
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Triagem para Funcional não-codificante variantes genéticas Usando eletroforética Mobilidade Mudança Assay (EMSA) e DNA afinidade Precipitation Assay (DAPA)
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
We present a strategic plan and protocol for identifying non-coding genetic variants affecting transcription factor (TF) DNA binding. A detailed experimental protocol is provided for electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genotype-dependent TF DNA binding.
Resumo
Population and family-based genetic studies typically result in the identification of genetic variants that are statistically associated with a clinical disease or phenotype. For many diseases and traits, most variants are non-coding, and are thus likely to act by impacting subtle, comparatively hard to predict mechanisms controlling gene expression. Here, we describe a general strategic approach to prioritize non-coding variants, and screen them for their function. This approach involves computational prioritization using functional genomic databases followed by experimental analysis of differential binding of transcription factors (TFs) to risk and non-risk alleles. For both electrophoretic mobility shift assay (EMSA) and DNA affinity precipitation assay (DAPA) analysis of genetic variants, a synthetic DNA oligonucleotide (oligo) is used to identify factors in the nuclear lysate of disease or phenotype-relevant cells. For EMSA, the oligonucleotides with or without bound nuclear factors (often TFs) are analyzed by non-denaturing electrophoresis on a tris-borate-EDTA (TBE) polyacrylamide gel. For DAPA, the oligonucleotides are bound to a magnetic column and the nuclear factors that specifically bind the DNA sequence are eluted and analyzed through mass spectrometry or with a reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analysis. This general approach can be widely used to study the function of non-coding genetic variants associated with any disease, trait, or phenotype.
Introdução
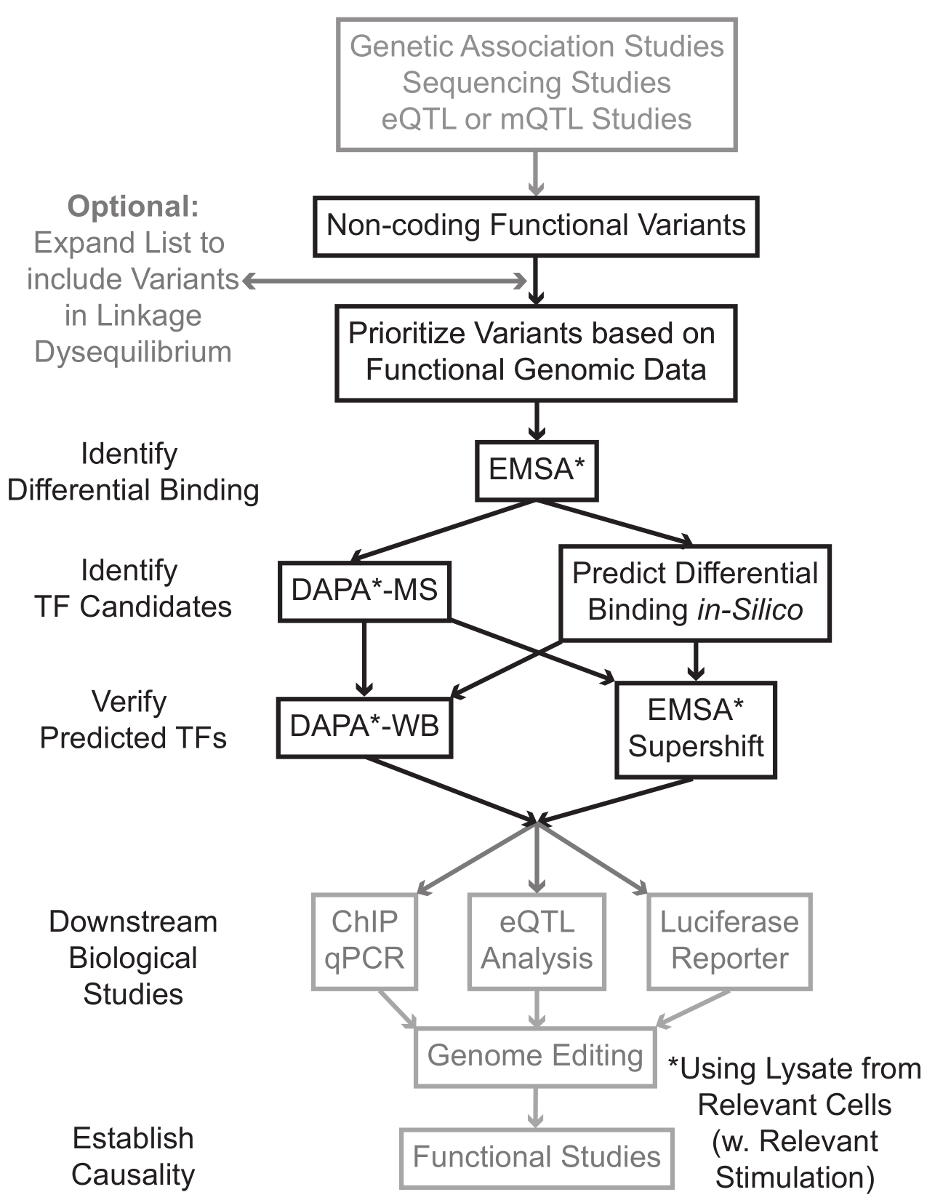
Sequenciação e estudos de genotipagem baseados, incluindo Estudos do Genoma-Wide Association (GWAS), estudos locus candidato, e deep-sequenciação estudos, identificamos muitas variantes genéticas que estão estatisticamente associados com uma doença, característica ou fenótipo. Ao contrário do que as previsões iniciais, a maior parte destas variantes (85-93%) estão localizados em regiões não codificantes e não alteram a sequência de aminoácidos de proteínas de 1,2. Interpretando a função destas variantes não-codificantes e determinar os mecanismos biológicos que ligam-los para a doença associada, traço, ou fenótipo provou desafiador 3-6. Nós desenvolvemos uma estratégia geral para identificar os mecanismos moleculares que ligam variantes para um importante intermediário fenótipo - a expressão do gene. Esse gasoduto é projetado especificamente para identificar a modulação da ligação de variantes genéticas TF. Esta estratégia combina abordagens computacionais e técnicas de biologia molecular voltadas para preverefeitos biológicos das variantes candidatos in silico e verifique estas previsões empiricamente (Figura 1).

Figura 1:.. Uma abordagem estratégica para a análise de Passos não-codificantes variantes genéticas que não estão incluídos no protocolo detalhado associado a este manuscrito estão com fundo cinza Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Em muitos casos, é importante começar pela expansão da lista de variantes para incluir todos aqueles em alta linkage-desequilíbrio (LD), com cada variante estatisticamente associada. LD é uma medida da associação não aleatória de alelos em duas posições cromossómica diferente, o que pode ser medido pela estatística de R 2 7. R2 é uma medida da linkage desequilíbrio entre duas variantes, com um r 2 = 1 denotando ligação perfeita entre duas variantes. Os alelos em alta LD são encontrados a co-segregar no cromossoma através de populações ancestrais. matrizes de genotipagem circulante não inclui todas as variantes conhecidas do genoma humano. Em vez disso, eles explorar o LD dentro do genoma humano e incluem um subconjunto das variantes conhecidas que funcionam como proxies para outras variantes dentro de uma região particular de LD 8. Assim, uma variante sem qualquer consequência biológica pode estar associada com uma doença em particular, porque é em LD com a variante a-causal variante com um efeito biológico significativo. Processualmente, recomenda-se para converter a versão mais recente dos 1.000 genomas projetar 9 arquivos de chamada variante (VCF) em arquivos binários compatíveis com Plink 10,11, uma ferramenta de código aberto para análise de associação do genoma inteiro. Posteriormente, todas as outras variantes genéticas com LD r 2> 0,8 com cada va genética de entradaRiant podem ser identificados como candidatos. É importante usar a população de referência apropriado para este passo-por exemplo, se uma variante foi identificada em indivíduos de ascendência europeia, dados de indivíduos de ascendência similar deve ser usado para a expansão LD.
LD expansão muitas vezes resulta em dezenas de variantes de candidatos, e é provável que apenas uma pequena fracção delas contribuir para mecanismo da doença. Muitas vezes, isto não é exequível para examinar experimentalmente cada uma destas variantes individualmente. Portanto, é útil para alavancar os milhares de conjuntos de dados genômicos funcionais publicamente disponíveis como um filtro para priorizar as variantes. Por exemplo, o consórcio CODIFICAR 12 realizou milhares de experiências Chip-seguintes que descrevem a ligação de TF e co-factores, e marcas de histona numa vasta variedade de contextos, juntamente com os dados de acessibilidade cromatina de tecnologias tais como a DNase-SEQ 13, ATAC -seq 14 e FAIRE-seq 15. databases e servidores web, tais como o navegador UCSC Genome 16, Roteiro Epigenomics 17, Blueprint Epigenome 18, Cistrome 19, e remapear 20 permitem o acesso gratuito aos dados produzidos por essas e outras técnicas experimentais através de uma ampla gama de tipos e condições celulares. Quando existem muitas variantes para examinar experimentalmente, estes dados podem ser utilizadas para hierarquizar os localizados dentro das regiões reguladoras prováveis em tipos de células e de tecidos relevantes. Além disso, nos casos em que uma variante está dentro de um pico ChIP-seq para uma proteína específica, esses dados podem fornecer potenciais clientes em potencial quanto à TF específico (s) ou co-fatores cuja ligação pode estar afetando.
Em seguida, as variantes resultantes priorizadas são testadas experimentalmente para validar proteína dependente do genótipo previu ligação usando EMSA 21,22. EMSA mede a mudança na migração do oligo num gel de TBE não redutoras. oligo marcado com fluorescência é incubada com olisado nuclear, e ligação de factores nucleares irá retardar o movimento do oligo no gel. Desta maneira, oligo que se tenha ligado mais factores nucleares vai apresentar-se como um sinal de fluorescência mais forte sobre a digitalização. Notavelmente, a EMSA não requer previsões sobre as proteínas específicas cuja ligação será afetado.
Uma vez que as variantes são identificados que estão localizados dentro das regiões reguladoras previstos e são capazes de factores nucleares de ligação diferencialmente, métodos computacionais são utilizados para prever o TF específico (s) cuja ligação que possa afectar. Nós preferimos usar CIS-BP 23,24, RegulomeDB 25, UNIProbe 26, e Jaspar 27. Uma vez candidato TFs são identificados, essas previsões podem ser testados especificamente utilizando anticorpos contra esses TFs (EMSA-supershifts e DAPA-Westerns). Um EMSA-supershift envolve a adição de um anticorpo específico para o TF ao lisado nuclear e oligo. Um resultado positivo em um EMSA-supershift é represented como mais um desvio na banda de EMSA, ou uma perda da banda (revisto em referência 28). No DAPA complementar, uma cadeia dupla oligo 5'-biotinilado contendo a variante e a 20 pares de bases que flanqueia nucleótidos são incubadas com lisado nuclear do tipo de célula relevante (s) para capturar quaisquer factores nucleares que se ligam especificamente os oligos. O complexo do factor nuclear-duplex oligonucleótido é imobilizado por estreptavidina micropérolas em uma coluna magnética. Os fatores nucleares ligados são recolhidos directamente através de eluição 29,48. previsões de ligação pode então ser avaliada por uma transferência de Western utilizando anticorpos específicos para a proteína. Nos casos em que não existem previsões óbvias, ou muitas previsões, as eluições de variantes menus pendentes dos experimentos DAPA podem ser enviadas para um núcleo de proteômica para identificar TFs candidatos usando espectrometria de massa, que podem posteriormente ser validados usando estes previamente descrito métodos.
No restante do articlE, o protocolo detalhado para análise EMSA e DAPA de variantes genéticas é fornecido.
Protocolo
1. Preparação de soluções e Reagentes
- Encomendar sondas de oligonucleotídeos de DNA personalizados para uso em EMSA e DAPA.
- Para reduzir a proteína de ligação não específica, conceber oligonucleótidos curtos (entre 35-45 pares de bases (pb) de comprimento) 30, e colocar a variante de interesse directamente no centro flanqueado por sua sequência genómica endógena de 17 pb. Para oligos EMSA, adicione um 'fluoróforo 5. Para oligos DAPA, adicionar uma marca 5 'biotina.
- Pedir tanto a cadeia de sentido e a sua cadeia complementar reverso. Alternativamente, duplex ordem (pré-recozido) oligos. Ao nomear os oligos, basear a nomenclatura em um genoma de referência estabelecido.
Nota: "Risk" e designação "não-risco" pode ser doença e projeto específico, enquanto que "referência" e "não-referência" são mais universalmente relevante. - Após a chegada dos oligos, girar rapidamente para baixo o conteúdo e ressuspender em água isenta de nuclease, a uma concentração final de 100 μ; H. Loja ressuspenso estoque a -20 ° C. Proteger oligos marcados com um fluoróforo da luz por envolvimento com folha de alumínio.
| Nome | Seqüência |
| rs76562819_REF_FOR | GTAATGCCTTAATGAGAGAG A GTTAGTCATCTTCTCACTTC |
| rs76562819_REF_REV | GAAGTGAGAAGATGACTAAC T CTCTCTCATTAAGGCATTAC |
| rs76562819_NONREF_FOR | GTAATGCCTTAATGAGAGAG G GTTAGTCATCTTCTCACTTC |
| rs76562819_NONREF_REV | GAAGTGAGAAGATGACTAAC C CTCTCTCATTAAGGCATTAC |
Tabela 1: Projeto oligonucleotídeo Exemplo EMSA / DAPA para testar um SNP para bin diferencialDing. "REF" representa o alelo de referência, enquanto que "NONREF" representa o alelo não referência. "PARA" representa o fio para a frente, enquanto "REV" indica o seu complemento. O SNP é visto no vermelho.
- Preparar tampão de extracção citoplásmico (EC) com uma concentração final de 10 mM de HEPES (pH 7,9), KCl 10 mM, e EDTA 0,1 mM em água desionizada.
- Prepare extracção nuclear (NE) tampão com uma concentração final de 20 mM de HEPES (pH 7,9), NaCl 0,4 M, e EDTA 1 mM em água desionizada.
- Preparar tampão de recozimento com uma concentração final de Tris 10 mM (pH 7,5-8,0), NaCl 50 mM, e EDTA 1 mM em água desionizada.
2. Preparação de Lisado nuclear de células em cultura
Nota: O protocolo experimental foi optimizado utilizando linhas de células B linfoblastóides, mas foi testado em várias outras linhas de células aderentes / suspensão independentes e funciona universalmente.
- culture células B-linfoblastóides em Roswell Park Memorial Institute (RPMI) 1640 com 2 mM de L-glutamina, soro de bovino fetal a 10%, e 1 x antibiótico-antimicótico contendo 100 unidades / ml de penicilina, 100 ug / ml de estreptomicina, e 250 ng / ml de anfotericina B.
- Semente na faixa de 200,000-500,000 células viáveis / ml e incubar frascos a 37 ° C com dióxido de carbono a 5% numa posição vertical com tampas com respiradouros ou soltas.
Nota: O crescimento de células B-linfoblastóides diminui quando atingem mais de 1.000.000 de células / mL. Quebra-se blastos de células por pipetagem para cima e para baixo várias vezes e as células para voltar 200,000-500,000 células / ml para manter uma taxa de crescimento rápido.
- Semente na faixa de 200,000-500,000 células viáveis / ml e incubar frascos a 37 ° C com dióxido de carbono a 5% numa posição vertical com tampas com respiradouros ou soltas.
- Lavar as células cultivadas duas vezes com 10 mL de gelo frio de fosfato salina tamponada (PBS), girar para baixo a 4 ° C, 300 xg durante 5 min e remover o PBS através de aspiração.
- Contagem de células usando um hemocitômetro e ressuspender pellet como 1 ml PBS gelado por 10 7 células.
Nota: Por exemplo, se a lise de 2 x 10 7 </ Sup> células, ressuspender em 2 ml PBS. - Aliquota de 1 ml para tubos de microcentrífuga de 1,5 ml de modo a que cada tubo contém 10 7 células em PBS. Centrifugar a 3300 xg durante 2 min a 4 ° C e aspirado fora PBS.
- Antes da utilização, adicionar 1 mM de ditiotreitol (DTT), inibidor de fosfatase 1x, e inibidor de protease 1x para um estoque de trabalho de tampão CE. Ressuspender o sedimento celular com 400 ul de tampão de CE e incubar em gelo durante 15 minutos.
- Adicionar 25 ul de 10% de Nonidet P-40 e misturar por pipetagem. Centrifugar a 4 ° C, velocidade máxima durante 3 minutos. Decantar e descartar o sobrenadante.
- Antes da utilização, adicionar 1 mM DTT, inibidor de fosfatase 1x, 1x e inibidor de protease a um estoque de trabalho de tampão de NE. Ressuspender o sedimento de células com 30 uL de tampão de NE e misturar por vortex.
- Incubar a 4 ° C num rotor de tubo ou em gelo durante 10 min. Centrífuga 3300 xg durante 2 min a 4 ° C.
- Recolher o sobrenadante transparente (lisado nuclear) e alíquota antes de armazenar a -806; C para evitar ciclos múltiplos de congelação-descongelação que pode degradar a proteína. Deixar uma alíquota de 10 ul para medir a concentração de proteína utilizando o ensaio de ácido bichoninic (BCA) 31.
3. eletroforética Mobilidade Mudança Assay (EMSA)
- Prepare Trabalho Oligo da EMSA e Gel.
- Se oligos foram encomendados em duplex, descongelar o estoque de 100 M e diluir 1: 2000 em tampão de recozimento para alcançar um estoque de trabalho de 50 nM.
- Se oligos foram ordenados de cadeia simples, descongelar os 100 M stocks e diluir 1:10 em tampão de emparelhamento para atingir 100 Nm stocks de trabalho. Combinar 100 ul da solução de complemento Strand 100 nm com o outro num tubo de microcentrífuga.
- Colocar num bloco de calor a 95 ° C durante 5 min. Desligue o bloco de calor e permitir que os oligos a arrefecer lentamente até à temperatura ambiente durante pelo menos uma hora antes da utilização.
- Pré-executar o Gel EMSA.
- Remover o slide a partir de um pré-molde de 6% TBE gel e lavar com água desionizada várias vezes para remover qualquer tampão dos poços. Prepare 1 litro de tampão TBE 0,5x pela adição de 50 ml de 10x TBE a 950 ml de água desionizada.
- Montar o aparelho de eletroforese em gel e verificar se há vazamentos, preenchendo a câmara interna com tampão 0,5x TBE. Se nenhum buffer vaza para a câmara exterior, encher a câmara exterior cerca de dois terços do caminho.
- Pré-correr o gel a 100 V durante 60 min.
- Lave cada poço com 200 mL de tampão 0,5x TBE.
- Prepare Binding Buffer Mix Master.
- Prepare 10x tampão de ligação, com uma concentração final de Tris 100 mM, KCl a 500 mM, DTT a 10 mM; pH 7,5 em água desionizada.
- Num tubo de microcentrifuga, criar uma mistura principal que consiste dos reagentes comuns para todas as reacções (10 uL de tampão de ligação 10x, 10 ul de DTT / polissorbato, 5 ul de poli D (IC), e ADN de esperma de salmão 2,5 ul; Tabela 2). Prepare um adicional de 10%para contabilizar perda de volume devido à pipetagem.
| Reagente | Conc final. | Rxn # 1 | Rxn # 2 | Rxn # 3 | Rxn # 4 |
| água ultrapura | a 20 ul vol. | 13,5 ul | 11,98 mL | 13.5μl | 11,98 mL |
| 10x Binding Buffer | 1x | 2 ul | 2 ul | 2 ul | 2 ul |
| DTT / TW-20 | 1x | 2 ul | 2 ul | 2 ul | 2 ul |
| DNA de esperma de salmão | 500 ng / ul | 0,5 ul | 0,5 ul | 0,5 ul | 0,5 ul |
| 1 ug / mL Poly d (IC) | 1 ug | 1 ul | 1 ul | 1 ul | 1 ul |
| Extracto nuclear (5,26 ug / uL) | 8 ug | - | 1,52 mL | - | 1,52 mL |
| Tampão NE | 1,52 mL | - | 1,52 mL | - | |
| Referência oligo alelo | 50 fmol | 1 ul | 1 ul | - | - |
| Não Referência oligo alelo | 50 fmol | - | - | 1 ul | 1 ul |
Tabela 2: Exemplo EMSA setu reaçãop. A tabela ilustra um exemplo EMSA para testar a hipótese de que existe ligação de TFS para um SNP específico dependente do genótipo.
- Adicionar água isenta de nuclease a cada tubo de microcentrífuga de tal modo que o volume final após a adição de todos os reagentes será de 20 ul.
- Adicionar a quantidade apropriada (5,5 ul) de mistura principal a cada tubo de microcentrífuga.
- Adicionar 8 ug de lisado nuclear para os tubos de microcentrífuga adequadas. Incluem tubos contendo o oligo sem extracto nuclear como controlos negativos (por exemplo, Tabela 2, Rxn # 1 e # 3 Rxn).
Nota: A quantidade óptima de lisado por reacção deve ser determinada experimentalmente por meio de titulação. Geralmente, titulando uma gama de 2-10 ug de lisado é suficiente. - Adicionar 50 fmol de oligonucleótido para os tubos de microcentrífuga adequadas. Flick e misturar brevemente girar o conteúdo para o fundo do tubo. Incubar durante 20 minutos à temperatura ambiente.
Nota: Se attempting um supershift, incubar a mistura de lisado com anticorpos durante 20 minutos à temperatura ambiente antes da adição de oligos. Recomenda-se utilizar 1 pg de um anticorpo de ChIP de grau para obter os melhores resultados. - Adicionar 2 mL de 10x laranja corante a cada tubo de microcentrífuga. Pipeta cima e para baixo para misturar.
- Coloque as amostras para o pré-run gel TBE 6% pipetando cima e para baixo para misturar e, em seguida, expulsando cada amostra em um poço separado. Submeter o gel a 80 V até que o corante laranja migrou 2/3 a 3/4 do caminho para baixo do gel. Isso deve levar cerca de 60-75 min.
- Remover o gel da gaveta de plástico, retirando-a aberta com uma faca gel e colocar o gel em um recipiente com tampão TBE 0,5% para mantê-lo de secar.
- Coloque o gel sobre a superfície de um sistema de imagem de infravermelhos e quimiluminescência, tendo a certeza de eliminar quaisquer bolhas ou contaminantes que destroem a imagem.
- Usando o software de sistema de digitalização, clique na guia "Adquirir"e, em seguida, seleccionar "desenhar Novo" para desenhar uma caixa ao redor da área correspondente para onde o gel é localizado na superfície do scanner.
- Na seção "Canais" do separador "Adquirir", selecione o canal correspondente ao comprimento de onda do tag fluoróforo na oligo. Na seção "Scanner", clique em "preview" para obter uma digitalização de visualização de baixa qualidade. Ajustar a área de digitalização arrastando a caixa azul em torno da imagem de visualização adquiridos até a porção do gel a ser trabalhada.
Nota: Por exemplo, se estiver usando oligos marcados com um fluoróforo nm 700, certifique-se o canal "700 nm" é selecionado antes da digitalização. - Na seção "Controles de digitalização", selecione a opção de resolução "84? M" e a opção de qualidade "Medium". Definir o foco para compensar a metade da espessura do gel. Nota: Por exemplo, um gel de 1 milímetro seria usar um foco 0,5 milímetros de deslocamento.
- Na seção "Scanner", clique em "Iniciar" para beGIN a varredura.
Nota: Durante a verificação, o esquema de brilho, contraste, cor e muitas vezes pode ser ajustado manualmente, dependendo do fabricante do sistema de digitalização. - Após a verificação terminar, selecione a guia "Imagem" e clique em "rodar ou inverter" na seção "Criar" para corrigir a orientação. Salve o arquivo de imagem, clicando em "Exportar" no menu principal e, em seguida, selecione "Imagem Single View".
4. ADN de afinidade Purificação Ensaio (DAPA)
- Preparação de 5? M Oligo de trabalho Stock.
- Se oligos foram encomendados em duplex, descongelar o estoque de 100 M e diluir 1:20 em tampão de emparelhamento para alcançar um estoque de trabalho 5? M.
- Se oligos foram ordenados de cadeia simples, descongelar os 100 M stocks e diluir 1:10 em tampão de emparelhamento para atingir 10 � stocks de trabalho. Combinar 10 ul dos 10 uM cadeias complementares uns com os outros. Colocar num bloco de calor a 95 ° C durante5 min. Desligue o bloco de calor e permitir que os oligos a arrefecer lentamente até à temperatura ambiente antes de usar.
- Antes de começar, aquecer o tampão de ligação, tampão de lavagem de baixo rigor, tampão de lavagem de elevado rigor, e tampão de eluição à temperatura ambiente.
Nota: A concentração final de 50 ng / ml de poli d (IC) pode ser adicionado ao tampão de ligação, tampão de lavagem de baixo rigor, e tampão de lavagem de elevado rigor para reduzir o potencial de ligação não específica de proteínas para os oligos. - Prepare as misturas de ligação para cada variante.
- Misturar um volume de lisado nuclear com 2 volumes de tampão de ligação.
Nota: A quantidade necessária de lisado deve ser determinada experimentalmente, devido à variação abundância de FT. Usando entre 100-250 mg de lisado nuclear por coluna é suficiente na maioria dos casos. - Adicionar inibidor 1x fosfatase, inibidor de protease 1x, e potenciador de ligação 1x (opcional) e misture sacudindo o tubo várias vezes.
Nota: 100X vinculativointensif icador consiste em 750 mM de MgCl2 e 300 mM de ZnCl2. Adicionar intensificador de ligação, se a ligação de TF a ADN depende da co-factores ou agentes redutores. Se esta informação não é conhecida, adicione o potenciador de ligação. - Adicionar 10 ul de ADN de captura biotinilado 5 uM (50 pmol) de cada mistura de ligação respectivo. Incubar durante 20 minutos à temperatura ambiente.
Nota: O tempo de incubação e a temperatura pode variar, dependendo do TF. Os valores óptimos necessita de ser determinado experimentalmente.
- Misturar um volume de lisado nuclear com 2 volumes de tampão de ligação.
- Adicionar 100 ul de microesferas de estreptavidina. Incubar durante 10 minutos à temperatura ambiente.
- Para cada uma das sondas de oligo ser testado, colocar uma coluna de ligação no separador magnético. Coloque um tubo de microcentrífuga directamente sob cada coluna de ligação e aplicar 100 ul de tampão de ligação para lavar a coluna.
- Pipetar o conteúdo de cada mistura de ligação em colunas separadas e permitir que o líquido a fluir completamente através da coluna para o microcentrífugatubo antes de prosseguir. Certifique-se de rotular as colunas com o oligo variante que foi usado na mistura de ligação. Rotular as amostras de escoamento e substituir por novos tubos de microcentrífuga para recolher as lavagens de baixo rigor.
- Adicionar 100 ul de tampão de lavagem de baixo rigor com a coluna; esperar até que o reservatório está vazio da coluna. Repita lavagem de 4x. Rotular as amostras de lavagem de baixo rigor e substituir por novos tubos de microcentrífuga para recolher as lavagens de elevado rigor.
- Adicionar 100 ul de tampão de lavagem de elevado rigor para a coluna; esperar até que o reservatório está vazio da coluna. Repita lavagem de 4x. Rotular as amostras de lavagem de elevado rigor e substituir por novos tubos de microcentrífuga para recolher o pré-eluição.
- Adicionar 30 ul de tampão de eluição à coluna nativa e deixar repousar durante 5 min. Rotular as amostras de pré-eluição e substituir por novos tubos de microcentrífuga para recolher a eluição.
Nota: Isto não eluir a proteína ligada; ele lava o alto rigor restantestampão da coluna e substitui-lo com tampão de eluição para maximizar a eficiência da eluição. - Adicionar um tampão de eluição nativa adicional de 50 ul para eluir os TFs vinculados. Para um rendimento mais elevado, mas menos eluato concentrado, adicionar um adicional de 50 ul de tampão de eluição nativa e recolher o fluxo de passagem.
Nota: a análise de amostras de eluição através de espectrometria de massa para determinar a identidade do TF ligado 32. Em seguida, verificar os resultados proteomic através de dodecilo de sódio sulfito de electroforese em gel de poliacrilamida (SDS-PAGE) seguida de um Western blot 33. Se espectrometria de massa não está disponível, executar uma coloração de prata utilizando a técnica padrão em vez de uma transferência de Western para determinar o tamanho da proteína (s) que mostra o genótipo-dependente de ligação. Use essas informações para diminuir a lista de TFs previstos a partir das abordagens computacionais detalhados na introdução.
Resultados
Nesta seção, os resultados representativos do que esperar são fornecidos ao executar uma EMSA ou DAPA, ea variabilidade no que diz respeito à qualidade do ligado é caracterizado. Por exemplo, tem sido sugerido que a congelação e descongelação de amostras de proteína várias vezes pode resultar na desnaturação. A fim de explorar a reprodutibilidade das análises EMSA no contexto destes ciclos de "congelamento-descongelamento", dois 35 oligos bp que diferem em uma var...
Discussão
Apesar de avanços em tecnologias de sequenciamento e genotipagem tem bastante reforçada a nossa capacidade para identificar variantes genéticas associadas à doença, a nossa capacidade de compreender os mecanismos funcionais impactados por estas variantes está atrasado. Uma das principais fontes do problema é que muitas variantes associados à doença estão localizados em n-codificante em regiões do genoma, que provavelmente afectam mais difícil de prever mecanismos que controlam a expressão do gene. Aqu...
Divulgações
Os autores não têm nada para revelar.
Agradecimentos
We thank Erin Zoller, Jessica Bene, and Lindsey Hays for input and direction in protocol development. MTW was supported in part by NIH R21 HG008186 and a Trustee Award grant from the Cincinnati Children's Hospital Research Foundation. ZHP was supported in part by T32 GM063483-13.
Materiais
| Name | Company | Catalog Number | Comments |
| Custom DNA Oligonucleotides | Integrated DNA Technologies | http://www.idtdna.com/site/order/oligoentry | |
| Potassium Chloride | Fisher Scientific | BP366-500 | KCl, for CE buffer |
| HEPES (1 M) | Fisher Scientific | 15630-080 | For CE and NE buffer |
| EDTA (0.5M), pH 8.0 | Life Technologies | R1021 | For CE, NE, and annealing buffer |
| Sodium Chloride | Fisher Scientific | BP358-1 | NaCl, for NE buffer |
| Tris-HCl (1M), pH 8.0 | Invitrogen | BP1756-100 | For annealing buffer |
| Phosphate Buffered Saline (1x) | Fisher Scientific | MT21040CM | PBS, for cell wash |
| DL-Dithiothreitol solution (1 M) | Sigma | 646563 | Reducing agent |
| Protease Inhibitor Cocktail | Thermo Scientific | 87786 | Prevents catabolism of TFs |
| Phosphatase Inhibitor Cocktail | Thermo Scientific | 78420 | Prevents dephosphorylation of TFs |
| Nonidet P-40 Substitute | IBI Scientific | IB01140 | NP-40, for nuclear extraction |
| BCA Protein Assay Kit | Thermo Scientific | 23225 | For measuring protein concentration |
| Odyssey EMSA Buffer Kit | Licor | 829-07910 | Contains all necessary EMSA buffers |
| TBE Gels, 6%, 12 Wells | Invitrogen | EC6265BOX | For EMSA |
| TBE Buffer (10x) | Thermo Scientific | B52 | For EMSA |
| FactorFinder Starting Kit | Miltenyi Biotec | 130-092-318 | Contains all necessary DAPA buffers |
| Licor Odyssey CLx | Licor | Recommended scanner for DAPA/EMSA | |
| Antibiotic-Antimycotic | Gibco | 15240-062 | Contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Fungizone® Antimycotic |
| Fetal Bovine Serum | Gibco | 26140-079 | FBS, for culture media |
| RPMI 1640 Medium | Gibco | 22400-071 | Contains L-glutamine and 25 mM HEPES |
Referências
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ward, L. D., Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 30 (11), 1095-1106 (2012).
- Paul, D. S., Soranzo, N., Beck, S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 36 (2), 191-199 (2014).
- Zhang, F., Lupski, J. R. Non-coding genetic variants in human disease. Hum Mol Genet. , (2015).
- Lee, T. I., Young, R. A. Transcriptional regulation and its misregulation in disease. Cell. 152 (6), 1237-1251 (2013).
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 9 (6), 477-485 (2008).
- Bush, W. S., Moore, J. H. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 8 (12), e1002822 (2012).
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 491 (7422), 56-65 (2012).
- Chang, C. C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
- Purcell, S., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81 (3), 559-575 (2007).
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Crawford, G. E., et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16 (1), 123-131 (2006).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10 (12), 1213-1218 (2013).
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., Lieb, J. D. FAIRE Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877-885 (2007).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 518 (7539), 317-330 (2015).
- Martens, J. H., Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica. 98 (10), 1487-1489 (2013).
- Liu, T., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12 (8), R83 (2011).
- Griffon, A., et al. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 43 (4), e27 (2015).
- Staudt, L. M., et al. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 323 (6089), 640-643 (1986).
- Singh, H., Sen, R., Baltimore, D., Sharp, P. A. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 319 (6049), 154-158 (1986).
- Weirauch, M. T., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158 (6), 1431-1443 (2014).
- Ward, L. D., Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40 (Database issue), D930-D934 (2012).
- Boyle, A. P., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22 (9), 1790-1797 (2012).
- Hume, M. A., Barrera, L. A., Gisselbrecht, S. S., Bulyk, M. L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43 (Database issue), D117-D122 (2015).
- Mathelier, A., et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42 (Database issue), 142-147 (2014).
- Smith, M. F., Delbary-Gossart, S. Electrophoretic Mobility Shift Assay (EMSA). Methods Mol Med. 50, 249-257 (2001).
- Franza, B. R., Josephs, S. F., Gilman, M. Z., Ryan, W., Clarkson, B. Characterization of cellular proteins recognizing the HIV enhancer using a microscale DNA-affinity precipitation assay. Nature. 330 (6146), 391-395 (1987).
- . BCA Protein Assay Kit: User Guide Available from: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (2014)
- Wijeratne, A. B., et al. Phosphopeptide separation using radially aligned titania nanotubes on titanium wire. ACS Appl Mater Interfaces. 7 (21), 11155-11164 (2015).
- Silva, J. M., McMahon, M. The Fastest Western in Town: A Contemporary Twist on the Classic Western Blot Analysis. J. Vis. Exp. (84), (2014).
- Lu, X., et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet. 96 (5), 731-739 (2015).
- Ramana, C. V., Chatterjee-Kishore, M., Nguyen, H., Stark, G. R. Complex roles of Stat1 in regulating gene expression. Oncogene. 19 (21), 2619-2627 (2000).
- Fillebeen, C., Wilkinson, N., Pantopoulos, K. Electrophoretic Mobility Shift Assay (EMSA) for the Study of RNA-Protein Interactions: The IRE/IRP Example. J. Vis. Exp. (94), e52230 (2014).
- Heng, T. S., Painter, M. W. Immunological Genome Project, C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 9 (10), 1091-1094 (2008).
- Wu, C., et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10 (11), R130 (2009).
- Wu, C., Macleod, I., Su, A. I. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41 (Database issue), D561-D565 (2013).
- Wang, J., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22 (9), 1798-1812 (2012).
- Holden, N. S., Tacon, C. E. Principles and problems of the electrophoretic mobility shift assay. J Pharmacol Toxicol Methods. 63 (1), 7-14 (2011).
- Xu, J., Liu, H., Park, J. S., Lan, Y., Jiang, R. Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development. 141 (7), 1442-1452 (2014).
- Yang, T. -. P., et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 26 (19), 2474-2476 (2010).
- Fort, A., et al. A liver enhancer in the fibrinogen gene cluster. Blood. 117 (1), 276-282 (2011).
- Solberg, N., Krauss, S. Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol. 977, 65-78 (2013).
- Rahman, M., et al. A repressor element in the 5'-untranslated region of human Pax5 exon 1A. Gene. 263 (1-2), 59-66 (2001).
- Mali, P., et al. RNA-Guided Human Genome Engineering via Cas9. Science. 339 (6121), 823-826 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados