Method Article
Análisis de arquitecturas de proteínas y complejos de proteína-ligando por espectrometría de masa estructural integrativo
En este artículo
Resumen
Espectrometría de masas (MS) se ha convertido en una herramienta importante para la investigación de la estructura y la dinámica de asambleas macromoleculares. Aquí, integramos los enfoques basados en la MS para interrogar la formación de complejos de proteína y de ligando.
Resumen
Las proteínas son una clase importante de biológicas macromoléculas que desempeñan muchas funciones clave en las funciones celulares, incluyendo genes, catalizar reacciones metabólicas, replicación y reparación del ADN. Por lo tanto, una comprensión detallada de estos procesos proporciona información crítica sobre cómo las células de función. Integradora estructural MS métodos ofrecen información estructural y dinámica en conjunto complejo de proteínas, conectividad compleja, estequiometría de subunidad, Oligomerización de proteínas y ligando. Avances recientes en integrante MS estructurales han permitido la caracterización de los sistemas biológicos difíciles incluyendo grandes proteínas de unión de ADN y proteínas de membrana. Este protocolo describe cómo integrar diversos datos de la MS como MS nativa y ion movilidad-espectrometría de masas (IM-MS) con simulaciones de dinámica molecular para ganar penetraciones en una DNA helicase-nucleasa reparación del complejo de la proteína. El enfoque resultante proporciona un marco para estudios detallados de ligando a otros complejos proteicos implicados en procesos biológicos importantes.
Introducción
Análisis de espectrometría de masas nativas de proteínas intactas y sus complejos se lleva a cabo mediante electrospray y nano-electrospray ionización (nESI), que preservan el plegamiento de la proteína y las interacciones no-covalentes durante el proceso de ionización1, 2. En MS nativa, la estructura de las proteínas y sus complejos son retenidos en un estado casi nativo en la fase de gas de3,4. MS nativa detecta múltiples iones de proteínas cargadas, separadas según su masa para cargar cociente (m/z) permitiendo que la masa de la proteína o proteína-ligando compleja de calcular. Esta información permite la determinación de la estequiometría de la proteína intacta, composición de la subunidad, ligando e interacción redes3,4,5,6. MS nativa tiene varias ventajas en comparación con otras técnicas tales Cristalografía de rayos x y resonancia magnética nuclear espectroscopia5. En primer lugar, nativa MS es una técnica rápida y muy sensible, que requiere sólo unos pocos microlitros (μL de 2-3) de muestra a relativamente bajas concentraciones complejo finales nm alta baja μm Rango6. En segundo lugar, puede utilizarse MS nativa para interrogar las muestras de proteína heterogénea lo que es posible analizar las proteínas y los Estados múltiples simultáneamente. En tercer lugar, MS nativa no requiere muestras de proteína a ser modificados antes del análisis por reticulación química o proteína etiquetado. Estas ventajas han hecho MS estructural una poderosa herramienta para la investigación estructural de complejos de la proteína.
MS nativa puede combinarse con movilidad de iones (IM), una técnica que mide el tiempo que un ion de proteína lleva a viajar a través de un campo eléctrico, que permite la sección colisión (CCS) a determinar. El CCS informa baja resolución estructural, que permite topología e información heterogeneidad conformacional de las proteínas para obtener. Además, permite el examen de modelos estructurales de la proteína generada por métodos computacionales.
Estabilidad de la proteína en fase gaseosa puede ser investigado utilizando colisión inducida por despliegue (CIU) medido IM-MS. Durante el proceso CIU, los iones de proteínas son acelerados y activados a través del aumento aceleradoras colisiones con un gas buffer inerte dentro de un espectrómetro de masas7,8,9. Este proceso de activación colisión causa la proteína revelar parcialmente, que se traduce en un aumento en CCS. Este cambio en CCS y la energía necesaria para desdoblar la proteína puede ser medida IM-Sra. usando este enfoque, el efecto del ligando en la estabilidad de la proteína puede ser medido10. Subcomplejos de mando pueden ser generadas en solución utilizando métodos de interrupción en solución tales como la adición de solventes orgánicos para monitor nativo topologías de complejos de la proteína. Perturbación de los complejos de la proteína es principalmente debido a la interrupción de interacciones no-covalentes intra. Los complejos secundarios mantienen nativo topologías y, tras la detección de la MS, revelan información sobre inter-subunidad conectividad.
Enfoques integrales en biología estructural combinan diversos métodos para estudiar la estructura y dinámica de las proteínas y sus complejos3,4,5,6. Nativo MS y IM-MS se han utilizado para descubrir los detalles moleculares de sistemas biológicos desafiantes. Ha habido varios ejemplos de aplicaciones, incluyendo el estudio de la proteína Asamblea vías11,12,13,14, estudio de redes de interacción proteína-proteína15 , 16 , 17, membrana proteínas6,18,19,20,21y las interacciones proteína-ligando como ácidos nucleicos22,23 ,24.
Sin embargo, MS nativa también tiene sus limitaciones. Medidas MS nativas se realizan a menudo en tampones volátiles como el acetato de amonio acuoso en el que algunas proteínas no conservará su estado nativo doblado3,25. Sin embargo, trabajos recientes han demostrado que esta limitación puede ser superada por la optimización de la rociadura diámetro de punta de aguja (puntas de 0,5 mm) que proteínas e iones complejos de proteína se pueden formar directamente de búferes no volátil con alta fuerza iónica que mejor imitar el entorno fisiológico26. Además, MS nativa utiliza electrospray para ionizar y transferir conjuntos no covalentes de solución a la fase gas; por lo tanto, la abundancia relativa de complejos detectados puede no totalmente representar en la solución5,27. Además, en comparación a en solución, las interacciones hidrofóbicas de la fase de gas se vuelven más débiles e interacciones electrostáticas se vuelven más fuertes y por lo tanto favorecieron3,28.
En este artículo, ofrecemos protocolos, análisis de datos e interpretación para la identificación de la proteína y ligando vinculante utilizando MS nativa, IM-MS, CIU, en la solución de interrupción y la modelización. El complejo de reparación del ADN, HerA-NurA, es utilizado como sistema modelo. Roturas de doble hebra de DNA (distritales) son una de las formas más citotóxicas y deletéreas de daño de la DNA, dando por resultado inestabilidad genética y el eventual desarrollo de cáncer en seres humanos. Recombinación homóloga es el mecanismo de reparación que erradica distritales, un proceso que está orquestado por el complejo, de HerA-NurA ATP dependiente helicase-nucleasa22.
Combina nativas MS y IM-MS con el análisis funcionales y modelado permitieron la investigación de: i) el papel de NurA en la Asamblea, la conformación y la estabilidad del complejo, ii) la interacción entre dsDNA y el complejo y su influencia en la general estabilidad del complejo y iii) la estequiometría y el impacto de ATP binding en el montaje de22. En general, este trabajo condujo a una mejor comprensión de la base molecular del complejo de HerA-NurA uniendo los complejos cambios conformacionales de la proteína y estabilidad con enlace nucleótido. Este protocolo es genérico para cualquier complex(es) de la proteína que interactúa con uno o varios tipos de ligand(s).
Protocolo
1. preparación para MS nativa de proteínas y complejos de proteína-ligando
Nota: Para obtener una comprensión de la base molecular de un complejo proteico y ligando utilizando MS nativas, preparación de la muestra adecuada es clave. El objetivo de esta sección es destacar los pasos de preparación esencial muestra antes del análisis de MS con el complejo de HerA-NurA que ata la DNA y los nucleotides como ejemplo.
- Preparar 20 alícuotas μl del concentrado purificado de la proteína (típicamente 15-30 μm) en un tubo de 1,5 mL.
-
Para el análisis de unión de ATP o ADP
Nota: Agregar concentraciones cada vez mayor de los no hidrolizables ATP adenosina analógica 5′-O-(3-thiotriphosphate), sal tetralithium (ATP-γ-S) o 5-difosfato de adenosina (ADP). No hidrolizables ATP generen un complejo estable que permitiría la proteína ATP-limite ser capturado. Otros análogos no hidrolizables de ATP que podrían analizarse incluyen PNP AMP y ATP-γ-S-Mg2 +.- Estudios de HerA-NurA, mezclar 5 μm de proteína purificada con ATP-γ-S y ADP en concentraciones que van desde 0-1 mM.
- Para capturar a simultáneo enlace ATP-γ-S y ADP, añadir dos nucleótidos en las misma o diferentes concentraciones.
- Añadir 2 mM de MgCl2 e incubar a 25 ° C en una incubadora de baño seco para 1 h.
Nota: Análisis de unión de nucleótidos mediante nESI que MS nativa puede causar atascamiento artefactual en altas concentraciones, por lo tanto fijación no específica debe tenerse en cuenta29. Para investigar el atascamiento no específico, añadir una mayor concentración de nucleótidos de entre 2-5 mM).
-
Para el análisis de ADN vinculante
- Mezclar la proteína y el ADN en una proporción molar que permite la formación de complejos proteína ADN. Para HerA y HerA-NurA, mezcle 5 μm de proteico purificado con el ADN en una proporción 1:1.
- Incubar la HerA-NurA o HerA - mezcla de ADN durante 30 min a 25 ° C en una incubadora de baño seco hasta que alcanza el equilibrio. La duración y la temperatura de incubación pueden variar dependiendo de la proteína bajo investigación.
- Búfer intercambio proteína muestras MS compatible con buffers. Comúnmente, se utilizan solución de acetato de amonio acuoso entre 5 mM-1 M a pH 7-8. Otros búferes compatibles MS incluyen etilendiamonio diacetato (EDDA) y Trietilamonio acetato (TEAA)30. Estudios de HerA-NurA, use 200 mM amonio acetato pH 7.
Nota: Existen varios métodos para cambio de tampón antes del análisis por MS como el uso de un concentrador centrifugado o columnas de cromatografía. MS nativa está limitada sobre todo por la calidad de la muestra tales como tampones y aductores utilizados durante la purificación. Por lo tanto, es imprescindible realizar la desalación suficiente para obtener picos resueltos. - Para estudios de unión de ligando HerA-NurA, tampón intercambio muestras de 6 a 8 veces en acetato de amonio 200 mM utilizando un concentrador. Aunque este método es más lento, asegura que resolvió se alcanzan picos y permite la determinación de masa exacta de especie dependiente de ATP/ADP.
2. nativos de MS de adquisición y análisis para la investigación de complejos de proteínas y complejos de proteína-ligando
Nota: MS condiciones deberían optimizarse para alcanzar picos de alta resolución para las medidas exactas de la masa. Los datos de esta sección optimizan los parámetros en un espectrómetro de masas de Q-ToF con un quadrupole de m/z 32 k límite superior.
- Preparar los capilares internos para nano-electrospray y realizar la calibración masa instrumento para las medidas exactas de masas según lo detallado por Kirshenbaum et al. 1.
- Seleccione movilidad TOF modos, adquisición de iones positivos y sensibilidad.
- Encender los gases trampa, API y IMS. Para la separación de la IM, use nitrógeno (60 mL/min) y argón (8,4 mL/min para la región de la trampa) como puntos de partida y ajuste.
- Coloque una adquisición adecuada de m/z. Para una proteína desconocida, pasos de optimización inicial deben utilizar una amplia gama como 32.000 500 m/z.
- Carga 2-3 μl de la solución compleja de proteínas a analizar en un tubo capilar recubierto oro e insertarla en un soporte de tubo capilar.
- Suavemente apriete el tubo capilar y coloque el capilar en la etapa de fuente electrospray y deslice la etapa en posición para empezar a adquirir datos.
- Aplique una presión de gas de nano-flujo bajo (0.00-0.05 Bar) hasta que una gota se forma en el extremo del tubo capilar. Nano-presión puede descartarse hasta que el chorro se mantiene.
- Ajustar el tubo capilar en relación con el cono moviendo el tubo capilar en x, y, z las posiciones y controlar el ion actual para lograr un ion estable actual. Aplicar una tensión capilar en el rango de 0.9-1.6 kV.
- Ajuste el cono de muestreo (50-120 V), offset fuente (60.0), temperatura (25 ° C) de la fuente y caudal de gas de cono (0.0 L/h). Éstos sugirieron las condiciones iniciales se pueden ajustar.
- Para la adquisición de un espectro de masa bien resuelto y para maximizar la transmisión de iones, ajustar parámetros de MS y monitorear el cambio resultante en los espectros. Se trata de ajustar el caudal de gas de la trampa (2-8 mL/min), él celular (180 mL/min) y célula IMS (90 mL/min) para lograr la mejor separación de transmisión máxima.
- Ajustar las energías de colisión de trampa si desvíos de tensión no son suficientes. Un punto de partida óptimo es entre 10-50 V.
Nota: Aumentar la energía de la trampa puede quitar no-covalentemente aductos. Sin embargo, tenga cuidado para evitar la disociación inducida por colisión y despliegue del complejo proteína-ligando. Realizar mediciones de movilidad de iones para comprobar si las condiciones del instrumento mantienen la proteína en el estado doblado nativo (paso 3). - Mejorar desolvation optimizando el voltaje de bias de trampa. Un óptimo punto de partida es el V 20-45.
- Optimizar la velocidad de las ondas y la altura de ola para lograr la mejor separación de movilidad. Una explicación detallada y protocolo pueden encontrarse aquí31. Para los estudios de HerA-NurA, usar la velocidad de las ondas de 40 (m/s) y altura de onda de 550-650 (V).
- Utilizar otros parámetros como valores por defecto de instrumento.
- Preparar una muestra libre de ligando para el análisis como un control para cada serie (figura 1). Para los experimentos de unión de ligando, realizar al menos tres mediciones independientes.
- Utilice el software Masslynx para medir masas de especies generadas e identificar el atascamiento del ligand, como enlace de ATP y ADP y los Estados (figura 2 y 3). Otros software disponible incluyen UniDec32, PULSAR33 y34de Anfitrite.
- Para cuantificar la abundancia relativa de especies, utilizar las correspondientes intensidades de iones observadas en el espectro ESI-MS crudo (por ejemplo límite del ligando diferentes oligómeros, etcetera). Como alternativa, realizar cuantificación usando software especializado como UniDec y Massign35 (figura 1 y 3 ).
3. adquisición y análisis de IM-MS
Nota: IM-MS separa los iones en la fase de gas basado en su tamaño (masa), la forma y la carga. Cada característica resuelto en espectro de m/z se asocia una distribución del tiempo de deriva. IM-MS mide el tiempo de la deriva de un ion que puede ser utilizado para calcular la sección de colisión (CCS). Valores de tiempo deriva medición desde IM-MS datos adquiridos mediante que un tubo de la deriva puede ser linealmente correlacionado con CCS valores36. Para viajar las medidas IM-MS (TWIMS) de la onda, calculando valores CCS requiere una curva de calibración obtenida a partir de estándares de proteína con el conocido CCS valores37. Estructuras compactas viajan más rápido que extendido o reducción de estructuras alargadas debido a interacciones con el gas de búfer en la movilidad celular38. Por lo tanto, IM-MS puede usarse para detectar si la estructura nativa plegada se ha mantenido en la fase de gas39,40. Esta sección describe cómo medir IM-MS y calcular la CCS de proteína TWIMS.
- Después de optimizar las condiciones del instrumento de transmisión estable (paso 2), reducir la energía de colisión y el cono de muestreo tan baja como sea posible manteniendo calidad buena espectros.
- Utilice la velocidad de las ondas optimizado y altura de ola para adquirir IM-MS (paso 2).
- Medir la deriva de ion el tiempo con IM-MS a tres velocidades diferentes de la onda (por ejemplo, 550, 600 y 650 m/s), manteniendo la misma altura de onda (por ejemplo, 40 V).
- Para determinar los iones de proteínas CCS, calibradores de proteína medida bajo las mismas condiciones del instrumento utilizadas para la proteína bajo investigación. Una óptima calibración de tiempo deriva requiere medición de las proteínas con CCS conocidas.
- Seleccione cuatro calibradores, dos con una masa por encima y dos con una masa inferior a la de la proteína bajo investigación37. Lo más importante, asegúrese de que la altura de ola y la velocidad de la onda son los mismos que los registrados para la proteína bajo investigación.
- Calcular manualmente CCS41 o usando un software especializado como PULSAR33 y Amphitrite34 (figura 5).
- Para comprobar si la proteína es nativo-como en la fase gaseosa, CCS experimental compara a CCS teóricos obtenidos de estructuras de alta resolución. Para HerA-NurA, calcular CCS teórica utilizando el método de aproximación de proyección (PA) en MOBCAL42. Otros métodos incluyen trayectoria método (TM)42 y exacto ámbito difícil dispersión (EHS)43.
4. solución en interrupción de complejos de la proteína nativa MS y IM-MS Led determinación estructural
Nota: Los complejos de la proteína pueden en algunos casos identificar de la misma solución como complejo intacto. Sin embargo, puede lograrse estructural información adicional como la inter-subunidad conectividad y complejo conjunto de interrumpir las interacciones de la proteína en solución, para formar los complejos. Esto puede lograrse de varias maneras tales como la adición de solvente orgánico, aumentando la fuerza iónica o manipulando el pH. Para conocer de la HerA-NurA subunidad compleja conectividad y complejo conjunto, los complejos se generaban en solución mediante la adición de disolventes que perturban las interacciones de la subunidad.
- Preparar el cambio de muestra y tampón de proteínas en acetato de amonio como se describe en el paso 1.
- Añadir 10-40% de solvente en incrementos del 10%. Disolventes normalmente utilizados son el dimetil sulfóxido (DMSO), metanol (MeOH) y acetonitrilo (ACN).
Nota: Esto puede realizarse dentro de un tubo de microcentrífuga de polipropileno. - Incubar la mezcla en hielo durante 1 hora.
- Adquirir un espectro IM-MS para cada condición (pasos 2 y 3) (figura 4).
- Utilice el software Cumbre del44 para asignar los complejos de la proteína y generar redes de interacción de proteínas. Como alternativa, manualmente generar una lista de masas teóricas de las especies esperadas.
- Para asegurar se dobla Subcomplejos mando, calcular los valores experimentales de CCS para los complejos secundarios y comparar a CCS teórica como se explica en el paso 3 (tabla 1, figura 5 y figura 6).
5. investigar la proteína compleja estabilidad mediante colisión inducida por despliegue (CIU)
Nota: Se puede utilizar CIU sonda la estabilidad estructural de las proteínas y sus complejos con el ligando. Paquetes de software especializados como PULSAR33, Amphitrite34 y CIU suite9 pueden utilizarse entonces para modelar el despliegue de la fase gaseosa de la proteína bajo investigación con y sin ligando. Por ejemplo, esta sección describe el procedimiento de control de fase gaseosa desarrollando trayectorias e investigar el efecto que se estabiliza del atascamiento de la DNA y ATP en el complejo de HerA-NurA.
- Registrar datos de IM-MS mientras que aumenta el voltaje de aceleración de trampa de 10 V a 200 V en incrementos de 2-10 V a revelar progresivamente la proteína en la fase gaseosa.
Nota: Registro de resultados de incrementos más pequeños en más archivos de datos para procesar, sin embargo este enfoque proporciona el diagrama de despliegue más resuelto, que es importante para el análisis de los puntos de transición entre las especies de plegado/desplegado. - Analizar los datos adquiridos mediante PULSAR33, Amphitrite34 o CIU suite9 y generar diagramas de despliegue bidimensionales en unidades de CCS en función de la aceleración de tensión (paso 3). Para cada estado de carga, este se crea apilando las distribuciones CCS intensidad-normalizado en cada voltaje de aceleración (figura 7 A-Bi).
- Generar un diagrama de despliegue teórico utilizando uno de los paquetes de software. Se instalarán los datos a un modelo de despliegue. Esto hace posible cuantificar la energía de colisión en que despliegue las transiciones ocurren y determinan la estabilidad de las proteínas con y sin ligandos consolidados33. Una transición que es cuando las transiciones de una especie de un estado (basado en sus valores experimentales de CCS) a otro estado con un CCS más grande.
- Para cuantificar las transiciones, calcular la midpoint(s) transición entre Estados mediante algoritmos y software como PULSAR33. Esto se divulga comúnmente como CV50, cual es el valor de tensión de choque (trampa) en que se agote el 50% de un estado específico.
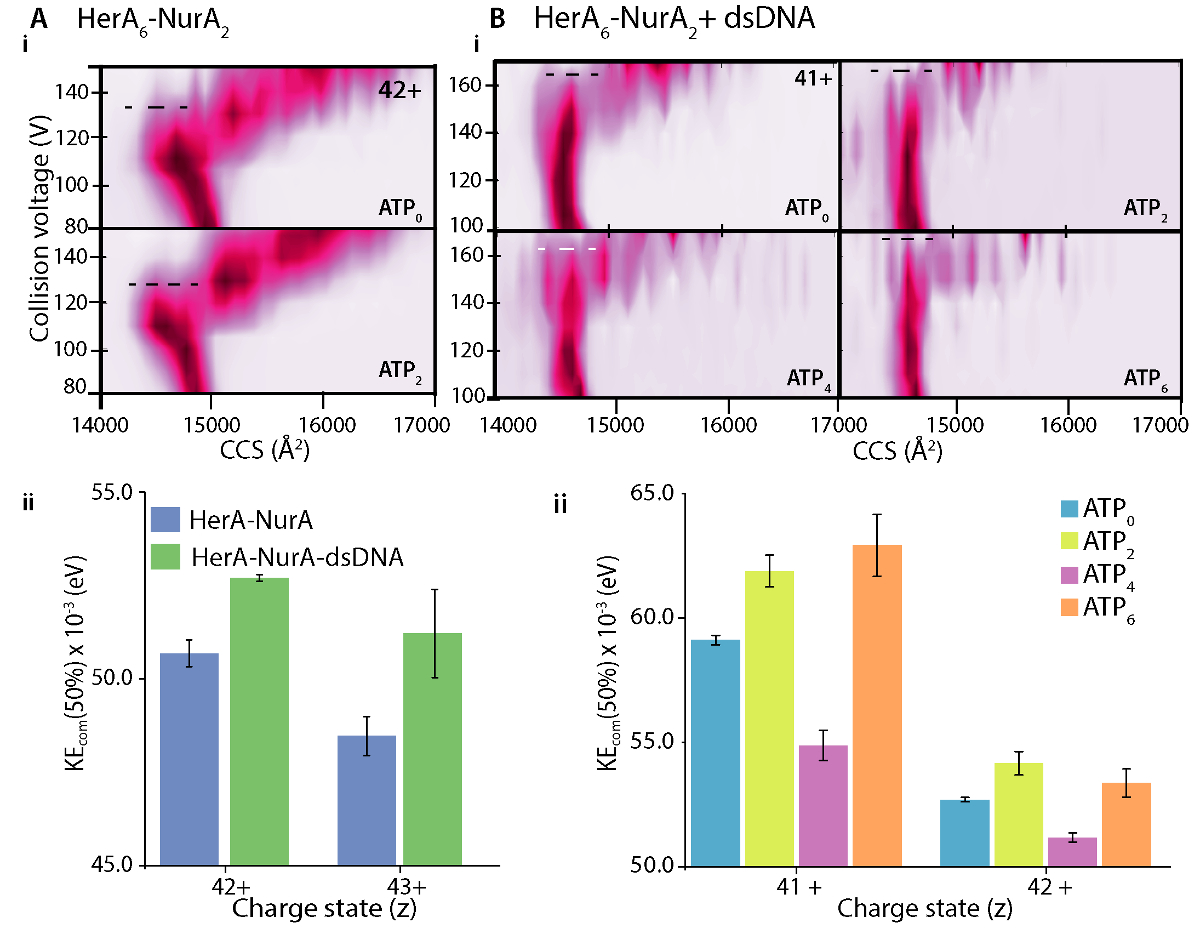
- El valor de50 CV, calcular la energía interna total de un ion con la colisión del centro de masa energía (KECOM)45. KECOM se define por la energía interna total disponible para la transición de despliegue de un ion y se calcula a partir de la energía cinética y las Misas de los socios de colisión (ion proteína y gas neutro) tal como se describe en la ecuación (1)10
KEcom (eV) = (ecuación 1).
(ecuación 1).
Donde Z es la carga de iones, MN es la masa de los gases neutros yiones de M es la masa del ion de la proteína.
Nota: Esto es porque CIU de proteínas es cargo dependent46, 47. Se recomienda realizar el análisis KECOM a más de un estado de carga ( figura 7Aii).
6. procedimientos de modelado para simulaciones de dinámica Molecular diferencial utilizado en MS Integrativa
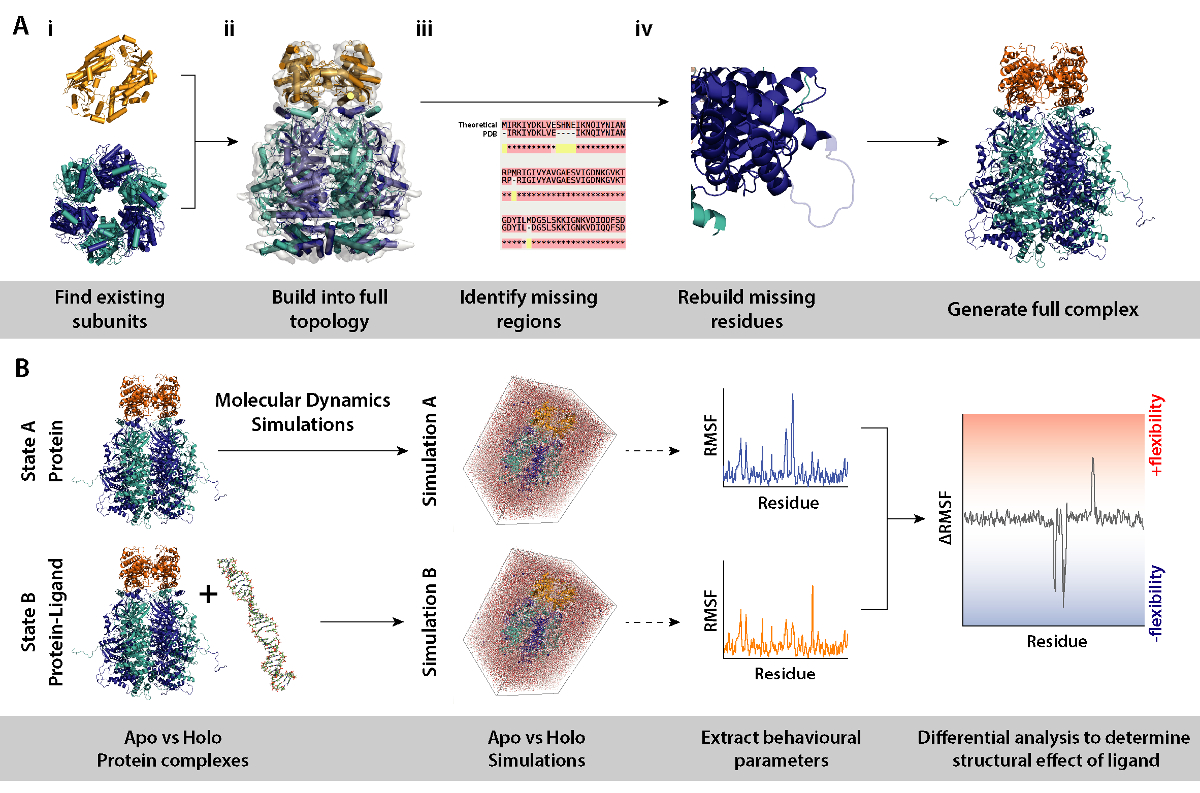
Nota: Usar modelos de subunidades de proteínas o de complejos como de las estructuras cristalinas, simulaciones diferencial (complejo con y sin ligando de la proteína) pueden utilizarse para determinar efectos de por ejemplo presencia de ligando en la dinámica y estructura de la proteína. Esta sección detalla un flujo de trabajo y las herramientas necesarias para el modelado de procedimientos necesarios para simulaciones de dinámica molecular diferencial de la instalación.
- Identificar las subunidades que componen el complejo (figura 8A, en los pasos 2 y 3). Fuente de los modelos existentes de subunidades, por ejemplo, las estructuras cristalinas de la base de datos de copia (https://www.rcsb.org). El UniProt entrada de la proteína contiene una lista de conocer estructuras cristalográficas/NMR (http://www.uniprot.org). Si estos no están disponibles, la secuencia teórica puede entrar a la explosión para identificar plantillas convenientes para modelado de homología (http://blast.ncbi.nlm.nih.gov/).
- Montar el conjunto de la topología correcta (figura 8A-ii). Esto puede hacerse a través de varios métodos. Las subunidades individuales pueden montarse en mapas de la microscopia electrónica disponible encontradas el EMDB para armar el complejo intacto (https://www.ebi.ac.uk/pdbe/emdb/). Un tutorial para el montaje de PDBs en EM mapas usando Molecular dinámica Flexible montaje (MDFF) puede encontrarse aquí: http://www.ks.uiuc.edu/Training/Tutorials/science/mdff/tutorial_mdff-html/.
- Identificar las regiones que faltan del complejo (figura 8A-iii). Realizar alineamiento múltiple de secuencias (MSA) entre el PDB y secuencia teórica para identificar residuos que pueden ser preparados en las estructuras cristalinas, o ninguna mutaciones heredadas de experimentos cristalográficos. MSA se puede realizar utilizando los servidores de web tales como T-café (http://tcoffee.crg.cat/apps/tcoffee/do:regular).
- Residuos que falta regenerados mediante homología modelado (figura 8A-iv). Falta de residuos de la proteína del complejo se puede construir usando el programa MODELLER (https://salilab.org/modeller/). MODELLER puede salir un conjunto de n los modelos en diferentes configuraciones regenerados. Buenos modelos se pueden identificar partiendo su calificación de energía proteína optimización discreta (droga). Se proporciona un tutorial completo en el sitio web de software (https://salilab.org/modeller/tutorial/).
- Realizar simulaciones de dinámica molecular diferencial (MD) de la proteína del complejo (figura 8B) para identificar las regiones de las proteínas que responden a un cambio ambiental en particular, por ejemplo, la presencia de un ligando. En tales simulaciones, parámetros de comportamiento de la simulación A (sólo proteínas) que actúa como referencia, se resta de B simulación (proteína + ligando). La fluctuación diferencial cuadrático (RMSF) calculada entre simulaciones A y B puede informar en las regiones de la proteína que aumentan o disminuyen en la flexibilidad de una manera dependiente de ligando.
- Realizar simulaciones y análisis descendente con GROMACS (http://www.gromacs.org). Un tutorial se puede encontrar en: http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin/gmx-tutorials/Lysozyme/index.html. Para eliminar sesgos de modelo, se debe generar primero la estructura del complejo ligando-limite. La proteína se copiará de esto sin el ligando, para producir un modelo de proteína idéntico al complejo ligando enlazado.
Resultados
Nativo MS de resultados revelaron el estado oligomérico, composición y topología del HerA-NurA complejo (figura 1). Como las interacciones no covalentes se conservan en la fase gaseosa, MS nativa de experimentos de valoraciones de ADP y ATP-γ-S determina la Unión de nucleótidos pares a HerA-NurA (figura 2) y aumentando la concentración de ATP-γ-S aumenta el pariente intensidad del hexámero HerA (figura 3). Con respecto a las interacciones de la subunidad se obtuvieron de la interrupción de la solución en seguida MS nativa y estaban de acuerdo con la información estructural y masas teóricas (figura 4 y tabla 1).
Los valores experimentales de CCS de las proteínas y sus complejos se derivan de experimentos IM-MS (figura 5). Estos valores son promedio rotatorio de fase de gas transversales cálculos de la forma molecular y describen el estado tridimensional de la proteína. Se comparan valores de CCS a las mediciones teóricas de Cristalografía de rayos x y un buen acuerdo se deduce que la forma nativa de retenido en la fase gaseosa (tabla 1). Esto valida utilizando valores de CCS para la construcción de modelos de baja resolución de la Asamblea de proteína48.
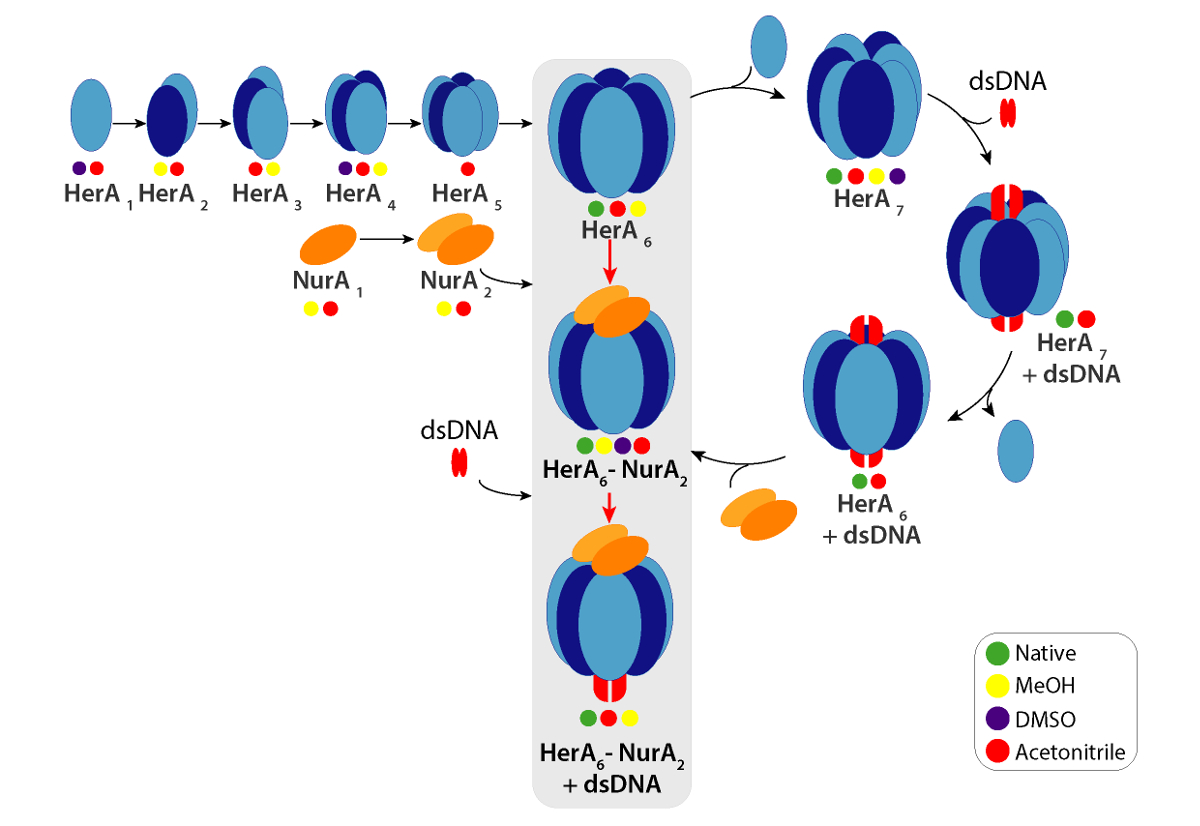
CCSs experimentales pueden calcularse para cada ión de estado de carga. Un conformer nativo-como la proteína puede dar lugar a iones de estado de carga con valores similares de CCS. Sin embargo, más iones de estado de carga aumentaron repulsiones coulombic que pueden llevar a fase gaseosa despliegue y más grandes CCS valores de proteína comparados con la CCSs teórica. El valor de CCS del más bajo estado de carga los iones suelen ser por lo tanto usa49. Para HerA-NurA, experimentos en la solución de interrupción sobre HerA y HerA-NurA con y sin ADN impulsó la generación de un camino conjunto a partir de monómeros y luego formando el hexámero todo HerA (HerA6)-complejo dimérico de NurA (anexo2) ADN (figura 6).
Diferencias en las parcelas que CIU entre la apo (ligand-libre) y el ligando enlazado definen el cambio en estabilidad compleja al atascamiento del ligand. Un CV más alto50 o KECOM valor implica un ión más estabilizado en la fase gaseosa. CIU y KECOM el análisis reveló que ADN enlazado HerA-NurA es más estable que el complejo de ADN libre (figura 7Aii). De análisis del CIU-MS en los respectivos Estados de unión a ATP, el estado consolidado de cuatro-ATP-γ-S reducida estabilidad compleja en la fase de gas y seis estado encuadernado -ATP-γ-S donde se ocupan todos los sitios fue la más estable (figura 7Bii). MS nativa puede revelar el nucleótido discreto enlace Estados de HerA; sin embargo, no puede distinguir qué subunidades de HerA son vinculantes ATP y donde realiza este enlace. Esta información se puede derivar de simulaciones solvente explícitos sobre el hexámero HerA y HerA-NurA siguiendo el flujo de trabajo resumido (figura 8).

Figura 1. Interrogar el estado oligomérico, composición y topología del complejo no covalente HerA-NurA. (A) espectrométricos de HerA, NurA HerA y HerA-NurA en presencia de DNA (ADN de doble hebra 15,4 kDa 25 bp). El complejo sub HerA existe como un hexamer y un heptamer. NurA de dímero se une y un hexamer HerA imponer conversión oligomérica. El ADN une el complejo formado de HerA - NurA (adaptados de Ahdash Z. et al., 201722resultados). (B) intensidades relativas de las especies identificadas se calculan utilizando UniDec32. Haga clic aquí para ver una versión más grande de esta figura.
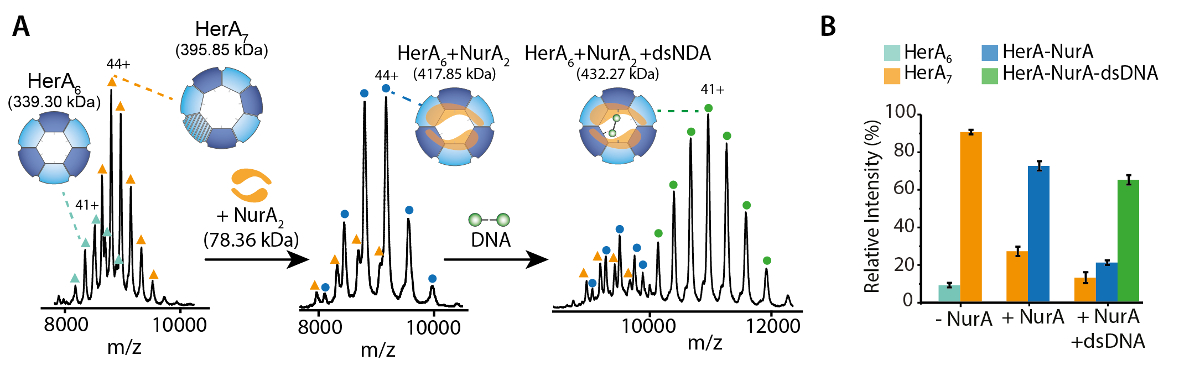{kind=link}

Figura 2. ESI-MS nativo revela el mecanismo de unión de nucleótido a HerA-NurA. Espectros de masas (A) HerA-NurA y (B) ADN HerA-NurA con el aumento de las concentraciones de (i) ATP-γ-S y (ii) ADP. Medida masas se comparan masas teóricas y la cantidad de ATP-γ-S o ADP limitado se determinan. Medida masas y el número de nucleótidos enlazados aparecen en los espectros. Experimentos de titulaciones ATP-γ-S y ADP determinan la Unión de pares de nucleótidos a HerA-NurA solo y cuando en complejo con el ADN que indica un mecanismo de reacción cíclica (adaptados de Ahdash Z. et al., 201722resultados). Haga clic aquí para ver una versión más grande de esta figura.
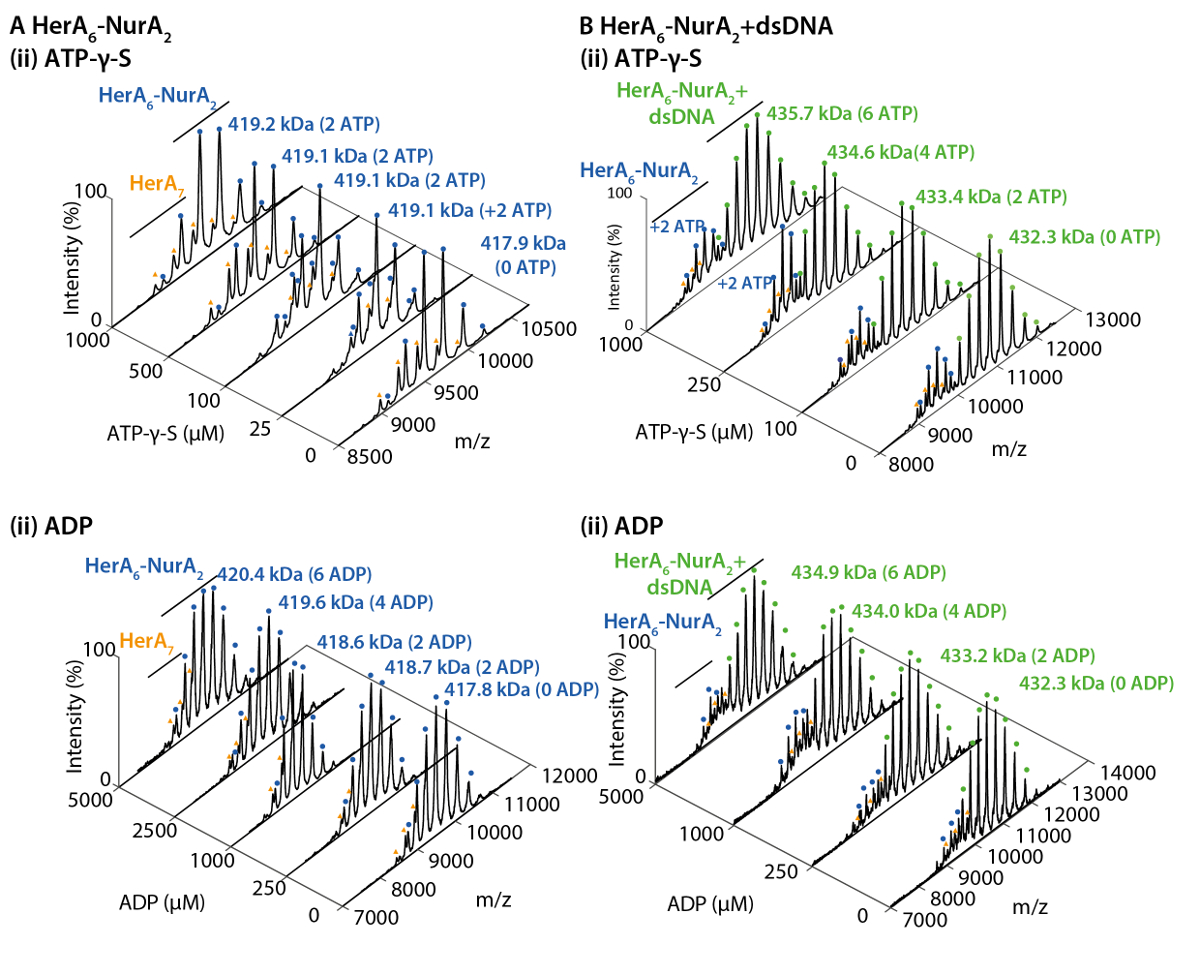{kind=link}

Figura 3. Medir el efecto de aumentar las concentraciones de ATP-γ-S en estado oligomérico de HerA. (A) espectrométricos de HerA en el aumento de las concentraciones de ATP-γ-S. (B) gráfico que muestra las intensidades relativas de diferentes especies de nativas MS calculada usando UniDec deconvolución software32. Como la concentración de ATP-γ-S aumenta, también aumenta la intensidad relativa del hexámero HerA. Número de moléculas de ATP-γ-S límite se muestra en los espectros (adaptados de Ahdash Z. et al., 201722resultados). Haga clic aquí para ver una versión más grande de esta figura.
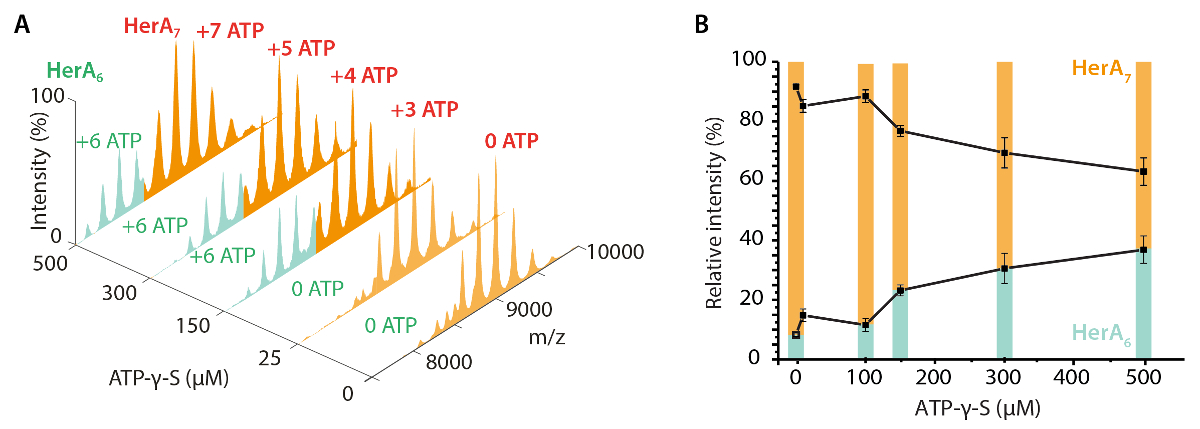{kind=link}

Figura 4. Masa de espectros y productos de la disociación del complejo de HerA (A) y (B) (i) HerA-NurA solo y en presencia del siguiente (ii) ADN de la solución en interrupción. Se realizaron experimentos de alteración en la solución con 10-40% de acetonitrilo, metanol (MeOH) o dimetilsulfóxido (DMSO) y dio lugar a la formación de varios Subcomplejos de mando. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5. Ion movilidad llegada tiempo distribuciones que se muestra en un eje de CCS para complejos y complejos secundarios generados. Icono para cada sub-complejo correlato con los anotados en los espectros en la figura 4. Experimentales y calculadas las masas y valores de CCS de los complejos aparecen en la tabla 1 los cuales mostraron un acuerdo entre los valores experimentales y calculados (después de considerar la incertidumbre típica en la resolución de viajar ion de onda masa espectrometría de movilidad de ± 5 - 8%37). Haga clic aquí para ver una versión más grande de esta figura.
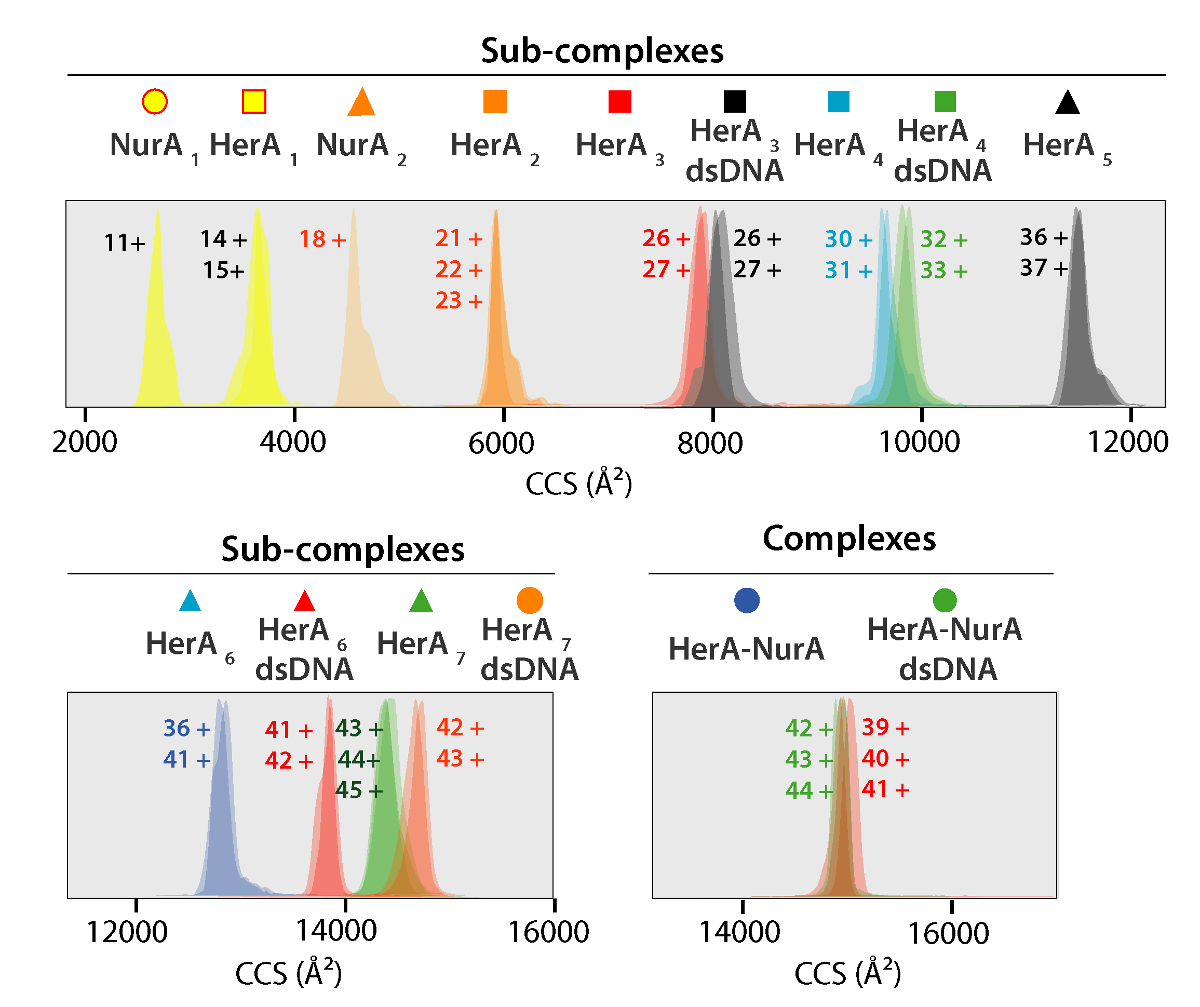{kind=link}

Figura 6. Camino de montaje del complejo de HerA-NurA generado a partir de la interrupción de la solución en círculos nativos de MS y IM-Sra. color indican condiciones donde cada sub complejo observó: nativo (verde, antes de la interrupción), metanol (amarillo), DMSO (púrpura) o Acetonitrilo (rojo). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7. Investigando el efecto estabilizador del (A) ADN en HerA-NurA y (B) ATP binding a HerA-NurA-DNA. (i) parcelas fase gas CIU-MS y el cálculo de energías (KEcom) colisión (ii) centro de masa muestran que la presencia de dsDNA estabiliza el complejo de HerA-NurA y que el ATP-γ-S 6 había limitado estado es la más estable. Estabilización de Estados de carga distintos se muestra. Parcelas fueron generadas usando PULSAR 33. Resultados de la (A) adaptados de Ahdash Z. et al., 201722. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8. Flujo de trabajo para los procedimientos de modelado para simulaciones de dinámica molecular diferenciada. (A) generar el complejo objeto de investigación mediante la construcción de la topología de existentes subunidades y la reconstrucción de restos desaparecidos. (B) flujo de trabajo para la ejecución de simulaciones de dinámica molecular en el complejo con y sin ligando de la proteína. Simulaciones de dinámica molecular funcionó para la proteína sólo que actúa como referencia y que se resta de la simulación de proteína y de ligando. Esto es seguido por calcular la fluctuación diferencial cuadrático (RMSF) entre las simulaciones y determinar el efecto del ligando. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Complejos / el complejo | Masa teórica (kDa) | Masa experimental (kDa) | Teórico CCS (Å2) | Experimental CCS (Å2) [cargo] | Condición de HN |
| HerA6- anexo2 | 416.22 | 417.85 | 14531 | 14577 [42 +] 14599 [43 +] 14608 [44 +] 14637 [45 +] | EFE, 10-40% de DMSO, de 10-20% 10% MeOH |
| HerA6- anexo2- dsDNA | 431.72 | 432.27 | - | 14661 [+ 39] 14728 [40 +] 14781 [41 +] 14837 [42 +] | ACN de 10%, 10% MeOH |
| Anexo1 | 39.12 | 38.18 | 3254 | 2618 [10 +] 2746 [11 +] 2878 [12 +] | 10-40% ACN, 10% MeOH, 20-40% DMSO |
| Anexo2 | 78.24 | 78,36 | 4890 | 4903 [16 +] 4614 [+ 17] 4537 [18 +] 4666 [+ 19] | 10-40% MeOH, 20-40% de DMSO |
| HerA1 | 56.33 | 56.32 | 4131 | 3647 [+ 14] 3792 [15 años] 3950 [16 +] | 10-40% ACN, DMSO 40% |
| HerA2 | 112.66 | 112.95 | 6475 | 5648 [20 +] 5747 [21 +] 5842 [+ 22] 5996 [23 +] | Metanfetamina 40%, 10-40% ACN |
| HerA3 | 168.99 | 169.39 | 8607 | 7501 [25 años] 7616 [26 +] 7717 [+ 27] 7867 [28 +] | 10-40% MeOH, CAN 10-40%, 40% DMSO |
| HerA3 + ADN | 183.99 | 184.976 | - | 7655 [26 +] 7990 [+ 27] 8107 [28 +] | 10-30% ACN |
| HerA4 | 225.32 | 226.2 | 10477 | 9205 [30 +] 9287 [31] 9493 [32 +] 9961 [33 +] | 10-40% MeOH, 10-40% ACN |
| HerA4 + ADN | 240.82 | 241.33 | - | 9637 [31 +] 9756 [32 +] 9830 [33 +] | 10-30% ACN |
| HerA5 | 281.65 | 282.75 | 11853 | 10847 [36 +] 10958 [37 +] 11161 [38 +] | 30-40% ACN |
| HerA6 | 337.98 | 339.3 | 12517 | 12335 [38 +] 12386 [+ 39] 12498 [40 +] 12590 [41 +] 12676 [42 +] 13019 [43 +] | 10-40% MeOH, 10-40% ACN |
| HerA6 + ADN | 353.48 | 354.626 | - | 12890 [40 +] 13081 [41 +] 13184 [42 +] 13273 [43 +] 13463 [44 +] 13576 [45 +] | 30% ACN |
| HerA7 | 394.3 | 395.85 | 13901 | 14154 [42 +] 14219 [43 +] 14261 [44 +] 14285 [45 +] 14335 [46 +] | 10-40% MeOH, 10-40% ACN, 10-40% DMSO |
| HerA7 + ADN | 409.8 | 410.62 | - | 14414 [41 +] 14510 [42 +] 14558 [43 +] 14598 [44 +] 14630 [45 +] 14641 [46 +] | 10% ACN |
Tabla 1. Experimentales y calculadas las masas y valores de CCS de HerA-NurA y sus sub-complejos generan estudios de interrupción de la solución en forma.
Discusión
MS está jugando un papel cada vez más importante en la caracterización de la estequiometría, la interacción y arquitectura de subunidades de complejos de la proteína. IM-MS datos pueden usarse para definir arreglos topológicos de subunidades dentro de complejos de múltiples componentes. En comparación con otros métodos existentes de la biología estructural, MS tiene varias ventajas. Nativa es una técnica altamente sensible y rápida y puede usarse para muestras de proteína heterogénea. Cuando se combina con experimentos en la solución de interrupción, caminos de la disociación de los conjuntos de proteínas pueden controlarse. Junto con las estructuras cristalinas o modelos de la homología, la información ofrecida por MS estructural ofrece una herramienta para la investigación de las interacciones proteína-ligando y proporcionar modelos de materno y Asamblea vías11.
Aquí, describimos los procedimientos experimentales necesarios para analizar la estequiometría y la composición de las interacciones proteína-ligando, con uno o más ligandos, utilizando MS integradora. Esto incluye la preparación de la muestra MS, adquisición de datos, análisis de datos y la integración de datos de MS usando herramientas computacionales. Para ello, se utilizó el ADN-resección HerA-NurA hetero-oligoméricos complejo de la proteína, limitado a tres ligands (ADN, ATP y ADP), como nuestro sistema modelo. El protocolo muestra el uso de lo software disponible actualmente para facilitar la presentación y análisis de datos.
Adquisición de espectros de alta calidad es importante para el análisis de unión de ligando, por lo tanto, muestra cuidado crítico, incluyendo purificación de proteínas, valoración de ligando y cambio de buffer. Una limitación de nESI MS nativa al estudiar ligando es fijación no específica. Unión no específica ocurre durante desolvation gota durante todo el proceso electrospray. Esto aumenta las concentraciones de ligando y por lo tanto altera el cociente proteína/ligando del29. La Unión de nucleótidos se traduce en una relativamente pequeña masa diferencia apo proteína nucleótido-limite que no altera la eficiencia de ionización50,51.
Utilizamos el sistema Synapt G2-Si MS para nuestro trabajo, pero los protocolos son aplicables para diversas investigaciones de otros complejos de proteína-ligando usando otros espectrómetros de masas de nano-electrospray comercialmente disponibles. Integrante MS estructural cada vez más está jugando un papel importante en el tratamiento de problemas biológicos de mayor complejidad. El flujo de trabajo y técnicas que se describen aquí son adecuadas para entender las consecuencias estructurales y construcción de mecanismos de formación de proteína-ligando y complejo de la proteína que de lo contrario son difíciles de estudiar con las técnicas estructurales convencionales .
Divulgaciones
No hay conflicto de interés declarado.
Agradecimientos
Nos gustaría agradecer a Peter Karl Hopfner y Robert Thomas Byrne por favor muestras de proteínas proporciona HerA y HerA-NurA y por su ayuda con el diseño experimental. También agradecemos a Dr. Eamonn Reading por su revisión del manuscrito. Agradecemos a nuestros organismos: Wellcome Trust [109854/Z/15/Z] y sociedad real [RG150216 a A.P.].
Materiales
| Name | Company | Catalog Number | Comments |
| Adenosine 5′-(3-thiotriphosphate) tetralithium salt | Merck Millipore | 119120-25MG | |
| Adenosine 5′-diphosphate | Sigma-Aldrich | 20398-34-9 | |
| Ammonium acetate solution | Sigma-Aldrich | A2706 | |
| Micro Bio-Spin Chromatography Columns | Bio-Rad | 7326204 | |
| Vivaspin concentrator | Sartorius | Z614041-25EA | |
| Magnesium chloride hexahydrate | Sigma-Aldrich | 246964 | |
| Water TraceSelect | Sigma-Aldrich | 95305 | |
| Borosilicate Capillaries | Harvard Apparatus | 300060 |
Referencias
- Wilm, M., Mann, M. Analytical Properties of the Nanoelectrospray Ion Source. Analytical Chemistry. 68 (1), 1-8 (1996).
- Fenn, J. B., Mann, M., Meng, C. K., Wong, S. F., Whitehouse, C. M. Electrospray ionization for mass spectrometry of large biomolecule. Science. 246 (4926), 64-71 (1989).
- Hernandez, H., Robinson, C. V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nature Protocols. 2 (3), 715-726 (2007).
- Heck, A. J., Van Den Heuvel, R. H. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom Review. 23 (5), 368-389 (2004).
- Boeri Erba, E., Petosa, C. The emerging role of native mass spectrometry in characterizing the structure and dynamics of macromolecular complexes. Protein Science. 24 (8), 1176-1192 (2015).
- Laganowsky, A., Reading, E., Hopper, J. T. S., Robinson, C. V. Mass spectrometry of intact membrane protein complexes. Nature. 8 (4), 639-651 (2013).
- Benesch, J. L. Collisional activation of protein complexes: picking up the pieces. Journal of the American Society for Mass Spectrometry. 20 (3), 341-348 (2009).
- Tian, Y., Han, L., Buckner, A. C., Ruotolo, B. T. Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Analytical Chemistry. 87 (22), 11509-11515 (2015).
- Eschweiler, J. D., Rabuck-Gibbons, J. N., Tian, Y., Ruotolo, B. T. CIUSuite: A Quantitative Analysis Package for Collision Induced Unfolding Measurements of Gas-Phase Protein Ions. Analytical Chemistry. 87 (22), 11516-11522 (2015).
- Hopper, J. T., Oldham, N. J. Collision induced unfolding of protein ions in the gas phase studied by ion mobility-mass spectrometry: the effect of ligand binding on conformational stability. Journal of the American Society for Mass Spectrometry. 20 (10), 1851-1858 (2009).
- Politis, A., et al. Topological models of heteromeric protein assemblies from mass spectrometry: application to the yeast eIF3:eIF5 complex. Chemical Biology. 22 (1), 117-128 (2015).
- Morgner, N., et al. Hsp70 forms antiparallel dimers stabilized by post-translational modifications to position clients for transfer to Hsp90. Cell Reports. 11 (5), 759-769 (2015).
- Lai, Y. T., et al. Structure of a designed protein cage that self-assembles into a highly porous cube. Nature Chemistry. 6 (12), 1065-1071 (2014).
- Levy, E. D., Boeri Erba, E., Robinson, C. V., Teichmann, S. A. Assembly reflects evolution of protein complexes. Nature. 453 (7199), 1262-1265 (2008).
- Sharon, M., et al. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure. 17 (1), 31-40 (2009).
- Jurneczko, E., Barran, P. E. How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst. 136 (1), 20-28 (2011).
- Politis, A., Schmidt, C. Structural characterisation of medically relevant protein assemblies by integrating mass spectrometry with computational modelling. Journal of Proteomics. 175, 34-41 (2017).
- Zhou, M., et al. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 334 (6054), 380-385 (2011).
- Barrera, N. P., Booth, P. J., Robinson, C. V. Micelles Protect Membrane Complexes from Solution to Vacuum. Science. 321 (5886), 243-246 (2008).
- Konijnenberg, A., et al. Global structural changes of an ion channel during its gating are followed by ion mobility mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 111 (48), 17170-17175 (2014).
- Schmidt, C., Beilsten-Edmands, V., Mohammed, S., Robinson, C. V. Acetylation and phosphorylation control both local and global stability of the chloroplast F1 ATP synthase. Scientific Reports. 7, 44068 (2017).
- Ahdash, Z., et al. Mechanistic insight into the assembly of the HerA-NurA helicase-nuclease DNA end resection complex. Nucleic Acids Research. 45 (20), 12025-12038 (2017).
- Pagel, K., Natan, E., Hall, Z., Fersht, A. R., Robinson, C. V. Intrinsically disordered p53 and its complexes populate compact conformations in the gas phase. Angewandte Chemie. 52 (1), 361-365 (2013).
- Afonso, J. P., et al. Insights into the structure and assembly of the Bacillus subtilis clamp-loader complex and its interaction with the replicative helicase. Nucleic Acids Research. 41 (9), 5115-5126 (2013).
- van Duijn, E. Current limitations in native mass spectrometry based structural biology. Journal of the American Society for Mass Spectrometry. 21 (6), 971-978 (2010).
- Susa, A. C., Xia, Z., Williams, E. R. Native Mass Spectrometry from Common Buffers with Salts That Mimic the Extracellular Environment. Angewandte Chemie. 56 (27), 7912-7915 (2017).
- Breukera, K., McLafferty, F. W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10 to 10 s. Proceedings of the National Academy of Sciences of the United States of America. 105 (47), 18145-18152 (2008).
- Robinson, C. V., et al. Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein-CoA Ligand Binding and Assembly. Journal of the American Chemical Society. 118 (36), 8646-8653 (1996).
- Shimon, L., Sharon, M., Horovitz, A. A method for removing effects of nonspecific binding on the distribution of binding stoichiometries: application to mass spectroscopy data. Biophysical Journal. 99 (5), 1645-1649 (2010).
- Dyachenko, A., Gruber, R., Shimon, L., Horovitz, A., Sharon, M. Allosteric mechanisms can be distinguished using structural mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 110 (18), 7235-7239 (2013).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. 19 (40), (2010).
- Marty, M. T., et al. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Analytical Chemistry. 87 (8), 4370-4376 (2015).
- Allison, T. M., et al. Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nature Communications. 6, 8551-8561 (2015).
- Sivalingam, G. N., Yan, J., Sahota, H., Thalassinos, K. Amphitrite: A program for processing travelling wave ion mobility mass spectrometry data. International Journal of Mass Spectrometry. 345-347, 54-62 (2013).
- Morgner, N., Robinson, C. V. Massign: an assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Analytical Chemistry. 84 (6), 2939-2948 (2012).
- Bush, M. F., et al. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Analytical Chemistry. 82, 9557-9565 (2010).
- Ruotolo, B. T., Benesch, J. L., Sandercock, A. M., Hyung, S. J., Robinson, C. V. Ion mobility-mass spectrometry analysis of large protein complexes. Nature Protocols. 3 (7), 1139-1152 (2008).
- Lanucara, F., Holman, S. W., Gray, C. J., Eyers, C. E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nature Chemistry. 6 (4), 281-294 (2014).
- Wyttenbach, T., Bowers, M. T. Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. Journal of Physical Chemistry B. 115 (42), 12266-12275 (2011).
- Marcoux, J., Robinson, C. V. Twenty years of gas phase structural biology. Structure. 21 (9), 1541-1550 (2013).
- Michaelevski, I., Kirshenbaum, N., Sharon, M. T-wave ion mobility-mass spectrometry: basic experimental procedures for protein complex analysis. Journal of Visualized Experiments. (41), (2010).
- Mesleh, M. F., Hunter, J. M., Shvartsburg, A. A., Schatz, G. C., Jarrold, M. F. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. Journal of Physical Chemistry A. 100, 16082-16086 (1996).
- Shvartsburg, A. A., Jarrold, M. F. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chemical Physics Letters. 261, 86-91 (1996).
- Taverner, T., Hernández, H., Sharon, M., Ruotolo, B. T., Matak-Vinković, D., Devos, D., Russell, R. B., Robinson, C. V. Subunit Architecture of Intact Protein Complexes from Mass Spectrometry and Homology Modeling. Accounts of Chemical Research. 41 (5), 617-627 (2008).
- Wysocki, V. H., Joyce, K. E., Jones, C. M., Beardsley, R. L. Surface-induced dissociation of small molecules, peptides, and non-covalent protein complexes. Journal of the American Society for Mass Spectrometry. 19 (2), 190-208 (2008).
- Hall, Z., Politis, A., Bush, M. F., Smith, L. J., Robinson, C. V. Charge-state dependent compaction and dissociation of protein complexes: insights from ion mobility and molecular dynamics. Journal of the American Chemical Society. 134 (7), 3429-3438 (2012).
- Hyung, S. J., Robinsons, C. V., Ruotolo, B. T. Gas-Phase Unfolding and Disassembly Reveals Stability Differences in Ligand-Bound Multiprotein Complexes. Chemistry & Biology. 16, 382-390 (2009).
- Hall, Z., Politis, A., Robinson, C. V. Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure. 20 (9), 1596-1609 (2012).
- Woods, L. A., Radford, S. E., Ashcroft, A. E. Advances in ion mobility spectrometry-mass spectrometry reveal key insights into amyloid assembly. Biochimica et Biophysica Acta. 1834 (6), 1257-1268 (2013).
- Daubenfeld, T., Bouin, A. P., van der Rest, G. A deconvolution method for the separation of specific versus nonspecific interactions in noncovalent protein-ligand complexes analyzed by ESI-FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 17 (9), 1239-1248 (2006).
- Loo, J. A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrometry Reviews. 16, 1-23 (1997).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados