
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Screening per Funzionale non codificante Varianti genetiche Utilizzando Electrophoretic Mobility Shift Assay (EMSA) e DNA-affinità precipitazioni Assay (DAPA)
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
Abstract
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
Introduzione
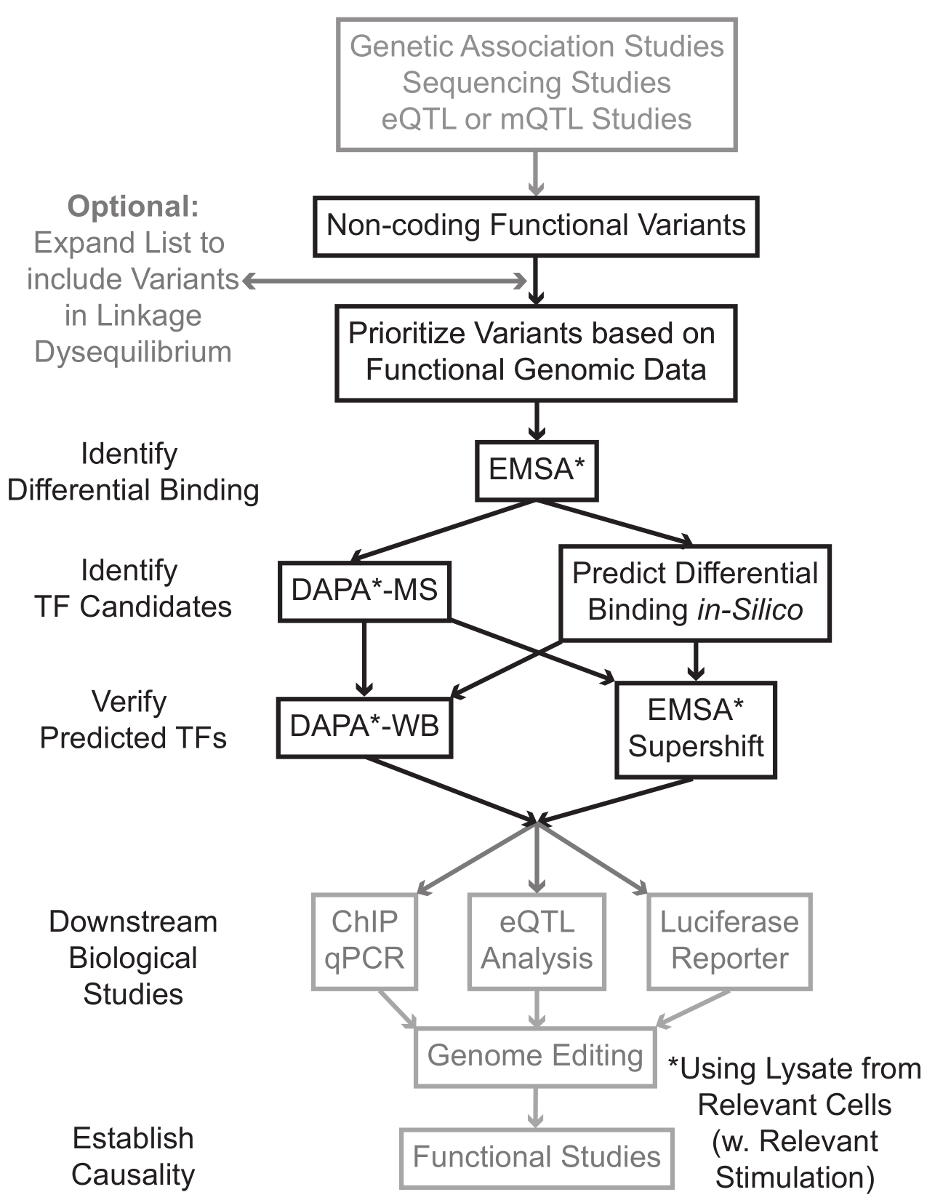
Sequencing e studi di genotipizzazione basato, compresi gli studi Genome-Wide Association (GWAS), studi locus candidato, e profondo-sequenziamento studi, hanno identificato molte varianti genetiche che sono statisticamente associati con una malattia, caratteristica, o fenotipo. Contrariamente alle previsioni iniziali, la maggior parte di queste varianti (85-93%) si trovano in regioni non codificanti e non cambiano la sequenza di amminoacidi delle proteine 1,2. Interpretando la funzione di queste varianti non codificanti e determinare i meccanismi biologici che li collegano alla malattia associata, tratto, o fenotipo è dimostrato impegnativo 3-6. Abbiamo sviluppato una strategia generale di identificare i meccanismi molecolari che collegano le varianti di un importante fenotipo intermedio - l'espressione genica. Questo gasdotto è specificamente progettato per identificare la modulazione di legare da varianti genetiche TF. Questa strategia combina approcci computazionali e tecniche di biologia molecolare finalizzate a prevedereeffetti biologici delle varianti candidati in silico e verificare empiricamente queste previsioni (Figura 1).

Figura 1:.. Approccio strategico per l'analisi dei passaggi non codificanti varianti genetiche che non sono inclusi nel protocollo dettagliato associato a questo manoscritto sono di colore grigio Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
In molti casi, è importante iniziare ampliare l'elenco delle varianti di includere tutti quelli in alta linkage disequilibrium (LD) con ciascuna variante statisticamente associato. LD è una misura di associazione non casuale di alleli in due differenti posizioni cromosomiche, che può essere misurata dal r 2 statistica 7. r 2 è una misura del linKage squilibrio fra due varianti, con una R 2 = 1 denota perfetto collegamento tra due varianti. Alleli in alta LD si trovano a co-separare sul cromosoma attraverso popolazioni ancestrali. array di genotipizzazione attuali non includono tutte le varianti conosciute nel genoma umano. Invece, sfruttano il LD all'interno del genoma umano e comprendono un sottoinsieme delle varianti note che agiscono come proxy per altre varianti di una particolare regione del LD 8. Così, una variante senza alcuna conseguenza biologica può essere associato ad una particolare malattia perché è in LD con causale variant-variante con un effetto biologico significativo. Procedurale, si consiglia di convertire l'ultima versione dei 1.000 genomi Project 9 file di chiamata variante (VCF) in file binari compatibili con PLINK 10,11, uno strumento open-source per l'intera analisi di associazione genoma. In seguito, tutte le altre varianti genetiche con LD r 2> 0.8 con ogni ingresso va geneticariant può essere identificato come candidati. E 'importante usare la popolazione di riferimento appropriato per questo passo- esempio, se una variante è stato identificato nei soggetti di origine europea, i dati provenienti da soggetti di origini simili dovrebbero essere utilizzati per l'espansione LD.
espansione LD si traduce spesso in decine di varianti candidati, ed è probabile che solo una piccola frazione di queste contribuiscono alla malattia meccanismo. Spesso, è praticamente impossibile esaminare sperimentalmente ciascuna di queste varianti singolarmente. E 'quindi utile per sfruttare le migliaia di pubblicamente disponibili i set di dati di genomica funzionale come filtro per dare priorità alle varianti. Ad esempio, il consorzio ENCODE 12 ha eseguito migliaia di esperimenti di ChIP-Seq che descrivono il legame del TF e co-fattori, e segni istoni in una vasta gamma di contesti, insieme ai dati della cromatina di accessibilità da tecnologie come DNasi-ss 13, ATAC -seq 14, e FAIRE-ss 15. databAsi e server web, come il browser UCSC Genome 16, tabella di marcia Epigenomics 17, Blueprint Epigenome 18, Cistrome 19, e Remap 20 fornire libero accesso ai dati prodotti da questi e altre tecniche sperimentali in una vasta gamma di tipi e condizioni di cellule. Quando ci sono troppe varianti per esaminare sperimentalmente, questi dati possono essere utilizzati per dare la priorità quelli situati all'interno probabili regioni regolatorie in materia tipi di cellule e tessuti. Inoltre, nei casi in cui una variante è all'interno di un picco ChIP-seq per una proteina specifica, questi dati possono fornire cavi potenziali per il TF specifico (s) o cofattori cui legame potrebbe essere che interessano.
Successivamente, le varianti risultanti priorità sono proiettati sperimentale per validare proteina genotipo-dipendente predetto legame con EMSA 21,22. EMSA misura la variazione della migrazione del oligo su un gel TBE non riducente. oligo fluorescente è incubato con illisato nucleare e legame di fattori nucleari ritardare il movimento del oligo sul gel. In questo modo, oligo che ha legato più fattori nucleari presenterà come un segnale fluorescente forte alla scansione. In particolare, l'EMSA non richiede previsioni circa le proteine specifiche il cui legame saranno interessati.
Una volta che le varianti sono identificati che si trovano all'interno di regioni regolatorie previsti e sono in grado di fattori nucleari differenziale vincolanti, metodi computazionali sono impiegati per predire il TF specifico (s), il cui legame che potrebbero influenzare. Noi preferiamo usare CIS-BP 23,24, RegulomeDB 25, UNIProbe 26, e JASPAR 27. Una volta che il candidato TF sono identificate, queste previsioni possono essere specificamente testati utilizzando anticorpi contro questi TF (EMSA-supershifts e DAPA-western). Un EMSA-supershift comporta l'aggiunta di un anticorpo specifico TF al lisato nucleare e oligo. Un risultato positivo in un EMSA-supershift è represented come un ulteriore spostamento nella banda EMSA, o una perdita di banda (valutata in riferimento 28). Nel DAPA complementare, un duplex oligo 5'-biotinilato contenente la variante e il 20 paia di basi di accompagnamento nucleotidi sono incubati con lisato nucleare dal tipo di cellula in questione (s) per catturare eventuali fattori nucleari legame specifico gli oligonucleotidi. Il complesso fattore duplex-nucleare oligo è immobilizzato dalla streptavidina microsfere in una colonna magnetica. I fattori nucleari legati vengono raccolti direttamente tramite eluizione 29,48. previsioni vincolanti possono poi essere valutati da un Western blot utilizzando anticorpi specifici per la proteina. Nei casi in cui non ci sono previsioni evidenti, o anche molte previsioni, i eluizioni dalla variante di pull-down degli esperimenti DAPA possono essere inviati a un nucleo di proteomica per identificare TF candidati mediante spettrometria di massa, che possono in seguito essere convalidati utilizzando questi precedentemente descritte metodi.
Nel resto della articlE, viene fornito il protocollo dettagliato per EMSA e DAPA analisi delle varianti genetiche.
Protocollo
1. preparazione di soluzioni e reagenti
- Ordinare sonde oligonucleotidiche DNA personalizzati per l'utilizzo in EMSA e DAPA.
- Per ridurre la proteina non specifica vincolante, progettare oligos brevi (tra i 35-45 coppie di basi (bp) di lunghezza) 30, e inserire la variante di interesse direttamente al centro affiancato da suo 17 bp endogena sequenza genomica. Per oligo EMSA, aggiungere un 'fluoroforo 5. Per oligo DAPA, aggiungere un tag 5 'biotina.
- Ordinare sia il filamento senso e il suo filone complemento inverso. In alternativa, l'ordine duplex (pre-ricotto) oligonucleotidi. Quando si nomina la oligo, basare la nomenclatura su un genoma di riferimento stabilito.
Nota: "Rischio" e la designazione "non a rischio" può essere la malattia e specifico progetto, mentre "di riferimento" e "non-reference" sono più universalmente rilevanti. - All'arrivo degli oligonucleotidi, far girare brevemente verso il basso i contenuti e risospendere in acqua priva di nucleasi ad una concentrazione finale di 100 μ; M. Conservare risospeso magazzino a -20 ° C. Proteggere oligo contrassegnati con un fluoroforo dalla luce avvolgendo con un foglio di alluminio.
| Nome | Sequenza |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG A GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
Tabella 1: disegno oligonucleotide Esempio EMSA / DAPA per testare un SNP per bin differenzialeding. "REF" sta per l'allele di riferimento, mentre "nonref" sta per l'allele non di riferimento. "PER" sta per il filo in avanti, mentre "REV" indica il suo complemento. Il SNP è visto in rosso.
- Preparare estrazione citoplasmatica (CE) tampone con una concentrazione finale di 10 mM HEPES (pH 7,9), 10 mM KCl, e 0,1 mM EDTA in acqua deionizzata.
- Preparare estrazione nucleare (NE) tampone con una concentrazione finale di 20 mM HEPES (pH 7,9), 0,4 M NaCl, e 1 mM EDTA in acqua deionizzata.
- Preparare tampone di ricottura con una concentrazione finale di 10 mM Tris (pH 7.5-8.0), 50 mM NaCl, e 1 mM EDTA in acqua deionizzata.
2. Preparazione di lisato nucleare da cellule in coltura
Nota: Questo protocollo sperimentale è stato ottimizzato utilizzando linee cellulari B-linfoblastoidi, ma è stato testato in diverse altre linee di cellule aderenti / sospensioni indipendenti e lavora universalmente.
- culturi cellule B-linfoblastoidi a Roswell Park Memorial Institute (RPMI) 1640 con 2 mM L-glutammina, siero fetale bovino 10%, e 1x antibiotico-antimicotico contenente 100 unità / ml di penicillina, 100 ug / ml di streptomicina e 250 ng / ml di amfotericina B.
- Seed ad una gamma di 200,000-500,000 vitali cellule / ml e incubare beute a 37 ° C con 5% di anidride carbonica in posizione verticale con tappi ventilati o allentati.
Nota: La crescita delle cellule B-linfoblastoidi rallenta quando raggiungono oltre 1.000.000 cellule / ml. Break up esplosioni delle cellule pipettando su e giù parecchie volte e tornare alle cellule di 200,000-500,000 cellule / ml a mantenere un rapido tasso di crescita.
- Seed ad una gamma di 200,000-500,000 vitali cellule / ml e incubare beute a 37 ° C con 5% di anidride carbonica in posizione verticale con tappi ventilati o allentati.
- Lavare le cellule in coltura due volte con 10 ml di ghiaccio freddo fosfato salina tamponata (PBS), centrifugare a 4 ° C, 300 xg per 5 min e rimuovere PBS tramite aspirazione.
- Contare le cellule utilizzando un emocitometro e risospendere pellet in 1 ml di PBS freddo ghiaccio per 10 7 cellule.
Nota: Ad esempio, se lisi 2 x 10 7 </ Sup> celle, risospendere in 2 ml di PBS. - Aliquota 1 ml a 1,5 ml provette da microcentrifuga in modo tale che ogni tubo contiene 10 7 cellule in PBS. Centrifugare a 3.300 xg per 2 minuti a 4 ° C e aspirare fuori PBS.
- Prima dell'uso, aggiungere 1 mM ditiotreitolo (DTT), inibitore della fosfatasi 1x, e l'inibitore della proteasi 1x a un ceppo di lavoro di tampone CE. Risospendere pellet con 400 microlitri di tampone CE e incubare su ghiaccio per 15 minuti.
- Aggiungere 25 ml di 10% Nonidet P-40 e mescolare pipettando. Centrifugare a 4 ° C, velocità massima per 3 min. Decantare e scartare il surnatante.
- Prima dell'uso, aggiungere 1 mM DTT, inibitore della fosfatasi 1x e 1x inibitore della proteasi per uno stock di lavoro di tampone NE. Risospendere pellet con 30 ml di buffer di NE e mescolare nel vortex.
- Incubare a 4 ° C in un rotore tubo o in ghiaccio per 10 min. Centrifuga 3.300 xg per 2 minuti a 4 ° C.
- Raccogliere il surnatante chiaro (lisato nucleare) e un'aliquota prima di riporlo a -806; C per evitare più cicli di gelo-disgelo che possono degradare la proteina. Lascia un aliquota di 10 microlitri per misurare la concentrazione di proteine usando il saggio di acido bichoninic (BCA) 31.
3. elettroforetica Mobility Shift Assay (EMSA)
- Preparare lavoro Oligo & Stock EMSA Gel.
- Se oligo sono state ordinate in duplex, scongelare il magazzino 100 micron e diluire 1: 2.000 in tampone ricottura per ottenere un titolo di lavoro 50 nm.
- Se oligonucleotidi sono state ordinate a singolo filamento, scongelare le scorte 100 micron e diluire 1:10 in tampone di ricottura per ottenere 100 titoli di lavoro nm. Combinare 100 microlitri della soluzione di complemento strand 100 nM con l'altro in una provetta.
- Mettere in un blocco di calore a 95 ° C per 5 min. Spegnere il blocco di calore e permettono oligonucleotidi raffreddare lentamente fino a temperatura ambiente per almeno un'ora prima dell'uso.
- Pre-eseguire il Gel EMSA.
- Rimuovere il vetrino da un pre-fusione 6% TBE gel e sciacquati con acqua deionizzata parecchie volte per rimuovere qualsiasi tampone dai pozzetti. Preparare 1 L di tampone 0,5x TBE con l'aggiunta di 50 ml di 10x TBE a 950 ml di acqua deionizzata.
- Montare l'apparecchiatura elettroforesi su gel e verificare eventuali perdite riempiendo la camera interna con tampone TBE 0.5x. Se il tampone non fuoriesce nella camera esterna, riempire la camera esterna circa due terzi della strada.
- Pre-eseguire il gel a 100 V per 60 min.
- Lavare ogni pozzetto con 200 ml di tampone 0,5x TBE.
- Preparare Binding Buffer Master Mix.
- Preparare 10x tampone di legame con una concentrazione finale di 100 mM Tris, 500 mM KCl, 10 mM DTT; pH 7,5 in acqua deionizzata.
- In una provetta, creare un master mix costituito dai reagenti comuni a tutte le reazioni (10 microlitri 10x tampone legante, 10 microlitri DTT / polisorbato, 5 microlitri Poly d (IC), e 2,5 ml di DNA di sperma di salmone; Tabella 2). Preparare un ulteriore 10%per tenere conto di perdita di volume a causa di pipettaggio.
| Reagente | Conc finale. | RXN 1 # | RXN 2 # | RXN 3 # | RXN 4 # |
| acqua ultrapura | a 20 microlitri vol. | 13,5 ml | 11.98 ml | 13.5μl | 11.98 ml |
| 10x Binding Buffer | 1x | 2 ml | 2 ml | 2 ml | 2 ml |
| DTT / TW-20 | 1x | 2 ml | 2 ml | 2 ml | 2 ml |
| DNA sperma di salmone | 500 ng / ml | 0,5 ml | 0,5 ml | 0,5 ml | 0,5 ml |
| 1 ug / ml poli d (IC) | 1 mcg | 1 ml | 1 ml | 1 ml | 1 ml |
| Estratto nucleare (5,26 ug / ml) | 8 mcg | - | 1,52 ml | - | 1,52 ml |
| NE Buffer | 1,52 ml | - | 1,52 ml | - | |
| Riferimento allele oligo | 50 fmol | 1 ml | 1 ml | - | - |
| Non Riferimento allele oligo | 50 fmol | - | - | 1 ml | 1 ml |
Tabella 2: Esempio EMSA reazione Setup. La tabella illustra un esempio EMSA per verificare l'ipotesi che non ci sia il genotipo-dipendente legame di TFS a uno specifico SNP.
- Aggiungere acqua priva di nucleasi a ciascuna provetta tale che il volume finale seguente aggiunta di tutti i reagenti sarà di 20 microlitri.
- Aggiungere la quantità appropriata (5.5 ml) di master mix per ogni provetta.
- Aggiungere 8 mg di lisato nucleare per le provette da microcentrifuga appropriate. Includere provette contenenti il oligo senza estratto nucleare come controlli negativi (ad esempio, tabella 2, Rxn # 1 e # Rxn 3).
Nota: La quantità ottimale di lisato per reazione deve essere determinato sperimentalmente mediante titolazione. Generalmente, titolando un intervallo di 2-10 mg di lisato è sufficiente. - Aggiungere 50 fmol di oligo ai tubi microcentrifuga appropriate. Flick per mescolare e brevemente girare il contenuto sul fondo della provetta. Incubare per 20 minuti a temperatura ambiente.
Nota: Se attempting un supershift, incubare la miscela lisato con l'anticorpo per 20 minuti a temperatura ambiente prima dell'aggiunta di oligo. Si consiglia di utilizzare 1 mg di un anticorpo ChIP-grade per i migliori risultati. - Aggiungere 2 ml di 10x Arancione colorante di caricamento per ogni provetta. Pipetta su e giù per mescolare.
- Caricare i campioni nella pre-run gel TBE 6% pipettando su e giù per mescolare e poi espellendo ogni campione in un pozzo separato. Attivare il gel a 80 V fino a quando il colorante arancione è migrata 2/3 a 3/4 fino in fondo il gel. Questo dovrebbe prendere circa 60-75 min.
- Rimuovere il gel dalla cassetta di plastica facendo leva con un coltello gel e posizionare il gel in un contenitore con tampone TBE 0,5% per evitare che si secchi.
- Porre il gel sulla superficie di un sistema di imaging a infrarossi e chemiluminescenza, avendo cura di eliminare eventuali bolle o contaminanti che disturbare l'immagine.
- Utilizzando il software di sistema di scansione, fare clic sulla scheda "Acquisisci"quindi selezionare "Draw Nuovo" per disegnare una casella intorno all'area corrispondente in cui il gel si trova sulla superficie dello scanner.
- Nella sezione "canali" della scheda "Acquisisci", selezionare il canale che corrisponde alla lunghezza d'onda del tag fluoroforo sul oligo. Nella sezione "Scanner", fai clic su "Anteprima" per avere un'anteprima di scansione di bassa qualità. Regolare l'area di scansione trascinando il riquadro blu che circonda l'anteprima immagine acquisita verso la porzione del gel da acquisire.
Nota: Per esempio, se si utilizza oligonucleotidi marcati con un fluoroforo nm 700, assicurarsi che il canale "700 nm" è selezionato prima della scansione. - Nella sezione "Controlli di scansione", selezionare l'opzione di risoluzione "84 micron" e l'opzione di qualità "Medium". Impostare l'attivo a compensare alla metà dello spessore del gel. Nota: Ad esempio, un gel 1 millimetro userebbe un offset fuoco 0,5 mm.
- Nella sezione "Scanner", cliccare su "Start" per bEgin la scansione.
Nota: Durante la scansione, il regime di luminosità, contrasto e colore può essere spesso regolata manualmente a seconda del produttore del sistema di scansione. - Dopo la scansione è terminata, selezionare la scheda "Immagine" e fare clic su "ruotare o capovolgere" nella sezione "Crea" per correggere l'orientamento. Salvare il file immagine facendo clic su "Esporta" nel menu principale, quindi selezionare "Visualizzazione immagine singola".
4. DNA Affinity Purification Assay (DAPA)
- Preparazione di 5 micron Oligo funzionamento Stock.
- Se oligo sono state ordinate in duplex, scongelare il magazzino 100 micron e diluire 1:20 buffer di ricottura per ottenere un titolo di lavoro 5 micron.
- Se oligonucleotidi sono state ordinate a singolo filamento, scongelare le scorte 100 micron e diluire 1:10 in tampone di ricottura per ottenere 10 micron scorte di lavoro. Unire 10 ml di 10 mM filamenti complementari tra loro. Mettere in un blocco di calore a 95 ° C per5 minuti. Spegnere il blocco termico e permettere ai oligonucleotidi raffreddare lentamente fino a temperatura ambiente prima dell'uso.
- Prima di iniziare, riscaldare il tampone di legame, basso tampone di lavaggio rigore, tampone di lavaggio ad alta rigore, e tampone di eluizione a temperatura ambiente.
Nota: Una concentrazione finale di 50 ng / mL Poly d (IC) può essere aggiunto al buffer vincolante, bassa stringenza tampone di lavaggio e tampone di lavaggio di alta stringenza per ridurre il potenziale di legame non specifico di proteine agli oligo. - Preparare le miscele vincolanti per ogni variante.
- Mescolare 1 volume di lisato nucleare con 2 volumi di tampone di legame.
Nota: la quantità necessaria di lisato deve essere determinato in via sperimentale a causa di variazione abbondanza di TF. Usando tra 100-250 ug di lisato nucleare per colonna è sufficiente nella maggior parte dei casi. - Aggiungere inibitore 1x fosfatasi, inibitore della proteasi 1x e 1x enhancer binding (opzionale) e mescolare muovendo il tubo più volte.
Nota: 100x vincolanteenhancer è costituito da 750 mm MgCl 2 e 300mm ZnCl 2. Aggiungere enhancer binding se il legame di TF di DNA dipende cofattori o agenti riducenti. Se questa informazione non è nota, aggiungere il enhancer binding. - Aggiungere 10 ml di 5 micron DNA cattura biotinilato (50 pmol) per ciascuna rispettiva miscela vincolante. Incubare per 20 minuti a temperatura ambiente.
Nota: il tempo di incubazione e la temperatura possono variare a seconda del TF. I valori ottimali devono essere determinati sperimentalmente.
- Mescolare 1 volume di lisato nucleare con 2 volumi di tampone di legame.
- Aggiungere 100 ml di microsfere streptavidina. Incubare per 10 minuti a temperatura ambiente.
- Per ogni sonda oligo in fase di test, inserire una colonna vincolante nel separatore magnetico. Mettere una provetta direttamente sotto ogni colonna vincolante e applicare 100 ml di tampone di legame per lavare la colonna.
- Pipettare il contenuto di ciascuna miscela vincolante in colonne separate e consentire al liquido di fluire completamente attraverso la colonna in microcentrifugatubo prima di procedere. Assicurarsi di etichettare le colonne con l'oligo variante che è stato utilizzato nella miscela vincolante. Etichettare i campioni a flusso continuo e sostituirli con nuovi tubi microcentrifuga per raccogliere i lavaggi a bassa stringenza.
- Applicare 100 ml di bassa severità tampone di lavaggio alla colonna; attendere il serbatoio colonna è vuota. Ripetere il lavaggio 4x. Etichettare i campioni di lavaggio a bassa stringenza e sostituirli con nuovi tubi microcentrifuga per raccogliere i lavaggi ad alta stringenza.
- Applicare 100 ml di alta rigore tampone di lavaggio alla colonna; attendere il serbatoio colonna è vuota. Ripetere il lavaggio 4x. Etichettare i campioni di lavaggio ad alta stringenza e sostituirli con nuovi tubi microcentrifuga per raccogliere il pre-eluizione.
- Aggiungere 30 ml di tampone di eluizione nativo alla colonna e lasciar riposare per 5 minuti. Etichettare i campioni pre-eluizione e sostituirli con nuovi tubi microcentrifuga per raccogliere l'eluizione.
Nota: Questo non eluire le proteine; lava il restante alta rigoretampone della colonna e lo sostituisce con tampone di eluizione per massimizzare l'efficienza del eluizione. - Aggiungere un 50 microlitri di buffer aggiuntivo di eluizione nativo per eluire le TF legati. Per un rendimento superiore, ma eluato meno concentrata, aggiungere altri 50 ml di tampone di eluizione nativo e raccogliere il flusso continuo.
Nota: Analizzare i campioni di eluizione attraverso la spettrometria di massa per determinare l'identità del legato TF 32. In seguito, verificare i risultati di proteomica attraverso sodio dodecil solfito di gel di poliacrilammide (SDS-PAGE) seguita da una macchia occidentale 33. Se spettrometria di massa non è disponibile, eseguire una macchia d'argento con la tecnica standard invece di un Western blot per determinare le dimensioni della proteina (s) che mostra genotipo-dipendente vincolante. Utilizzare queste informazioni per ridurre l'elenco dei TF previsti dagli approcci computazionali dettagliati nell'introduzione.
Risultati
In questa sezione, i risultati rappresentativi di cosa aspettarsi sono forniti durante l'esecuzione di un EMSA o DAPA, e la variabilità per quanto riguarda la qualità del lisato è caratterizzata. Ad esempio, è stato suggerito che il congelamento e scongelamento campioni proteici più volte può causare denaturazione. Al fine di esplorare la riproducibilità delle analisi EMSA nel contesto di questi cicli "gelo-disgelo", due 35 oligo bp diverse in una sola variante genet...
Discussione
Nonostante i progressi nelle tecnologie di sequenziamento e genotipizzazione hanno notevolmente migliorato la nostra capacità di individuare le varianti genetiche associate alla malattia, la nostra capacità di comprendere i meccanismi funzionali colpite da queste varianti è in ritardo. Una fonte importante del problema è che molte varianti associate alla malattia sono situati in n on-regioni codificanti del genoma, che probabilmente incidere più difficile da prevedere meccanismi che controllano l'espressi...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
Materiali
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
Riferimenti
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati