Method Article
Analyse des Architectures de protéine et Complexes protéine-Ligand par spectrométrie de masse structurelle intégrative
Dans cet article
Résumé
Spectrométrie de masse (MS) est devenue un outil important pour l’enquête de structure et la dynamique des assemblages macromoléculaires. Ici, nous intégrons les approches axées sur les MS pour interroger la formation de complexes protéiques et de liaison du ligand.
Résumé
Les protéines sont une classe importante de macromolécules biologiques qui jouer de nombreux rôles principaux dans des fonctions cellulaires, y compris l’expression des gènes, catalyse des réactions métaboliques, la réplication et la réparation de l’ADN. Donc une compréhension détaillée de ces processus fournit des informations cruciales sur la fonction des cellules. Intégrative structurels MS méthodes offrent des informations structurales et dynamiques sur l’assemblage complexe de protéine, connectivité complexe, sous-unité stoechiométrie, oligomérisation de protéine et la liaison du ligand. Progrès récents en intégration MS structurelles ont permis pour la caractérisation des systèmes biologiques difficiles, y compris les grosses protéines de liaison d’ADN et de protéines membranaires. Ce protocole décrit comment intégrer diverses données MS comme native MS et ion mobility-spectrométrie de masse (IM-MS) avec des simulations de dynamique moléculaire pour nous éclairer dans un ADN hélicase-nucléase réparation complexe protéique. L’approche qui en résulte fournit un cadre pour des études détaillées de liaison du ligand à d’autres complexes de protéines impliquées dans des processus biologiques importants.
Introduction
Native analyse par spectrométrie de masse des protéines intactes et leurs complexes se fait avec l’électrospray et nano-electrospray ionisation (nESI), qui conservent le repliement des protéines et des interactions non covalentes pendant le processus d’ionisation de1, 2. Dans native MS, la structure des protéines et leurs complexes sont conservées dans un état quasi native dans la phase gazeuse3,4. Native MS détecte plusieurs ions chargés de protéines, qui sont séparées selon leur masse pour charger le ratio (m/z) permettant à la masse de la protéine ou protéine-ligand complexe à calculer. Cette information permet la détermination d’une protéine intacte stoechiométrie, la composition, liaison du ligand et interaction réseaux3,4,5,6. Native MS présente plusieurs avantages par rapport à d’autres techniques telles de spectroscopie résonance magnétique nucléaire et la cristallographie aux rayons x5. Tout d’abord, native MS est une technique rapide et hautement sensible, nécessitant seulement quelques microlitres (2-3 µL) de l’échantillon à des concentrations relativement faibles de complexes finales nm haute et basse µM rang6. Deuxièmement, native MS peut être utilisée pour interroger les échantillons de protéines hétérogènes ce qui permet d’analyser plusieurs protéines et oligomères États simultanément. Troisièmement, native MS ne nécessite pas d’échantillons de protéines pour être modifiés avant l’analyse par réticulation chimique ou marquage des protéines. Ces avantages ont fait MS structurelles un outil puissant pour l’étude structurale des complexes protéiques.
Native MS est cumulable avec la mobilité de l’ion (IM), une technique qui mesure le temps de qu'un ion de protéine prend de voyager à travers un champ électrique, permettant à la section efficace de collision (CCS) à déterminer. Le CCS renseigne basse résolution structurale, qui permet la topologie et les informations d’hétérogénéité conformationnelle des protéines qui doivent être obtenus. En outre, il permet l’examen des modèles structuraux de protéine produite par approches computationnelles.
Stabilité de phase gazeuse des protéines peut être étudiée à l’aide de collision induite déplier (CIU) mesurée par IM-SM. Au cours du processus CIU, ions de protéine sont accélérées et activées par le biais des collisions accélération accrues avec un gaz inerte tampon dans un spectromètre de masse de8,7,9. Ce processus d’activation par collision provoque la protéine se dérouler partiellement, qui se traduit par une augmentation du CCS. Ce changement de CCS et l’énergie nécessaire pour déplier la protéine peut être mesurée à l’aide de l’IM-Mme cette approche, l’effet de la liaison du ligand sur la stabilité des protéines peut être mesuré10. Subcomplexes peuvent être générés en solution en utilisant des méthodes de perturbation dans la solution telle que l’ajout de solvants organiques pour surveiller des topologies de type natif de complexes protéiques. Perturbation des complexes de protéine est principalement dû à la rupture des interactions non-covalentes intra. Les complexes sous maintiennent des topologies de type natif et, en cas de détection de MS, révèlent des informations sur la sous-unité inter connectivité.
Des approches intégratives en biologie structurale combinent diverses méthodes pour étudier la structure et la dynamique des protéines et leurs complexes3,4,5,6. Natif de MS et d’IM-MS ont été utilisés pour découvrir les détails moléculaires des systèmes biologiques difficiles. Il y a eu plusieurs exemples d’applications, y compris l’étude de la protéine Assemblée voies11,12,13,14, étudie l’interaction de protéine-protéine réseaux15 , 16 , 17, membrane protéines6,18,19,20,21et interactions de protéine-ligand comme les acides nucléiques22,23 ,,24.
Cependant, native MS a aussi ses limites. Native MS mesures sont souvent effectuées dans des tampons volatils tels que l’acétate d’ammonium aqueux dans lequel certaines protéines ne conservera pas leur état natif plié3,25. Néanmoins, les travaux récents ont montré que cette limitation peut être surmontée par l’optimisation de pulvérisation diamètre de pointe de l’aiguille (pointes de 0,5 mm), telle que la protéine et protéine ions complexes peuvent être formées directement à partir de tampons non volatile avec force ionique élevée que mieux imiter l' environnement physiologique26. En outre, native MS utilise electrospray pour ioniser et transférer des assemblys non covalents de solution dans la phase gazeuse ; par conséquent, l’abondance relative des complexes détectés ne peut-être entièrement représenter qui en solution5,27. Par ailleurs, en comparaison à en solution, les interactions hydrophobes de phase gaz deviennent plus faibles et les interactions électrostatiques deviennent plus fortes et donc favorisé3,28.
Dans cet article, nous fournissons des protocoles, l’analyse des données et interprétation pour l’identification de protéines et de ligand binding utilisant native MS, IM-MS, CIU, perturbation dans la solution et la modélisation. Le complexe de réparation d’ADN, HerA-NurA, est utilisé comme système modèle. Cassures double-brin d’ADN (CDB) sont une des formes plus cytotoxiques et délétères de dommages à l’ADN, ce qui entraîne une instabilité génétique et le développement ultérieur du cancer chez les humains. La recombinaison homologue est le mécanisme de réparation qui éradique la CDB, un processus qui est orchestrée par l’ATP dépendante helicase-nucléase complexe, HerA-NurA22.
Combinant native MS et IM-MS avec tests fonctionnels et de modélisation a permis à l’enquête de : i) le rôle de NurA dans l’Assemblée, conformation et la stabilité de l’immeuble, ii) l’interaction entre l’ADN double brin et le complexe et son influence sur l’ensemble stabilité de l’immeuble et iii) la stoechiométrie et l’impact de liaison de l’ATP sur l’Assemblée22. Dans l’ensemble, ce travail a conduit à une meilleure compréhension de la base moléculaire du complexe HerA-NurA en liant les changements conformationnels complexes protéiques et la stabilité avec la liaison de nucléotides. Ce protocole est générique pour n’importe quel complex(es) de protéine qui interagit avec un ou plusieurs types de toute.
Protocole
1. préparation pour MS Native des protéines et des Complexes protéine-Ligand
NOTE : Pour mieux comprendre les bases moléculaires d’une protéine complexe et liaison de ligand à l’aide de MS native, préparation de l’échantillon adéquat est la clé. Cette section vise à mettre en évidence les étapes de préparation indispensable échantillon avant l’analyse MS utilisant le complexe HerA-NurA qui lie l’ADN et des nucléotides à titre d’exemple.
- Préparer 20 µL d’extraits de concentré de protéine purifié (généralement 15 à 30 µM) dans un tube de 1,5 mL.
-
Pour les analyses de liaison ATP ou ADP
NOTE : Ajouter des concentrations croissantes de la non hydrolysables ATP adénosine analogique 5′-O-(3-thiotriphosphate), tetralithium sel (ATP-γ-S) ou 5′ adénosine-diphosphate (ADP). Dérivés de ATP non hydrolysables génèrent un complexe stable qui permettrait à la protéine de liaison ATP à capturer. Autres non hydrolysables analogues d’ATP qui pourraient être testés comprennent AMP-PNP et ATP-γ-S-Mg2 +.- Pour les études HerA-NurA, mélanger 5 µM de protéine purifiée avec ATP-γ-S et ADP à des concentrations allant de 0-1 mM.
- Pour capturer une liaison ATP-γ-S et ADP simultanée, ajouter les deux nucléotides à des concentrations identiques ou différentes.
- Ajouter 2 mM MgCl2 et incuber à 25 ° C dans une étuve à bain sec pendant 1 h.
Remarque : Analyse de liaison de nucléotides à l’aide de nESI que Native MS peut se traduire par liaison artéfactuelles à des concentrations élevées, donc non-spécifiques doit prendre en compte29. Afin d’étudier les liaisons non spécifiques, ajouter une concentration plus élevée des nucléotides entre 2-5 mM).
-
Pour analyse de liaison génétique
- Mélanger les protéines et l’ADN à un rapport molaire qui prévoit la formation d’un complexe protéine-ADN. Pour HerA et HerA-NurA, mélanger 5 µM de protéine purifiée avec de l’ADN à un ratio de 1:1.
- Incuber le HerA-NurA ou HerA - mélange d’ADN pendant 30 min à 25 ° C dans une étuve à bain sec jusqu'à ce qu’il atteint l’équilibre. La durée et la température d’incubation peuvent varier selon la protéine incriminés.
- Mémoire tampon échange des échantillons de protéines aux tampons compatibles MS. Solution d’acétate d’ammonium aqueux entre 5 mM-1 M à pH 7 et 8 sont généralement utilisés. Autres tampons compatibles MS comprennent ethylenediammonium diacétate (EDDA) et triéthylammonium acétate (TEAA)30. Pour les études HerA-NurA, utiliser 200 mM d’ammonium acétate pH 7.
Remarque : Il existe plusieurs méthodes pour l’échange de la mémoire tampon avant l’analyse par SM comme l’utilisation d’un concentrateur de spin ou colonnes de chromatographie. Native MS est principalement limitée par la qualité de l’échantillon, tels que les adduits et met en mémoire tampon utilisée durant la purification. Par conséquent, il est essentiel pour effectuer le dessalage suffisante pour obtenir des pics résolus. - Pour les études de liaison de ligand HerA-NurA, tampon échange échantillons 6 - 8 fois en acétate d’ammonium de 200 mM à l’aide d’un concentrateur. Bien que cette méthode est beaucoup plus de temps, il veille à ce que soit résolu sommets sont atteints et permettant la détermination de la masse exacte des espèces dépendant de l’ATP/ADP.
2. native MS Acquisition et analyse pour l’étude des Complexes protéiques et des Complexes protéine-Ligand
Remarque : Les conditions SM doivent être optimisées pour atteindre des pics très résolues afin de permettre des mesures précises de massives. Ce paragraphe détails optimisé paramètres sur un spectromètre de masse Q-ToF avec un quadripôle de m/z 32 k limite supérieure.
- Préparer les capillaires internes nano-electrospray et effectuer d’étalonnage des masses instrument pour des mesures précises de massives comme indiqué par Kirshenbaum et al. 1.
- Sélectionnez la sensibilité, l’acquisition de l’ion positif et modes TOF de mobilité.
- Allumez les gaz piège, API et IMS. Pour la séparation de l’IM, utilisent l’azote (60 mL/min) et argon (8,4 mL/min pour la région de piège) comme points de départ et ensuite ajuster.
- Définissez une plage d’acquisition approprié m/z. Pour une protéine inconnue, étapes d’optimisation initiale doivent utiliser une large gamme telle que 500-32 000 m/z.
- Charger 2 ou 3 µL de la solution complexe de protéines à doser dans un capillaire or couché et l’insérer dans un support capillaire.
- Doucement, serrer le tube capillaire et placez le tube capillaire au stade de la source électrospray et glissez la scène en position de commencer à acquérir des données.
- Appliquez une pression de gaz de nano-débit faible (0,00 à 0,05 Bar) jusqu'à ce qu’une goutte d’eau est formée à l’extrémité du capillaire. La pression d’alimentation-nano peut ensuite être supprimée jusqu'à ce que le spray est maintenu.
- Ajuster le capillaire en ce qui concerne le cône en déplaçant le capillaire x, y, z les positions et surveiller l’ion actuelle pour atteindre un ion stable actuel. Appliquer une tension capillaire de l’ordre de 0,9 à 1,6 kV.
- Définissez le cône d’échantillonnage (50-120 V), température (25 ° C) de la source et l’offset source (60,0), débit de gaz cône (0.0 L/h). Les suggérées conditions initiales peuvent être ajustées.
- Pour acquérir un spectre de masse bien résolu et maximiser la transmission de l’ion, régler les paramètres de MS et de surveiller le changement qui en résulte dans les spectres. Ceux-ci incluent le réglage du débit de gaz dans le piège (2 à 8 mL/min), la cellule il (180 mL/min) et la cellule IMS (90 mL/min) qui permet la meilleure séparation à transmission maximale.
- Ajuster les énergies de collision de piège si les offsets de tension sont insuffisantes. Un point de départ optimal est entre 10 et 50 V.
NOTE : Augmenter l’énergie de piège peut supprimer lié de façon non covalente adduits. Cependant, prendre soin d’éviter la dissociation induite par collision et déroulement du complexe protéine-ligand. Effectuer des mesures de mobilité ionique pour vérifier si des conditions aux instruments conservent la protéine à l’état natif plié (étape 3). - Améliorer la désolvatation en optimisant la tension de polarisation de piège. Un point de départ optimal est V 20-45.
- Optimiser la vitesse de l’onde et la hauteur des vagues qui permet la meilleure mobilité séparation. Une explication détaillée et protocole peuvent être trouvés ici31. Pour les études HerA-NurA, utiliser la vitesse de l’onde de 40 (m/s) et la hauteur des vagues de 550-650 (V).
- Utiliser tous les autres paramètres comme valeurs par défaut d’instrument.
- Préparer un échantillon exempt de ligand pour l’analyse comme un contrôle pour chaque série (Figure 1). Pour des expériences de liaison de ligand, effectuer au moins trois mesures indépendantes.
- Utilisez le logiciel Masslynx pour mesurer les masses des espèces générées et identifier la liaison du ligand, telles que la liaison de l’ATP et l’ADP et États oligomériques (Figure 2 et 3). Autres logiciels disponibles incluent UniDec32, PULSAR33 et Amphitrite,34.
- Afin de quantifier l’abondance relative des espèces, utiliser l’ion correspondant des intensités observées dans les spectres de ESI-MS bruts (par exemple ligand lié, oligomères différents, etc..). Vous pouvez également exécuter quantification à l’aide de logiciels spécialisés comme UniDec et Massign35 (Figure 1 et 3 ).
3. acquisition et l’analyse IM-MS
NOTE : IM-MS sépare les ions dans la phase gazeuse issu de leur taille (masse), la forme et la charge. Toutes les fonctionnalités résolue dans spectre de m/z sont associée à une distribution de temps de dérive. IM-MS mesure la durée de dérive d’un ion qui peut être utilisée pour calculer la section efficace de collision (CCS). Les valeurs de date dérive mesurées de données acquises IM-MS à l’aide de qu'un tube en dérive peut être linéairement corrélé à CCS valeurs36. Pour voyager de mesures d’ondes IM-MS (TWIMS), calculer des valeurs de CCS nécessite une courbe d’étalonnage obtenue à partir des normes de protéine connus CCS valeurs37. Structures compactes voyagent plus vite que l’étendue ou structures allongées due à réduit les interactions avec les gaz de tampon dans la mobilité cellulaire38. Par conséquent, IM-MS peut être utilisé pour détecter si la structure plissée native a été retenue dans le gaz phase39,40. Cette section décrit comment mesurer IM-MS et calculer le CCS de protéine à l’aide de TWIMS.
- Après l’optimisation des conditions d’instruments pour une transmission stable (étape 2), réduire l’énergie de collision et le cône d’échantillonnage aussi bas que possible tout en conservant une qualité bonne de spectres.
- Utiliser la vitesse de l’onde optimisée et la hauteur des vagues à acquérir IM-MS (étape 2).
- Mesurer la dérive d’ion temps avec IM-MS à trois vitesses des ondes différentes (par exemple, 550, 600 et 650 m/s) tout en conservant la même hauteur de vague (par exemple, 40 V).
- Pour déterminer les ions de protéines CCS, calibrants de protéine de mesure dans les mêmes conditions d’instrument utilisées pour la protéine incriminés. Calibration de dérive-temps optimale requiert la mesure des protéines avec CCS connus.
- Sélectionnez les quatre calibrants, deux avec une masse supérieure et deux avec une masse inférieure à celle de la protéine sous enquête37. Surtout, assurez-vous que la hauteur des vagues et la vitesse de l’onde sont les mêmes que ceux enregistrés pour la protéine incriminés.
- Calculer CCS manuellement41 ou à l’aide d’un logiciel spécialisé tel que le PULSAR33 et Amphitrite34 (Figure 5).
- Pour vérifier si la protéine est comme un natif dans la phase gazeuse, CCS expérimentale compare au CSC théorique obtenu à partir des structures de haute résolution. Pour HerA-NurA, calculer CCS théorique en utilisant la méthode de Projection rapprochement (PA) utilisée dans MOBCAL42. Autres méthodes incluent la trajectoire méthode (TM)42 et des sphères dures exacte diffusion (EHS)43.
4. en-Solution les perturbations des Complexes de protéine Native MS et IM-MS a conduit la détermination de la Structure
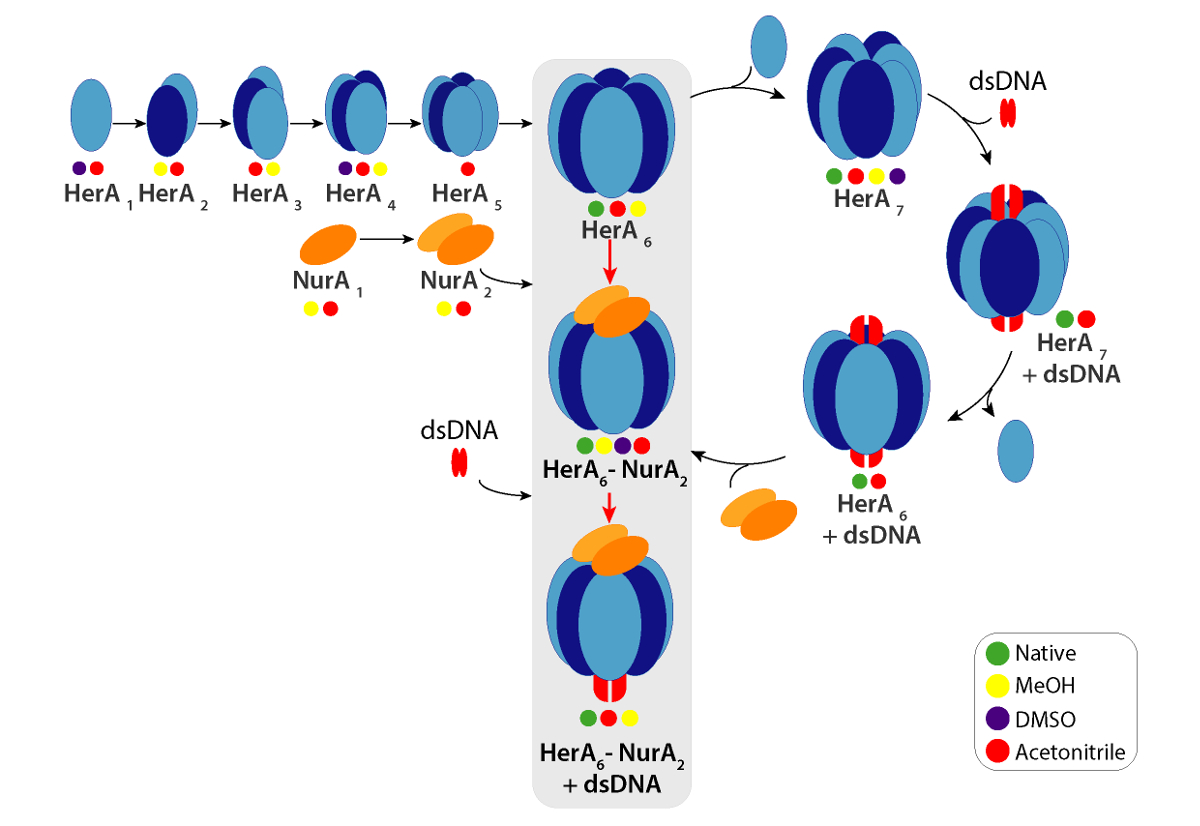
Remarque : Des complexes de protéine peuvent dans certains cas être identifiées à la même solution que le complexe intact. Toutefois, davantage d’informations structurelles telles que la sous-unité inter connectivité et complexes d’assemblage peuvent être assurées de perturber les interactions des protéines en solution, pour former des complexes sous. Ceci peut être réalisé de plusieurs façons, telles que l’ajout de solvant organique, augmente la force ionique ou en manipulant le pH. Pour avoir un aperçu de la connectivité de sous-unité complexe HerA-NurA et assemblage complexe, secondaires complexes ont été produites en solution en ajoutant des solvants qui perturbent les interactions de sous-unité.
- Préparer l’échange de sample et tampon de protéine dans l’acétate d’ammonium comme décrit à l’étape 1.
- Ajouter 10 à 40 % de solvant par incréments de 10 %. Solvants utilisés généralement sont le méthanol (MeOH), le diméthylsulfoxyde (DMSO) ou acétonitrile (ACN).
Remarque : Ceci peut être effectuée dans un tube en polypropylène microcentrifugeuse. - Incuber le mélange sur la glace pendant 1 h.
- Acquérir un spectre d’IM-MS pour chaque affection (étapes 2 et 3) (Figure 4).
- Utilisez le logiciel SUMMIT44 pour assigner des complexes de protéine et de générer des réseaux d’interactions protéiques. Vous pouvez également générer manuellement une liste de masses théoriques de l’espèce attendue.
- Afin de subcomplexes sont pliées, calculer les valeurs expérimentales de CCS pour les complexes sous et comparer à CCS théoriques, comme l’a expliqué à l' étape 3 (tableau 1, Figure 5 et Figure 6).
5. enquête sur la stabilité des protéines complexes à l’aide de dépliage induite par Collision (CIU)
Remarque : CIU permet la stabilité structurale des protéines et leurs complexes sur la liaison du ligand de la sonde. Spécialiste des progiciels tels que PULSAR33, Amphitrite34 et CIU suite9 puis peuvent être utilisés que pour modéliser le déroulement phase gazeuse de la protéine étudiée avec et sans ligand. À titre d’exemple, cette section décrit les modalités de la surveillance de phase gazeuse qui se déroulent de trajectoires et d’étudier l’effet stabilisant de liaison ADN et ATP sur le complexe de HerA-NurA.
- Enregistrer des données IM-MS tout en augmentant la tension d’accélération de piège de 10 V à 200 V en incréments de V 2-10 de se déployer progressivement de la protéine dans la phase gazeuse.
NOTE : Enregistrement petits incréments résultats dans plusieurs fichiers de données à traiter, cependant, cette approche fournit plus résolu intrigue qui se déroule, qui est important pour analyser les points de transition entre les espèces plié/déplié. - Analyser les données recueillies à l’aide de PULSAR33, Amphitrite34 ou SDI suite9 et générer des parcelles qui se déroule à deux dimensions en unités du CSC en fonction de la tension (étape 3) d’accélération. Pour chaque État de charge, celle-ci est créée par empilement les distributions de CCS intensité-normalisé à chaque tension d’accélération (Figure 7 A-Bi).
- Générer une intrigue qui se déroule théorique en utilisant l’un des progiciels. Les données seront dotées un modèle qui se déroule. Cela rend possible de quantifier l’énergie de collision de transitions qui se déroule se produisent et déterminer la stabilité des protéines avec et sans des ligands liés33. Une transition qui se déroule est lorsqu’une transition de l’espèce d’un État (selon leurs valeurs expérimentales de CCS) vers un autre État avec un CCS plus grande.
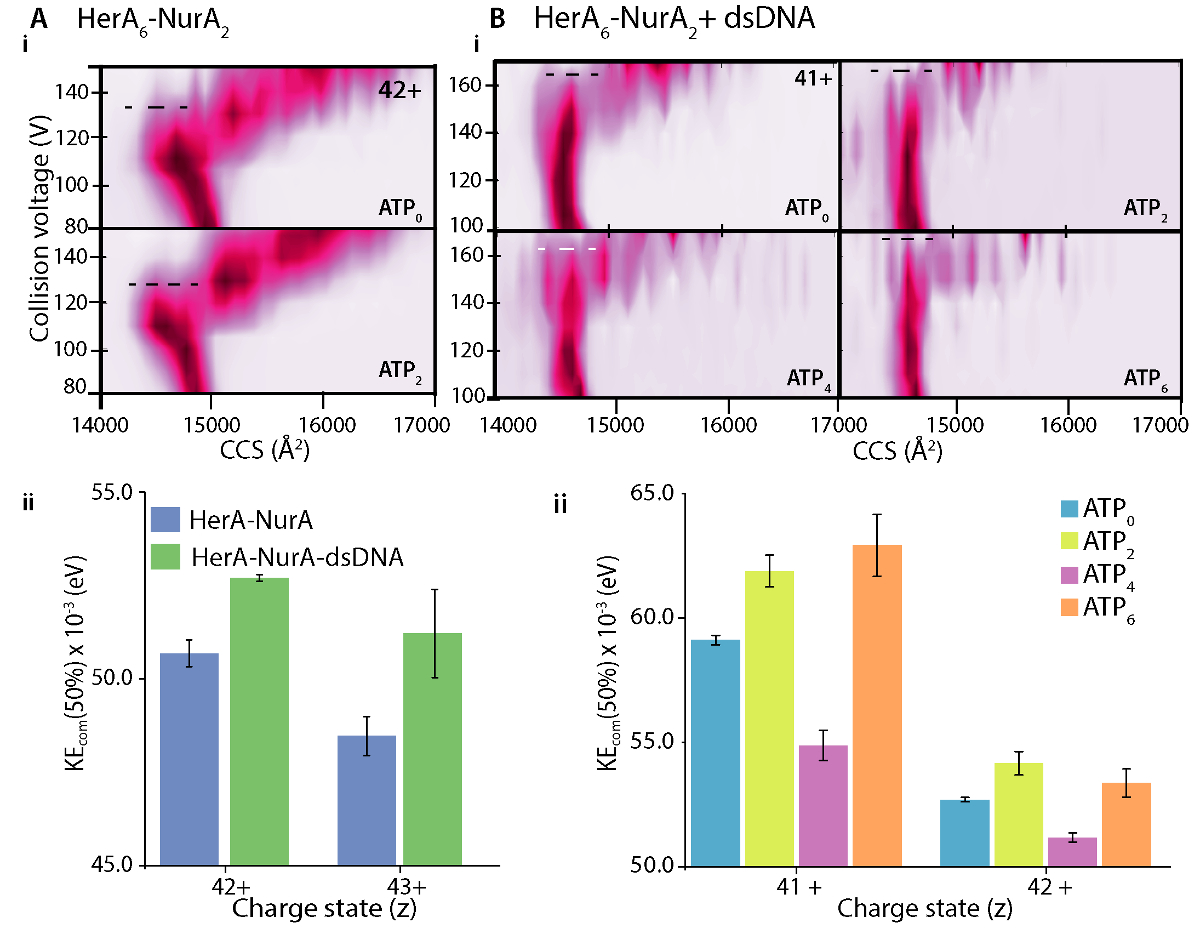
- Pour doser les transitions, calculer les midpoint(s) transition entre États à l’aide d’algorithmes et logiciels tels que PULSAR33. Ceci est fréquemment signalé comme CV50, qui est la valeur de tension de collision (trap) au cours de laquelle 50 % d’un État donné est épuisée.
- À l’aide de la valeur de50 CV, calculer l’énergie interne totale d’un ion en utilisant le centre de masse collision énergie (KECOM)45. KECOM est défini par l’énergie interne totale disponible pour la transition qui se déroule d’un ion et est calculée à partir l’énergie cinétique et les masses des partenaires collision (protéine ion et gaz neutre) comme décrit dans l’équation (1)10
KEcom (eV) = (équation 1).
(équation 1).
Où Z est la charge ionique,N est la masse de gaz neutre et MION est la masse de l’ion de protéine.
NOTE : C’est parce que CIU de protéines est frais dependent46, 47. Il est recommandé d’effectuer l’analyse KECOM à plus d’un état de charge ( Figure 7Aii).
6. modélisation des procédures pour des Simulations de dynamique moléculaire différentiels utilisés dans Integrative MS
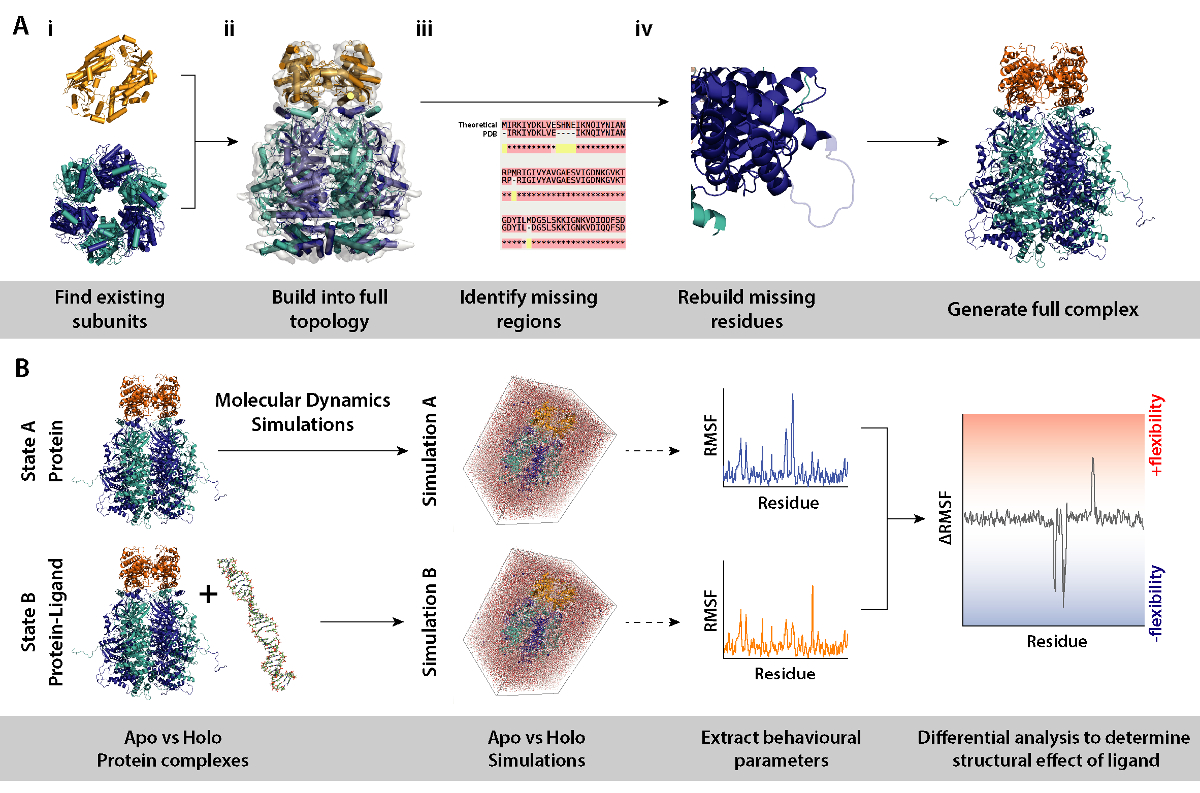
Remarque : L’utilisation de modèles de sous-unités protéiques ou complexes tels que des structures cristallines, différentiel MD simulations (protéines complexes avec et sans ligand) peuvent être utilisées pour déterminer les effets de par exemple présence de ligand sur la dynamique et la structure des protéines. Cette section décrit en détail un flux de travail et les outils nécessaires pour la modélisation des procédures nécessaires pour les simulations de dynamique moléculaire différentielle de Set-up.
- Identifier les sous-unités qui composent le complexe (Figure 8A, aux étapes 2 et 3). Source des modèles existants de sous-unités, par exemple, les structures cristallines de la Banque de données RCSB (https://www.rcsb.org). Le UniProt, entrée de la protéine contiendra une liste des savons structures cristallographiques/NMR (http://www.uniprot.org). Si ce ne sont pas disponibles, la séquence théorique peut être entrée à BLAST pour identifier des modèles appropriés d’homologie modélisation (http://blast.ncbi.nlm.nih.gov/).
- Assembler le complexe dans la topologie correct (Figure 8A-ii). Cela peut être fait par le biais de diverses méthodes. Les sous-unités individuelles peuvent être montées dans la microscopie électronique disponible cartes trouvées sur le EMDB pour assembler le complexe intact (https://www.ebi.ac.uk/pdbe/emdb/). Un tutoriel pour le montage des fichiers PDB EM des cartes à l’aide du raccord souple dynamique moléculaire (MDFF) peut être trouvé ici : http://www.ks.uiuc.edu/Training/Tutorials/science/mdff/tutorial_mdff-html/.
- Identifier les régions manquantes du complexe (Figure 8A-iii). Effectuer des alignements multiples de séquences (MSA) APB / séquence théorique visant à identifier les résidus qui peuvent être non ajustées dans les structures cristallines, ou n’importe quel mutations héritées d’expériences cristallographiques. MSA peut être effectuée en utilisant les serveurs Web tels que le T-café (http://tcoffee.crg.cat/apps/tcoffee/do:regular).
- Régénérer les résidus manquants par homologie de modélisation (Figure 8A-iv). Manque de résidus de la protéine complexe peut être construit en utilisant le programme modeleur (https://salilab.org/modeller/). Modeleur peut produire un ensemble de modèles n dans différentes configurations régénérées. Les bons modèles peuvent être identifiés selon leur score de discrètes optimisé protéine énergie (DOPE). Un tutoriel complet est disponible sur le site du logiciel (https://salilab.org/modeller/tutorial/).
- Réaliser des simulations de différentiel dynamique moléculaire (DM) de la protéine complexe (Figure 8B) pour identifier les régions de protéines qui répondent à un changement environnemental particulier, par exemple, la présence d’un ligand. Dans ces simulations, paramètres comportements de Simulation A (protéine uniquement) qui agit comme une référence, est soustraite de la Simulation B (protéine + ligand). La fluctuation de différentielle quadratique moyenne (FPMR) calculée entre les Simulations A et B peut informer sur les régions de la protéine qui augmentent ou diminuent sur la souplesse d’une manière dépendante du ligand.
- Effectuer des simulations de MD et analyse en aval à l’aide de GROMACS (http://www.gromacs.org). Un tutoriel peut être trouvé à : http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin/gmx-tutorials/Lysozyme/index.html. Biais d’elimate de modèle, la structure du complexe ligand-limite devrait être générée tout d’abord. La protéine est ensuite copiée à partir cela sans le ligand, pour produire un modèle de protéines identique au complexe ligand lié.
Résultats
Native MS résultats ont révélé l’État oligomère, la composition et la topologie du complexe HerA-NurA (Figure 1). Comme les interactions non covalentes sont conservées dans la phase gazeuse, native MS d’ATP-γ-S et des expériences de titrages ADP détermine la liaison par paires de nucléotides à HerA-NurA (Figure 2) et que l’augmentation de la concentration d’ATP-γ-S augmente la relative intensité de hexamérique HerA (Figure 3). Des informations structurelles concernant les interactions sous-unité provenaient de perturbation solution suivie native MS et étaient en accord avec et masses théoriques (Figure 4 et tableau 1).
Les valeurs expérimentales de CCS de protéines et de leurs complexes a été dérivé d’expériences IM-MS (Figure 5). Ces valeurs sont gazeux par rotation moyenne des calculs transversales de la forme moléculaire et décrivent l’État tridimensionnelle de la protéine. CCS valeurs sont comparées aux mesures théoriques de la cristallographie aux rayons x et un bon accord en déduit que la forme native dans retenus dans la phase gazeuse (tableau 1). Cela valide à l’aide de valeurs de CCS pour construire des modèles basse résolution de l' Assemblée de protéine48.
CCSs expérimentales peuvent être calculés pour chaque ion d’état de charge. Un conformère protéine native peut-être donner lieu à ions de l’état de charge avec des valeurs similaires de CCS. Cependant, les ions état frais plus élevées augmente répulsions coulombiennes pouvant conduire à la protéine phase gazeuse qui se déroule et plus CCS valeurs par rapport à la CCSs théorique. La valeur de CCS de l’état de charge le plus bas, les ions sont donc habituellement utilisée49. Pour HerA-NurA, expériences de la perturbation en solution sur HerA et HerA-NurA avec et sans ADN a incité la génération d’une voie d’assemblage à partir de monomères puis formant l’ensemble hexamérique HerA (HerA6)-complexe dimère de NurA (NurA2) avec l’ADN (Figure 6).
Différences dans les parcelles qui se déroule de CIU entre l’apo (ligand-libre) et un ligand lié définissent le changement en stabilité complexe sur la liaison du ligand. Un supérieur CV50 ou valeur KECOM implique un ion plus stable dans la phase gazeuse. CIU et KECOM analyse révèle que lié aux ADN HerA-NurA est plus stable que le complexe sans ADN (Figure 7Aii). De l’analyse de CIU-MS dans les États respectifs des ATP-binding, quatre-ATP-γ-S État lié réduite stabilité complexe dans la phase gazeuse et l’État lié de six -ATP-γ-S où sont tous les sites est occupés a été le plus stable (Figure 7Bii). Native MS peut révéler le nucléotide discrète, contraignant les États d’Héra ; Toutefois, il ne peut pas distinguer les sous-unités HerA lient ATP et où s’effectue cette liaison. Cette information peut être dérivée des simulations de MD solvants explicites sur la hexamérique HerA et le HerA-NurA suivant le flux de travail succinct (Figure 8).

La figure 1. Interroger l’État oligomère, la composition et la topologie du complexe non covalent HerA-NurA. (A) spectres de masse de Héra, HerA-NurA et HerA-NurA, en présence de l’ADN (ADN bicaténaire de 15,4 kDa 25 bp). Le complexe sous HerA existe en tant qu’un hexamère et un PA63. Dimère de NurA lie et à un hexamère de HerA imposant conversion oligomère. L’ADN lie le complexe formé de HerA - NurA (résultats adaptés de Z. Ahdash et al., 201722). (B) les intensités des espèces identifiées sont calculées au moyen de l’UniDec32. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
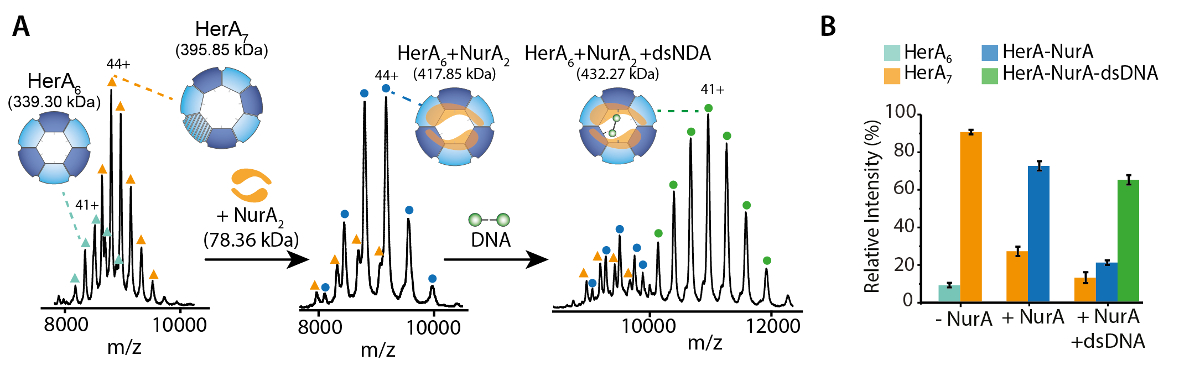{kind=link}

La figure 2. Natif de ESI-MS révèle le mécanisme de liaison de nucléotides à HerA-NurA. Spectres de masse (A) HerA-NurA et (B) HerA-NurA-ADN avec des concentrations croissantes de (i) ATP-γ-S et (ii) ADP. Les masses mesurées sont comparés aux masses théoriques et la quantité d’ATP-γ-S ou ADP lié sont déterminés. Les masses mesurées et nombre de nucléotides liés sont indiqués sur les spectres. Les expériences de titrages ATP-γ-S et ADP déterminé la liaison par paires de nucléotides d’HerA-NurA seul et en complexe avec l’ADN, ce qui indique un mécanisme de réaction cyclique (résultats adaptés de Z. Ahdash et al., 201722). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
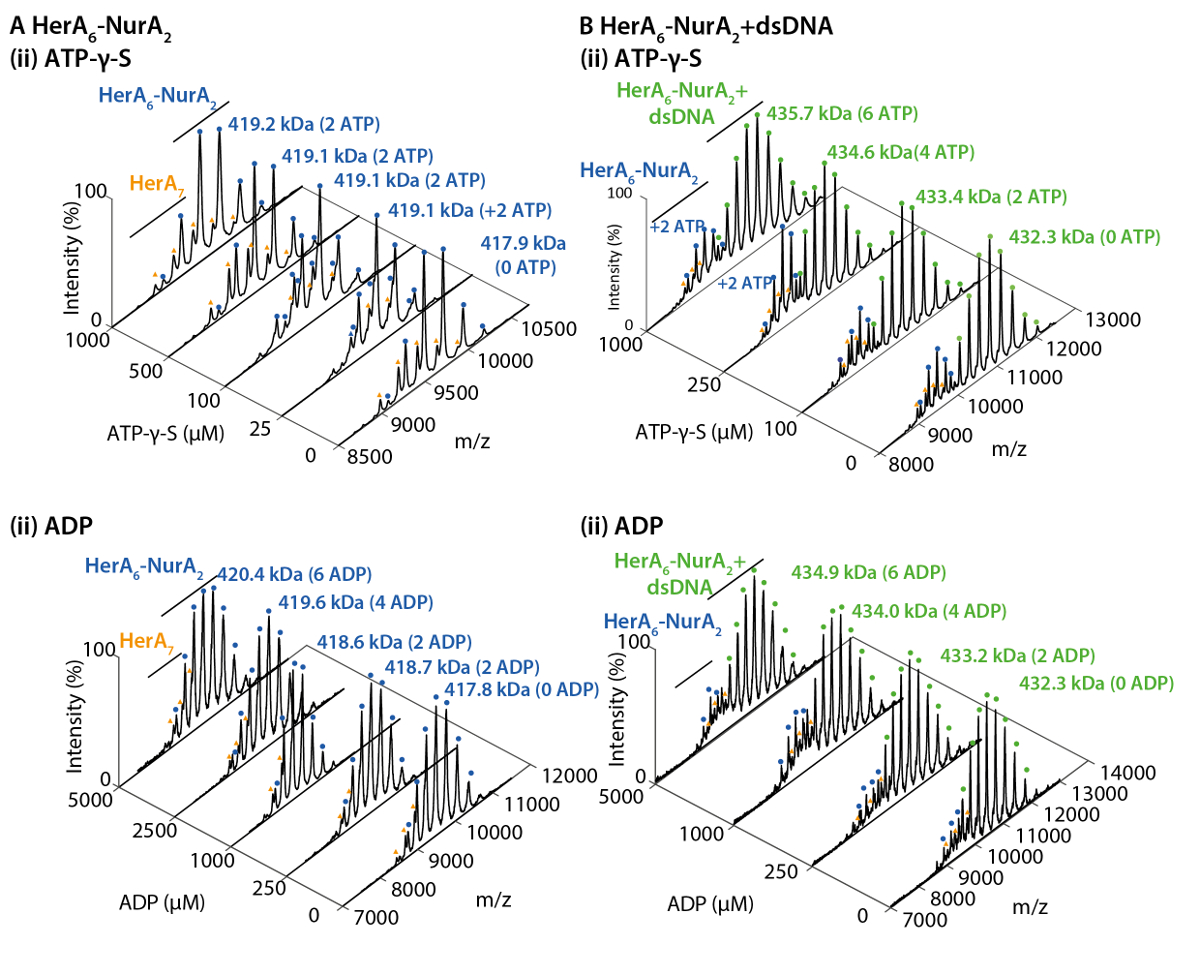{kind=link}

La figure 3. Mesurer l’effet de l’augmentation des concentrations d’ATP-γ-S État oligomère HerA. (A) les spectres de masse d’Héra à accroître les concentrations d’ATP-γ-S. (B) graphique illustrant les intensités relatives des différentes espèces de native MS calculées à l’aide de logiciels déconvolution UniDec32. Lorsque la concentration en ATP-γ-S augmente, l’intensité relative des hexamérique HerA augmente également. Nombre de molécules d’ATP-γ-S lié est indiqué sur les spectres (résultats adaptés de Z. Ahdash et al., 201722). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
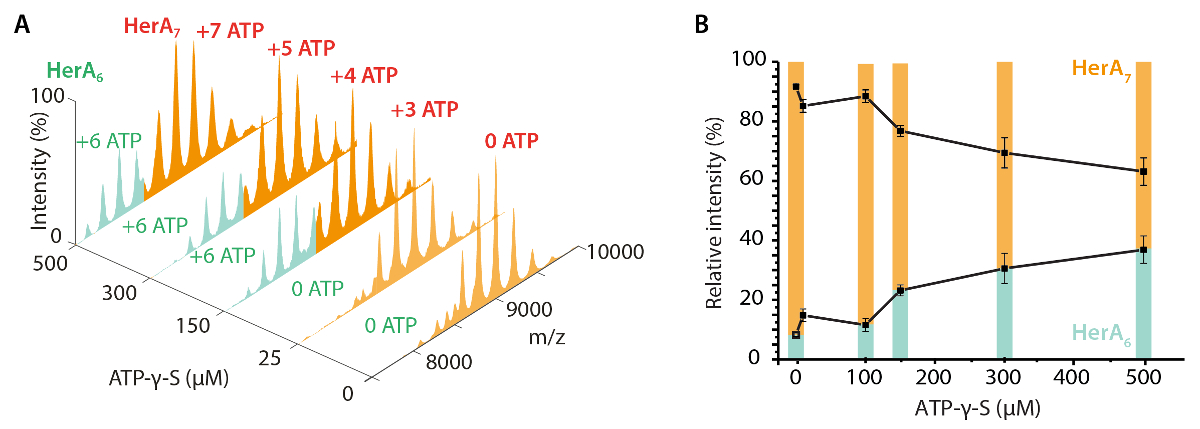{kind=link}

La figure 4. Masse de spectres et produits de dissociation des complexes de HerA (A) et (B) HerA-NurA (i) seul ou en présence de (ii) l’ADN qui suit d’une perturbation solution. A réalisé des expériences de perturbation dans la solution à l’aide de 10 à 40 % d’acétonitrile, méthanol (MeOH) ou le diméthylsulfoxyde (DMSO) et conduit à la formation de diverses subcomplexes. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5. Ion mobilité arrivée distributions de temps montrées sur un axe de CCS pour complexes et généré des complexes. Icône pour chaque Sub complexe en corrélation avec celles annotées sur les spectres à la Figure 4. Masses expérimentales et calculées et les valeurs de CCS de complexes sous figurent dans le tableau 1 , qui a montré un accord entre valeurs calculées et expérimentales (après compte tenu de l’incertitude typique dans la résolution de voyager ion vague mobilité la spectrométrie de masse de ± 5 - 8 %37). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
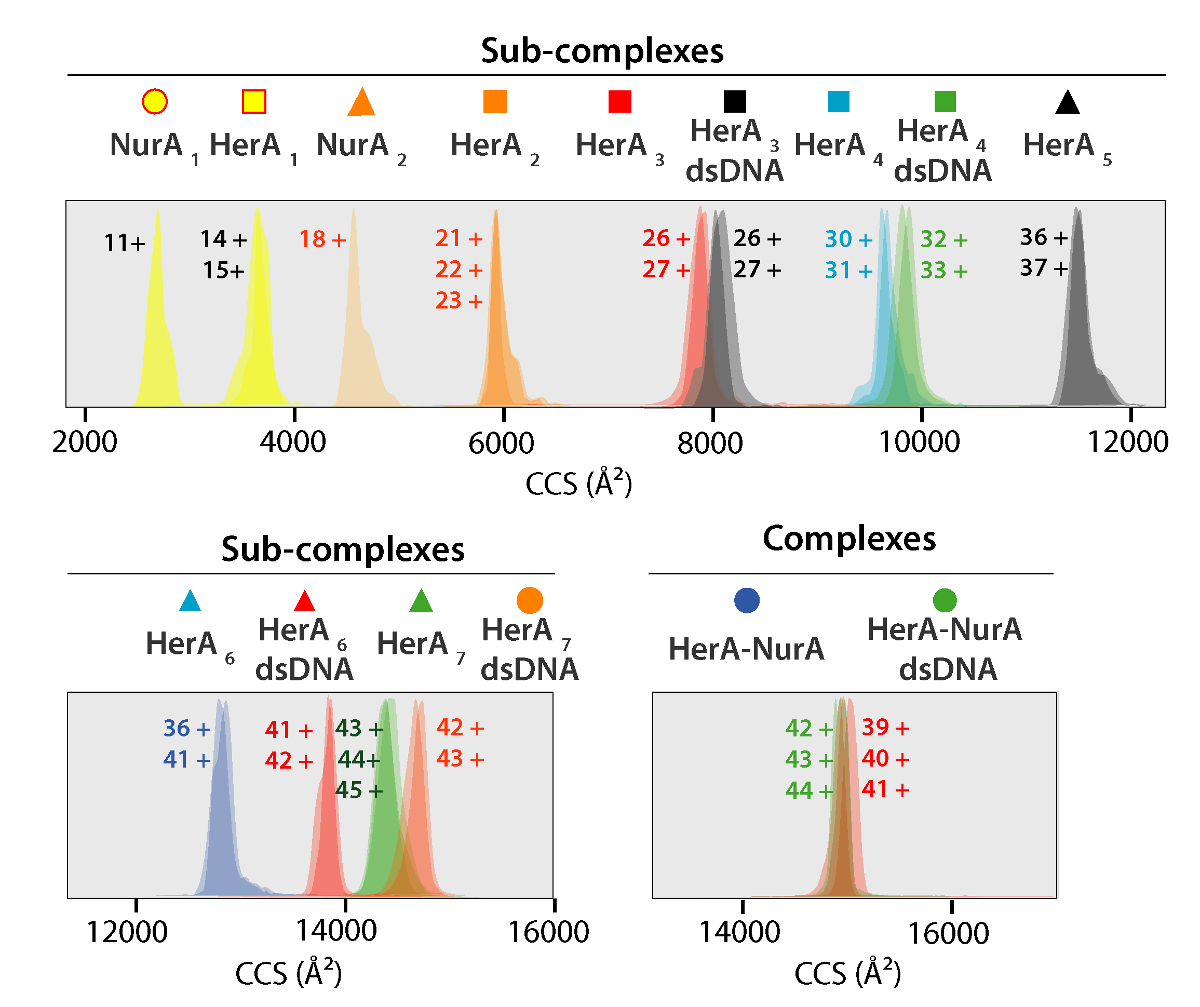{kind=link}

La figure 6. Voie de l’assemblage du complexe HerA-NurA généré de perturbation solution natives cercles MS et IM-Mme colorées indiquent des conditions où chaque complexe secondaire a été observé : natif (vert, avant l’interruption), méthanol (jaune), DMSO (violet) ou Acétonitrile (rouge). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

La figure 7. Enquête sur l’effet stabilisant de l’ADN (A) sur HerA-NurA et (B) ATP binding à HerA-NurA-ADN. i phase gazeuse CIU-MS parcelles et (ii) Centre-de-masse collision énergies (KEcom) calcul qui montrent que la présence d’ADN double brin stabilise le complexe HerA-NurA et que les six ATP-γ-S lié état est le plus stable. Stabilisation des États de charge différent est affichée. Parcelles ont été générés à l’aide de PULSAR 33. Résultats de (A) adaptés de Z. Ahdash et al., 201722. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

La figure 8. Flux de travail pour les procédures de modélisation pour les simulations de dynamique moléculaire différentielle. (A) générant le complexe sous enquête par la construction de la topologie des existantes sous-unités et reconstruction des résidus manquants. B flux de travail pour l’exécution de simulations de dynamique moléculaire sur la protéine complexe avec et sans ligand. Molecular dynamics simulations sont couru pour la protéine seulement qui agit comme une référence et qui est soustraite de la simulation de protéines plus ligand. Il est suivi par le calcul de la fluctuation de la différentielle quadratique moyenne (FPMR) entre les simulations et détermination de l’effet de liaison du ligand. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Complexes / Sub complexe | Masse théorique (kDa) | Masse expérimentale (kDa) | CCS théorique (Å2) | CCS expérimentale (Å2) [charge] | Condition HN |
| HerA6NurA -2 | 416,22 | 417.85 | 14531 | 14577 [42 +] 14599 [43 +] 14608 [44 +] 14637 [45 ans] | 10-20 % ACN, DMSO de 10 à 40 %, 10 % MeOH |
| HerA, NurA -62- ADN double brin | 431.72 | 432.27 | - | 14661 [39 +] 14728 [40 +] 14781 [41 +] 14837 [42 +] | ACN de 10 %, 10 % MeOH |
| NurA1 | 39.12 | 38.18 | 3254 | 2618 [10 +] 2746 [11 +] 2878 [12 ans et +] | 10-40 % ACN, MeOH de 10 %, 20-40 % DMSO |
| NurA2 | 78,24 | 78.36 | 4890 | 4903 [16 +] 4614 [17 +] 4537 [18 +] 4666 [19 +] | MeOH de 10 à 40 %, 20-40 % DMSO |
| HerA1 | 56.33 | 56.32 | 4131 | 3647 [14 +] 3792 [15 +] 3950 [16 +] | ACN DE 10 À 40 %, 40 % DMSO |
| HerA2 | 112.66 | 112,95 | 6475 | 5648 [20 +] 5747 [21 +] 5842 [22 +] 5996 [23 +] | 40 % meth, ACN de 10 à 40 % |
| HerA3 | 168,99 | 169,39 | 8607 | 7501 [25 ans] 7616 [26 +] 7717 [27 +] 7867 [28 +] | 10-40 % MeOH, bidon de 10 à 40 %, 40 % DMSO |
| HerA3 + ADN | 183,99 | 184.976 | - | 7655 [26 +] 7990 [27 +] 8107 [28 +] | 10-30 % ACN |
| HerA4 | 225.32 | 226,2 | 10477 | 9205 [30 +] 9287 [31] 9493 [32 +] 9961 [33 +] | 10-40 % MeOH, ACN de 10 à 40 % |
| HerA4 + ADN | 240.82 | 241.33 | - | 9637 [31 +] 9756 [32 +] 9830 [33 +] | 10-30 % ACN |
| HerA5 | 281,65 | 282.75 | 11853 | 10847 [36 +] 10958 [37 +] 11161 [38 +] | ACN DE 30-40 % |
| HerA6 | 337.98 | 339,3 | 12517 | 12335 mené [38 +] 12386 [39 +] 12498 [40 +] 12590 [41 +] 12676 [42 +] 13019 [43 +] | 10-40 % MeOH, ACN de 10 à 40 % |
| HerA6 + ADN | 353.48 | 354.626 | - | 12890 [40 +] 13081 [41 +] 13184 [42 +] 13273 [43 +] 13463 [44 +] 13576 [45 ans] | 30 % ACN |
| HerA,7 | 394,3 | 395.85 | 13901 | 14154 [42 +] 14219 [43 +] 14261 [44 +] 14285 [45 ans] 14335 [46 +] | 10-40 % MeOH, ACN de 10 à 40 %, 10-40 % DMSO |
| HerA,7 + ADN | 409,8 | 410.62 | - | 14414 [41 +] 14510 [42 +] 14558 [43 +] 14598 [44 +] 14630 [45 ans] 14641 [46 +] | 10 % ACN |
Table 1. Les masses expérimentales et calculées et les valeurs de CCS de HerA-NurA et ses complexes sous généré études de perturbation dans la solution de formulaire.
Discussion
MS joue un rôle de plus en plus important pour caractériser la stoechiométrie, les interactions et les architecture de sous-unité de complexes protéiques. IM-MS données peuvent servir à définir les modalités topologiques des sous-unités dans plusieurs composants complexes. Par rapport aux autres méthodes existantes de la biologie structurale, MS a plusieurs avantages. Native MS est une technique rapide et hautement sensible et peut être utilisée pour sonder les échantillons de protéines hétérogènes. Lorsqu’il est couplé avec des expériences de perturbation dans la solution, les voies de dissociation des ensembles de protéines peuvent être surveillés. Ainsi que des structures cristallines ou modèles d’homologie, l’information offerte par Mme structurelles offre un outil pour l’étude des interactions protéine-ligand et fournir des modèles quasi native et Assemblée voies11.
Nous décrivons ici les procédures expérimentales nécessaires pour analyser la stoechiométrie et la composition des interactions de protéine-ligand, avec un ou plusieurs ligands, à l’aide de MS intégrative. Cela comprend la préparation de l’échantillon MS, acquisition de données, analyse de données et l’intégration de données de MS à l’aide d’outils informatiques. Pour ce faire, nous avons utilisé l’ADN-résection HerA-NurA hétéro-oligomériques protéine complexe, lié aux trois ligands (ADN, l’ATP et ADP), que notre système de modèle. Le protocole montre l’utilisation du logiciel actuellement disponible pour faciliter la présentation et l’analyse des données.
Acquisition des spectres de haute qualité est important pour l’analyse de liaison de ligand, étapes de préparation d’échantillon prudent sont critiques, y compris purification de protéine, titrage de ligand et l’échange de mémoire tampon. Une des limitations de nESI native MS lorsque l'on étudie la liaison du ligand est non-spécifiques. Liaison non spécifique se produit au cours de la gouttelette désolvatation durant tout le processus electrospray. Cela augmente la concentration de ligand et modifie donc le ratio de protéine/Ligand29. La liaison des nucléotides se traduit par une différence de masse relativement faible entre apo et nucléotide lié aux protéines qui n’altère pas l’ionisation efficacité50,51.
Nous avons utilisé le système Synapt G2-Si SM pour notre travail, mais les protocoles sont applicables à différentes investigations d’autres complexes de protéine-ligand à l’aide d’autres spectromètres de masse de nano-electrospray disponibles dans le commerce. Intégration structurale MS plus en plus joue un rôle important dans la lutte contre les problèmes biologiques d’une plus grande complexité. Le flux de travail et les techniques décrites ici sont bien adaptées pour comprendre les conséquences structurelles et construire des mécanismes du complexe protéique et de la formation de la protéine-ligand, qui sont par ailleurs difficiles à étudier à l’aide de techniques structurelles classiques .
Déclarations de divulgation
Aucun conflit d’intérêts ne déclarés.
Remerciements
Nous tenons à remercier Karl-Peter Hopfner et Robert Thomas Byrne pour gentiment échantillons de protéines fournissant HerA et HerA-NurA et pour leur aide à la conception expérimentale. Nous remercions également m. Eamonn Reading pour son examen du manuscrit. Nous remercions sincèrement nos organismes de financement : Wellcome Trust [109854/Z/15/Z] et la Royal Society [RG150216 à A.P.].
matériels
| Name | Company | Catalog Number | Comments |
| Adenosine 5′-(3-thiotriphosphate) tetralithium salt | Merck Millipore | 119120-25MG | |
| Adenosine 5′-diphosphate | Sigma-Aldrich | 20398-34-9 | |
| Ammonium acetate solution | Sigma-Aldrich | A2706 | |
| Micro Bio-Spin Chromatography Columns | Bio-Rad | 7326204 | |
| Vivaspin concentrator | Sartorius | Z614041-25EA | |
| Magnesium chloride hexahydrate | Sigma-Aldrich | 246964 | |
| Water TraceSelect | Sigma-Aldrich | 95305 | |
| Borosilicate Capillaries | Harvard Apparatus | 300060 |
Références
- Wilm, M., Mann, M. Analytical Properties of the Nanoelectrospray Ion Source. Analytical Chemistry. 68 (1), 1-8 (1996).
- Fenn, J. B., Mann, M., Meng, C. K., Wong, S. F., Whitehouse, C. M. Electrospray ionization for mass spectrometry of large biomolecule. Science. 246 (4926), 64-71 (1989).
- Hernandez, H., Robinson, C. V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nature Protocols. 2 (3), 715-726 (2007).
- Heck, A. J., Van Den Heuvel, R. H. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom Review. 23 (5), 368-389 (2004).
- Boeri Erba, E., Petosa, C. The emerging role of native mass spectrometry in characterizing the structure and dynamics of macromolecular complexes. Protein Science. 24 (8), 1176-1192 (2015).
- Laganowsky, A., Reading, E., Hopper, J. T. S., Robinson, C. V. Mass spectrometry of intact membrane protein complexes. Nature. 8 (4), 639-651 (2013).
- Benesch, J. L. Collisional activation of protein complexes: picking up the pieces. Journal of the American Society for Mass Spectrometry. 20 (3), 341-348 (2009).
- Tian, Y., Han, L., Buckner, A. C., Ruotolo, B. T. Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Analytical Chemistry. 87 (22), 11509-11515 (2015).
- Eschweiler, J. D., Rabuck-Gibbons, J. N., Tian, Y., Ruotolo, B. T. CIUSuite: A Quantitative Analysis Package for Collision Induced Unfolding Measurements of Gas-Phase Protein Ions. Analytical Chemistry. 87 (22), 11516-11522 (2015).
- Hopper, J. T., Oldham, N. J. Collision induced unfolding of protein ions in the gas phase studied by ion mobility-mass spectrometry: the effect of ligand binding on conformational stability. Journal of the American Society for Mass Spectrometry. 20 (10), 1851-1858 (2009).
- Politis, A., et al. Topological models of heteromeric protein assemblies from mass spectrometry: application to the yeast eIF3:eIF5 complex. Chemical Biology. 22 (1), 117-128 (2015).
- Morgner, N., et al. Hsp70 forms antiparallel dimers stabilized by post-translational modifications to position clients for transfer to Hsp90. Cell Reports. 11 (5), 759-769 (2015).
- Lai, Y. T., et al. Structure of a designed protein cage that self-assembles into a highly porous cube. Nature Chemistry. 6 (12), 1065-1071 (2014).
- Levy, E. D., Boeri Erba, E., Robinson, C. V., Teichmann, S. A. Assembly reflects evolution of protein complexes. Nature. 453 (7199), 1262-1265 (2008).
- Sharon, M., et al. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure. 17 (1), 31-40 (2009).
- Jurneczko, E., Barran, P. E. How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst. 136 (1), 20-28 (2011).
- Politis, A., Schmidt, C. Structural characterisation of medically relevant protein assemblies by integrating mass spectrometry with computational modelling. Journal of Proteomics. 175, 34-41 (2017).
- Zhou, M., et al. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 334 (6054), 380-385 (2011).
- Barrera, N. P., Booth, P. J., Robinson, C. V. Micelles Protect Membrane Complexes from Solution to Vacuum. Science. 321 (5886), 243-246 (2008).
- Konijnenberg, A., et al. Global structural changes of an ion channel during its gating are followed by ion mobility mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 111 (48), 17170-17175 (2014).
- Schmidt, C., Beilsten-Edmands, V., Mohammed, S., Robinson, C. V. Acetylation and phosphorylation control both local and global stability of the chloroplast F1 ATP synthase. Scientific Reports. 7, 44068 (2017).
- Ahdash, Z., et al. Mechanistic insight into the assembly of the HerA-NurA helicase-nuclease DNA end resection complex. Nucleic Acids Research. 45 (20), 12025-12038 (2017).
- Pagel, K., Natan, E., Hall, Z., Fersht, A. R., Robinson, C. V. Intrinsically disordered p53 and its complexes populate compact conformations in the gas phase. Angewandte Chemie. 52 (1), 361-365 (2013).
- Afonso, J. P., et al. Insights into the structure and assembly of the Bacillus subtilis clamp-loader complex and its interaction with the replicative helicase. Nucleic Acids Research. 41 (9), 5115-5126 (2013).
- van Duijn, E. Current limitations in native mass spectrometry based structural biology. Journal of the American Society for Mass Spectrometry. 21 (6), 971-978 (2010).
- Susa, A. C., Xia, Z., Williams, E. R. Native Mass Spectrometry from Common Buffers with Salts That Mimic the Extracellular Environment. Angewandte Chemie. 56 (27), 7912-7915 (2017).
- Breukera, K., McLafferty, F. W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10 to 10 s. Proceedings of the National Academy of Sciences of the United States of America. 105 (47), 18145-18152 (2008).
- Robinson, C. V., et al. Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein-CoA Ligand Binding and Assembly. Journal of the American Chemical Society. 118 (36), 8646-8653 (1996).
- Shimon, L., Sharon, M., Horovitz, A. A method for removing effects of nonspecific binding on the distribution of binding stoichiometries: application to mass spectroscopy data. Biophysical Journal. 99 (5), 1645-1649 (2010).
- Dyachenko, A., Gruber, R., Shimon, L., Horovitz, A., Sharon, M. Allosteric mechanisms can be distinguished using structural mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 110 (18), 7235-7239 (2013).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. 19 (40), (2010).
- Marty, M. T., et al. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Analytical Chemistry. 87 (8), 4370-4376 (2015).
- Allison, T. M., et al. Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nature Communications. 6, 8551-8561 (2015).
- Sivalingam, G. N., Yan, J., Sahota, H., Thalassinos, K. Amphitrite: A program for processing travelling wave ion mobility mass spectrometry data. International Journal of Mass Spectrometry. 345-347, 54-62 (2013).
- Morgner, N., Robinson, C. V. Massign: an assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Analytical Chemistry. 84 (6), 2939-2948 (2012).
- Bush, M. F., et al. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Analytical Chemistry. 82, 9557-9565 (2010).
- Ruotolo, B. T., Benesch, J. L., Sandercock, A. M., Hyung, S. J., Robinson, C. V. Ion mobility-mass spectrometry analysis of large protein complexes. Nature Protocols. 3 (7), 1139-1152 (2008).
- Lanucara, F., Holman, S. W., Gray, C. J., Eyers, C. E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nature Chemistry. 6 (4), 281-294 (2014).
- Wyttenbach, T., Bowers, M. T. Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. Journal of Physical Chemistry B. 115 (42), 12266-12275 (2011).
- Marcoux, J., Robinson, C. V. Twenty years of gas phase structural biology. Structure. 21 (9), 1541-1550 (2013).
- Michaelevski, I., Kirshenbaum, N., Sharon, M. T-wave ion mobility-mass spectrometry: basic experimental procedures for protein complex analysis. Journal of Visualized Experiments. (41), (2010).
- Mesleh, M. F., Hunter, J. M., Shvartsburg, A. A., Schatz, G. C., Jarrold, M. F. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. Journal of Physical Chemistry A. 100, 16082-16086 (1996).
- Shvartsburg, A. A., Jarrold, M. F. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chemical Physics Letters. 261, 86-91 (1996).
- Taverner, T., Hernández, H., Sharon, M., Ruotolo, B. T., Matak-Vinković, D., Devos, D., Russell, R. B., Robinson, C. V. Subunit Architecture of Intact Protein Complexes from Mass Spectrometry and Homology Modeling. Accounts of Chemical Research. 41 (5), 617-627 (2008).
- Wysocki, V. H., Joyce, K. E., Jones, C. M., Beardsley, R. L. Surface-induced dissociation of small molecules, peptides, and non-covalent protein complexes. Journal of the American Society for Mass Spectrometry. 19 (2), 190-208 (2008).
- Hall, Z., Politis, A., Bush, M. F., Smith, L. J., Robinson, C. V. Charge-state dependent compaction and dissociation of protein complexes: insights from ion mobility and molecular dynamics. Journal of the American Chemical Society. 134 (7), 3429-3438 (2012).
- Hyung, S. J., Robinsons, C. V., Ruotolo, B. T. Gas-Phase Unfolding and Disassembly Reveals Stability Differences in Ligand-Bound Multiprotein Complexes. Chemistry & Biology. 16, 382-390 (2009).
- Hall, Z., Politis, A., Robinson, C. V. Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure. 20 (9), 1596-1609 (2012).
- Woods, L. A., Radford, S. E., Ashcroft, A. E. Advances in ion mobility spectrometry-mass spectrometry reveal key insights into amyloid assembly. Biochimica et Biophysica Acta. 1834 (6), 1257-1268 (2013).
- Daubenfeld, T., Bouin, A. P., van der Rest, G. A deconvolution method for the separation of specific versus nonspecific interactions in noncovalent protein-ligand complexes analyzed by ESI-FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 17 (9), 1239-1248 (2006).
- Loo, J. A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrometry Reviews. 16, 1-23 (1997).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.