Uniprobe 26 y 27 JASPAR. Una vez que se identifican candidato TFS, estas predicciones pueden ser probados específicamente el uso de anticuerpos contra estos TFS (EMSA-supershifts y DAPA-Oeste). Un EMSA-supershift implica la adición de un anticuerpo específico-TF al lisado nuclear y oligo. Un resultado positivo en un EMSA-supershift es represented como un cambio adicional en la banda de EMSA, o una pérdida de la banda (revisado en referencia 28). En la DAPA complementaria, un dúplex de oligo 5 'biotinilado que contiene la variante y la 20 pares de bases que flanquean nucleótidos se incuban con lisado nuclear de tipo de célula relevante (s) para capturar cualquier factores nucleares se unen específicamente a los oligos. El complejo del factor nuclear-duplex oligo es inmovilizado por estreptavidina microperlas en una columna magnética. Los factores nucleares unidas se recogieron directamente a través de la elución 29,48. predicciones de unión pueden ser evaluados por una transferencia de Western usando anticuerpos específicos para la proteína. En los casos en que no hay predicciones obvias, o demasiadas predicciones, las eluciones de variantes desplegables de los experimentos DAPA se pueden enviar a un núcleo de la proteómica para identificar TFS candidatos utilizando la espectrometría de masa, que posteriormente se pueden validar el uso de estos descritos anteriormente métodos.
En el resto de la article, se proporciona el protocolo detallado para el análisis EMSA y DAPA de variantes genéticas.
Protocolo
1. Preparación de soluciones y reactivos
- Ordenar sondas de oligonucleótidos de ADN personalizados para su uso en la EMSA y la DAPA.
- Para reducir la proteína de unión no específica, diseñar oligos cortos (entre 35-45 pares de bases (pb) de longitud) 30, y colocar la variante de interés directamente en el centro, flanqueado por su secuencia genómica endógena 17 pb. Para oligos EMSA, añadir un 'fluoróforo 5. Para oligos DAPA, añadir una etiqueta 5 'con biotina.
- Ordenar tanto la cadena sentido y su cadena de complemento inverso. Alternativamente, dúplex fin (pre-recocido) oligos. Al asignar nombres a los oligos, basar la nomenclatura en un genoma de referencia establecido.
Nota: "riesgo" y la designación de "no riesgo" puede ser la enfermedad y específica del proyecto, mientras que "referencia" y "no es de referencia" son más universalmente relevante. - A la llegada de los oligos, girar brevemente hacia abajo el contenido y volver a suspender en agua libre de nucleasa a una concentración final de 100 μ;METRO. Tienda volvió a suspender acciones a -20 ° C. Proteger los oligonucleótidos marcados con un fluoróforo de la luz envolviendo con papel de aluminio.
| Nombre | Secuencia |
| rs76562819_REF_FOR | Un GTAATGCCTTAATGAGAGAG GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
Tabla 1: Ejemplo de diseño de oligonucleótidos EMSA / DAPA para poner a prueba un SNP para Bin diferencialding. "REF" significa el alelo de referencia, mientras que "NONREF" significa el alelo no es de referencia. "A" representa el capítulo adelante, mientras que "REV" indica su complemento. El SNP se ve en rojo.
- Preparar tampón de extracción citoplasmática (CE) con una concentración final de HEPES 10 mM (pH 7,9), KCl 10 mM, y EDTA 0,1 mM en agua desionizada.
- Preparar extracción nuclear (NE) tampón con una concentración final de HEPES 20 mM (pH 7,9), 0,4 M NaCl, y EDTA 1 mM en agua desionizada.
- Preparar tampón de reasociación con una concentración final de Tris 10 mM (pH 7,5 a 8,0), NaCl 50 mM, y EDTA 1 mM en agua desionizada.
2. Preparación de lisado nuclear de células cultivadas
Nota: Este protocolo experimental se ha optimizado el uso de líneas de células linfoblastoides B, pero se ha probado en varias otras líneas de células adherentes / suspensión no relacionados y funciona universalmente.
- Culture células linfoblastoides B en Roswell Park Memorial Institute (RPMI) 1640 con 2 mM L-glutamina, suero bovino fetal 10%, y 1 x antibiótico-antimicótico que contiene 100 unidades / ml de penicilina, 100 mg / ml de estreptomicina y 250 ng / ml de anfotericina B.
- Seed en un rango de 200,000-500,000 células viables / ml y se incuban frascos a 37 ° C con dióxido de carbono al 5% en una posición vertical con tapas ventiladas o sueltas.
Nota: El crecimiento de las células linfoblastoides B se ralentiza cuando llegan a más de 1.000.000 células / ml. Romper blastos de células pipeteando arriba y abajo varias veces y las células volver a 200,000-500,000 células / ml para mantener una velocidad rápida de crecimiento.
- Seed en un rango de 200,000-500,000 células viables / ml y se incuban frascos a 37 ° C con dióxido de carbono al 5% en una posición vertical con tapas ventiladas o sueltas.
- Lavar las células cultivadas dos veces con solución salina tamponada con 10 ml de hielo frío de fosfato (PBS), girar hacia abajo a 4 ° C, 300 xg durante 5 min y retirar PBS a través de la aspiración.
- Recuento de células utilizando un hemocitómetro y se vuelve a suspender sedimento como 1 ml de PBS enfriado en hielo por 10 7 células.
Nota: Por ejemplo, si la lisis de 2 x 10 7 </ Sup> células, se vuelve a suspender en 2 ml de PBS. - Alícuota de 1 ml a tubos de 1,5 ml de microcentrífuga de modo que cada tubo contiene 10 7 células en PBS. Centrifugar a 3300 xg durante 2 min 4 ° C y aspirado fuera de PBS.
- Antes de usar, añadir ditiotreitol 1 mM (DTT), inhibidor de la fosfatasa 1x, y el inhibidor de la proteasa 1x a una madre de trabajo de tampón CE. Resuspender el sedimento celular con 400 l de tampón CE y se incuba en hielo durante 15 minutos.
- Añadir 25 l de 10% de Nonidet P-40 y se mezcla con la pipeta. Se centrifuga a 4 ° C, velocidad máxima durante 3 minutos. Decantar y desechar el sobrenadante.
- Antes de usar, añadir 1 mM de DTT, inhibidor de la fosfatasa 1x, 1x y inhibidor de la proteasa a una madre de trabajo de tampón NE. Resuspender el sedimento celular con 30 l de tampón NE y mezclar mediante agitación.
- Se incuba a 4 ° C en un rotador de tubo o en hielo durante 10 min. Centrífuga 3300 xg durante 2 min 4 ° C.
- Recoger el sobrenadante claro (lisado nuclear) y alícuota antes de almacenar a -806; C para evitar múltiples ciclos de congelación-descongelación que pueden degradar la proteína. Deja un alícuota de 10 l para medir la concentración de proteínas mediante el ensayo del ácido bichoninic (BCA) 31.
3. Desplazamiento de Movilidad electroforética de ensayo (EMSA)
- Preparar Oligo de Trabajo y EMSA gel.
- Si oligos fueron ordenados en dúplex, descongelar la madre 100 mM y se diluye 1: 2000 en tampón de recocido para conseguir una solución de trabajo de 50 nM.
- Si oligos fueron ordenados de cadena simple, descongelar los 100 M existencias y se diluye 1:10 en tampón de recocido para alcanzar 100 acciones de trabajo nm. Combinar 100 l de la solución de hebra de complemento 100 nM entre sí en un tubo de microcentrífuga.
- Colocar en un bloque de calor a 95 ° C durante 5 min. Apagar el bloque de calor y permitir que los oligos se enfríen lentamente hasta temperatura ambiente durante al menos una hora antes de su uso.
- Pre-correr el gel EMSA.
- Retire el portaobjetos de un prefabricado 6% TBE gel y enjuague con agua desionizada varias veces para eliminar cualquier tampón de los pocillos. Preparar 1 L de tampón TBE 0,5x mediante la adición de 50 ml de 10x TBE a 950 ml de agua desionizada.
- Montar el aparato de electroforesis en gel y comprobar si hay fugas llenando la cámara interior con tampón TBE 0,5x. Si no hay memoria intermedia se filtra en la cámara exterior, llenar la cámara exterior aproximadamente dos tercios del camino.
- Pre-correr el gel a 100 V durante 60 min.
- Lave cada pocillo con 200 l de tampón TBE 0,5x.
- Preparar tampón de unión Mix Master.
- Preparar 10x tampón de unión con una concentración final de Tris 100 mM, KCl 500 mM, DTT 10 mM; pH 7,5 en agua desionizada.
- En un tubo de microcentrífuga, crear una mezcla maestra que consiste en los reactivos comunes a todas las reacciones (10 l de 10x tampón de unión, 10 l de DTT / polisorbato, 5 l de poli d (IC), y ADN de esperma de salmón 2,5 l; tabla 2). Preparar un 10% adicionalpara tener en cuenta la pérdida de volumen debido a pipeteado.
| Reactivo | Conc final. | Rxn # 1 | Rxn # 2 | Rxn # 3 | Rxn # 4 |
| Agua ultra pura | a 20 l vol. | 13,5 l | 11,98 l | 13.5μl | 11,98 l |
| 10x tampón de unión | 1x | 2 l | 2 l | 2 l | 2 l |
| TDT / TW-20 | 1x | 2 l | 2 l | 2 l | 2 l |
| ADN de esperma de salmón | 500 ng / l | 0,5 l | 0,5 l | 0,5 l | 0,5 l |
| 1μg / l de poli d (IC) | 1 g | 1 l | 1 l | 1 l | 1 l |
| Extracto Nuclear (5.26 ug / l) | 8 mg | - | 1,52 l | - | 1,52 l |
| NE Buffer | 1,52 l | - | 1,52 l | - | |
| Referencia alelo oligo | 50 fmol | 1 l | 1 l | - | - |
| No es de referencia alelo oligo | 50 fmol | - | - | 1 l | 1 l |
Tabla 2: Ejemplo EMSA setu reacciónp. La tabla ilustra un ejemplo EMSA para probar la hipótesis de que existe el genotipo unión dependiente de TFS a un SNP específico.
- Añadir agua libre de nucleasa a cada tubo de microcentrífuga de tal manera que el volumen final después de la adición de todos los reactivos será de 20 l.
- Añadir la cantidad apropiada (5,5 l) de la mezcla maestra a cada tubo de microcentrífuga.
- Añadir 8 g de lisado nuclear para los tubos de microcentrífuga apropiadas. Incluyen tubos que contienen el oligo sin extracto nuclear como controles negativos (por ejemplo, la Tabla 2, Rxn # 1 y Rxn # 3).
Nota: La cantidad óptima de lisado por reacción debe determinarse experimentalmente mediante valoración. En general, valoración de una gama de 2 a 10 g de lisado es suficiente. - Añadir 50 fmol de oligo a los tubos de microcentrífuga apropiados. Flick para mezclar y brevemente girar el contenido al fondo del tubo. Incubar durante 20 min a temperatura ambiente.
Nota: Si attempting de un supershift, incubar la mezcla de lisado con el anticuerpo durante 20 minutos a temperatura ambiente antes de la adición de oligos. Se recomienda el uso de 1 mg de un anticuerpo chip-grado para mejores resultados. - Añadir 2 l de 10x naranja colorante de carga a cada tubo de microcentrífuga. Pipetear arriba y abajo para mezclar.
- Cargar las muestras en la previa al funcionamiento de gel de TBE 6% pipeteando arriba y abajo para mezclar y luego la expulsión de cada muestra en un pocillo separado. Correr el gel a 80 V hasta que el colorante naranja ha migrado de 2/3 a 3/4 del camino hacia abajo el gel. Esto debe tomar aproximadamente 60-75 minutos.
- Retire el gel de la casete de plástico haciendo palanca con un cuchillo de gel y colocar el gel en un recipiente con tampón TBE 0,5% para evitar que se reseque.
- Colocar el gel en la superficie de un sistema de imágenes de infrarrojos y de quimioluminiscencia, asegurándose de eliminar cualquier burbuja o contaminantes que contribuyen a perturbar la imagen.
- Utilizando el software de sistema de exploración, haga clic en la pestaña "Adquirir"y luego seleccionar "Dibujo Nuevo" para dibujar una caja alrededor de la zona correspondiente a donde el gel se encuentra en la superficie del escáner.
- En la sección "Canales" de la pestaña "Adquirir", seleccione el canal correspondiente a la longitud de onda de la etiqueta fluoróforo en el oligo. En la sección "Escáner", haga clic en "vista previa" para obtener una vista previa del escaneo de baja calidad. Ajustar el área de escaneado arrastrando el cuadro azul que rodea a la previsualización de la imagen adquirida hasta la porción del gel para formar una imagen.
Nota: Por ejemplo, si el uso de oligonucleótidos marcados con un fluoróforo 700 nm, asegúrese de que el canal "700 nm" se ha seleccionado antes de escanear. - En la sección "Controles de escaneo", seleccione la opción "84 M" resolución y la calidad de la opción "Medio". Establecer el foco para compensar a la mitad del grosor del gel. Nota: Por ejemplo, un gel de 1 mm utilizaría un 0,5 mm de desfase de enfoque.
- En la sección "Escáner", haga clic en "Inicio" para bEgin la exploración.
Nota: Durante la exploración, el esquema de brillo, contraste y color a menudo se puede ajustar manualmente en función del fabricante del sistema de exploración. - Después de finalizado el análisis, seleccione la pestaña "Imagen" y haga clic en "girar o voltear" en la sección "Crear" para corregir la orientación. Guarde el archivo de imagen haciendo clic en "Exportar" en el menú principal y luego seleccione "Imagen Individual con vistas."
4. ADN Purificación por Afinidad de ensayo (DAPA)
- Preparación de 5 M Oligo de trabajo Stock.
- Si oligos fueron ordenados en dúplex, descongelar la madre 100 mM y se diluye 1:20 en tampón de recocido para conseguir una solución de trabajo 5 micras.
- Si oligos fueron ordenados de cadena simple, descongelar los 100 M existencias y se diluye 1:10 en tampón de recocido para alcanzar 10 M reservas de trabajo. Combinar 10 l de los 10 M cadenas complementarias entre sí. Colocar en un bloque de calor a 95 ° C durante5 minutos. Apagar el bloque de calor y permitir que los oligos enfriar lentamente hasta la temperatura ambiente antes de su uso.
- Antes de comenzar, calentar el tampón de unión, el tampón de lavado de baja rigurosidad, tampón de lavado de alta rigurosidad, y tampón de elución a temperatura ambiente.
Nota: Una concentración final de 50 ng / ml de poli d (IC) puede ser añadido a la memoria intermedia de unión, el tampón de lavado de baja rigurosidad, y tampón de lavado de alta rigurosidad para reducir el potencial de unión no específica de proteínas a los oligos. - Preparar las mezclas de unión para cada variante.
- Mezclar 1 volumen de lisado nuclear con 2 volúmenes de tampón de unión.
Nota: La cantidad requerida de lisado debe determinarse experimentalmente debido a la variación abundancia de TFS. Uso de entre 100 a 250 g de lisado nuclear por columna es suficiente en la mayoría de los casos. - Añadir inhibidor de fosfatasa 1x, 1x inhibidor de la proteasa, y potenciador de unión 1x (opcional) y mezclar con un movimiento rápido el tubo varias veces.
Nota: 100X de uniónpotenciador consta de 750 mM MgCl 2 y 300 mM ZnCl 2. Añadir potenciador de unión si la unión del TF al ADN depende de cofactores o agentes reductores. Si esta información no se conoce, añadir el promotor de unión. - Añadir 10 l de 5 M de ADN de captura biotinilado (50 pmol) a cada mezcla de unión respectiva. Incubar durante 20 min a temperatura ambiente.
Nota: El tiempo de incubación y la temperatura pueden variar dependiendo de la TF. Los valores óptimos deben determinarse experimentalmente.
- Mezclar 1 volumen de lisado nuclear con 2 volúmenes de tampón de unión.
- Añadir 100 l de microperlas de estreptavidina. Incubar durante 10 min a temperatura ambiente.
- Para cada sonda oligo siendo probado, colocar una columna de unión en el separador magnético. Colocar un tubo de microcentrífuga directamente debajo de cada columna de unión y aplicar 100 l de tampón de unión para enjuagar la columna.
- Pipetear los contenidos de cada mezcla de unión en columnas separadas y permitir que el líquido fluya completamente a través de la columna en la microcentrífugatubo antes de continuar. Asegúrese de etiquetar las columnas con el oligo variante que se utilizó en la mezcla de unión. Etiquetar las muestras de flujo continuo y reemplazar con nuevos tubos de microcentrífuga para recoger las aguas de lavado de baja rigurosidad.
- Aplicar 100 l de tampón de lavado de baja rigurosidad a la columna; esperar hasta el depósito de la columna está vacía. Repetir el lavado de 4x. Etiquetar las muestras de lavado de baja rigurosidad y reemplazar con nuevos tubos de microcentrífuga para recoger las aguas de lavado de alta rigurosidad.
- Aplicar 100 l de tampón de lavado de alta rigurosidad a la columna; esperar hasta el depósito de la columna está vacía. Repetir el lavado de 4x. Etiquetar las muestras de lavado de alta rigurosidad y reemplazar con nuevos tubos de microcentrífuga para recoger el pre-elución.
- Añadir 30 l de tampón de elución nativa a la columna y dejar reposar durante 5 minutos. Etiquetar las muestras pre-elución y reemplazar con nuevos tubos de microcentrífuga para recoger la elución.
Nota: Esto no eluir la proteína unida; se lava la alta rigurosidad restantetampón de la columna y se lo reemplaza con tampón de elución para maximizar la eficiencia de la elución. - Añadir un 50 l de tampón de elución nativa adicional para eluir la TFS encuadernados. Para un mayor rendimiento, pero eluato menos concentrado, añadir un 50 l adicionales de tampón de elución nativa y recoger el flujo continuo.
Nota: Analizar las muestras de elución a través de espectrometría de masas para determinar la identidad de los TF unido 32. Después, verificar los resultados proteómicos a través de sodio dodecil electroforesis en gel de poliacrilamida al sulfito (SDS-PAGE), seguido de una transferencia de Western 33. Si la espectrometría de masas no está disponible, ejecutar una tinción de plata usando la técnica estándar en lugar de una transferencia Western para determinar el tamaño de la proteína (s) que muestra el genotipo dependiente de la unión. Utilice esta información para reducir la lista de TFS predichos de los métodos de cálculo que se detallan en la introducción.
Resultados
En esta sección, los resultados representativos de lo que cabe esperar se proporcionan al realizar una EMSA o DAPA, y la variabilidad en cuanto a se caracteriza la calidad de lisado. Por ejemplo, se ha sugerido que la congelación y descongelación muestras de proteínas varias veces puede dar lugar a la desnaturalización. Con el fin de explorar la reproducibilidad del análisis EMSA en el contexto de estos ciclos de "congelación-descongelación", dos 35 oligos pb que difier...
Discusión
A pesar de los avances en las tecnologías de secuenciación y genotipado han mejorado enormemente nuestra capacidad para identificar variantes genéticas asociadas con la enfermedad, nuestra capacidad para comprender los mecanismos funcionales afectadas por estas variantes se está quedando. Una fuente importante del problema es que muchas variantes asociados a la enfermedad se encuentran en n en la codificación de las regiones del genoma, lo que probablemente afectará más difícil de predecir mecanismos que c...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
Materiales
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
Referencias
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados