
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Proteómica de espectrometría de masas guiada por linaje celular en el embrión en desarrollo (rana)
En este artículo
Resumen
Aquí describimos una caracterización proteómica basada en espectrometría de masas de linajes celulares con destinos tisulares conocidos en el embrión vertebrado de Xenopus laevis .
Resumen
La caracterización de eventos moleculares a medida que las células dan lugar a tejidos y órganos plantea un potencial para comprender mejor el desarrollo normal y diseñar remedios eficientes para las enfermedades. Las tecnologías que permiten la identificación y cuantificación precisas de diversos tipos y un gran número de proteínas proporcionarían información aún faltante sobre los mecanismos moleculares que orquestan el desarrollo de tejidos y organismos en el espacio y el tiempo. Aquí, presentamos un protocolo basado en espectrometría de masas que permite la medición de miles de proteínas en linajes celulares identificados en embriones de Xenopus laevis (rana). El enfoque se basa en mapas reproducibles de destino celular y métodos establecidos para identificar, etiquetar fluorescentemente, rastrear y muestrear células y su progenie (clones) a partir de este modelo de desarrollo de vertebrados. Después de recolectar el contenido celular utilizando micromuestreo o aislar células por disección o clasificación celular activada por fluorescencia, las proteínas se extraen y procesan para el análisis proteómico ascendente. La cromatografía líquida y la electroforesis capilar se utilizan para proporcionar una separación escalable para la detección y cuantificación de proteínas con espectrometría de masas de alta resolución (HRMS). Se proporcionan ejemplos representativos para la caracterización proteómica de células destinadas al tejido neural. La proteómica HRMS guiada por linaje celular es adaptable a diferentes tejidos y organismos. Es lo suficientemente sensible, específico y cuantitativo para observar la dinámica espacio-temporal del proteoma durante el desarrollo de los vertebrados.
Introducción
Nuestra comprensión de la diferenciación celular y la génesis de tejidos y órganos es el resultado de décadas de elaboradas pantallas específicas de genes y sus productos. Aumentar nuestro conocimiento de todas las biomoléculas y sus cantidades durante eventos celulares importantes ayudaría a desentrañar los mecanismos moleculares que controlan el patrón espacial y temporal del plan corporal de los vertebrados. Las tecnologías que permiten la amplificación molecular y la secuenciación ahora pueden informar rutinariamente sobre un gran número de genes y transcripciones, apoyando estudios basados en hipótesis en investigación biológica básica y traslacional. Para comprender los sistemas en desarrollo, una relación compleja entre la transcripción y la traducción aboga por el análisis directo de múltiples proteínas y sus modificaciones postraduccionales. La proteómica global utilizando sistemas biológicos in vitro, como células madre pluripotentes inducidas, comenzó a delinear mecanismos de inducción tisular 1,2. En organismos complejos, como el embrión vertebrado, el desarrollo se basa en gradientes morfógenos en el contexto del espacio y el tiempo3. De ello se deduce que obtener conocimiento de los cambios proteómicos a medida que las células se diferencian para formar tejidos especializados, como los tejidos neurales, ofrece una clave para desbloquear programas moleculares que controlan el desarrollo normal y defectuoso y guiar la terapéutica de próxima generación.
La rana vertebrada sudafricana (Xenopus laevis) es un modelo bien establecido en biología celular y del desarrollo, neuro y regenerativa. El Premio Nobel de Fisiología o Medicina 2012 de Sir John Gurdon por el descubrimiento de la pluripotencia del núcleo somático destacó la importancia de este modelo para los descubrimientos en estudios básicos y traslacionales. Los embriones de Xenopus se desarrollan externamente a la madre, facilitando así la manipulación directa de células, clones celulares y expresión génica en varias etapas de desarrollo. La pigmentación asimétrica y las divisiones celulares estereotipadas permitieron el trazado de mapas de destino reproducibles del embrión de 7,8 células de 16-6 y 32 células. Para la proteómica basada en espectrometría de masas de alta resolución (HRMS), las ventajas adicionales del modelo incluyen un tamaño relativamente grande (~ 1 mm de diámetro), que produce un contenido abundante de proteínas para el análisis (~ 130 μg en embriones en etapa de escisión temprana, ~ 10 μg de contenido de proteína en células individuales del embrión de 16 células)9,10.
En la actualidad, HRMS es la tecnología líder de elección para detectar proteínas. Esta tecnología permite la detección y cuantificación directa, sensible y específica de múltiples, generalmente cientos a miles de proteínas diferentes11. La proteómica ascendente de HRMS implica una serie de pasos interconectados. Después de la extracción de la muestra de célula/tejido, las proteínas se digieren con una enzima proteolítica, como la tripsina (proteómica ascendente). Los péptidos resultantes se separan en función de sus diferentes propiedades fisicoquímicas, incluida la hidrofobicidad (cromatografía líquida de fase inversa, LC), la carga neta (cromatografía de intercambio iónico), el tamaño (cromatografía de exclusión de tamaño) o la movilidad electroforética (electroforesis capilar, CE). Los péptidos se cargan (ionizan), típicamente usando ionización por electrospray (ESI), y los iones peptídicos se detectan y secuencian a través de la fragmentación en fase gaseosa por HRMS en tándem. Los datos peptídicos resultantes se asignan al proteoma del organismo que se está estudiando. Con la intensidad de la señal de iones peptídicos específicos de proteínas (proteotípicos) que se correlacionan con la concentración, la cuantificación de proteínas se puede realizar sin etiqueta o basada en etiquetas (cuantificación de multiplexación). La proteómica HRMS proporciona un rico recurso de información sobre el estado molecular del sistema en estudio, lo que permite la generación de hipótesis y estudios funcionales de seguimiento.

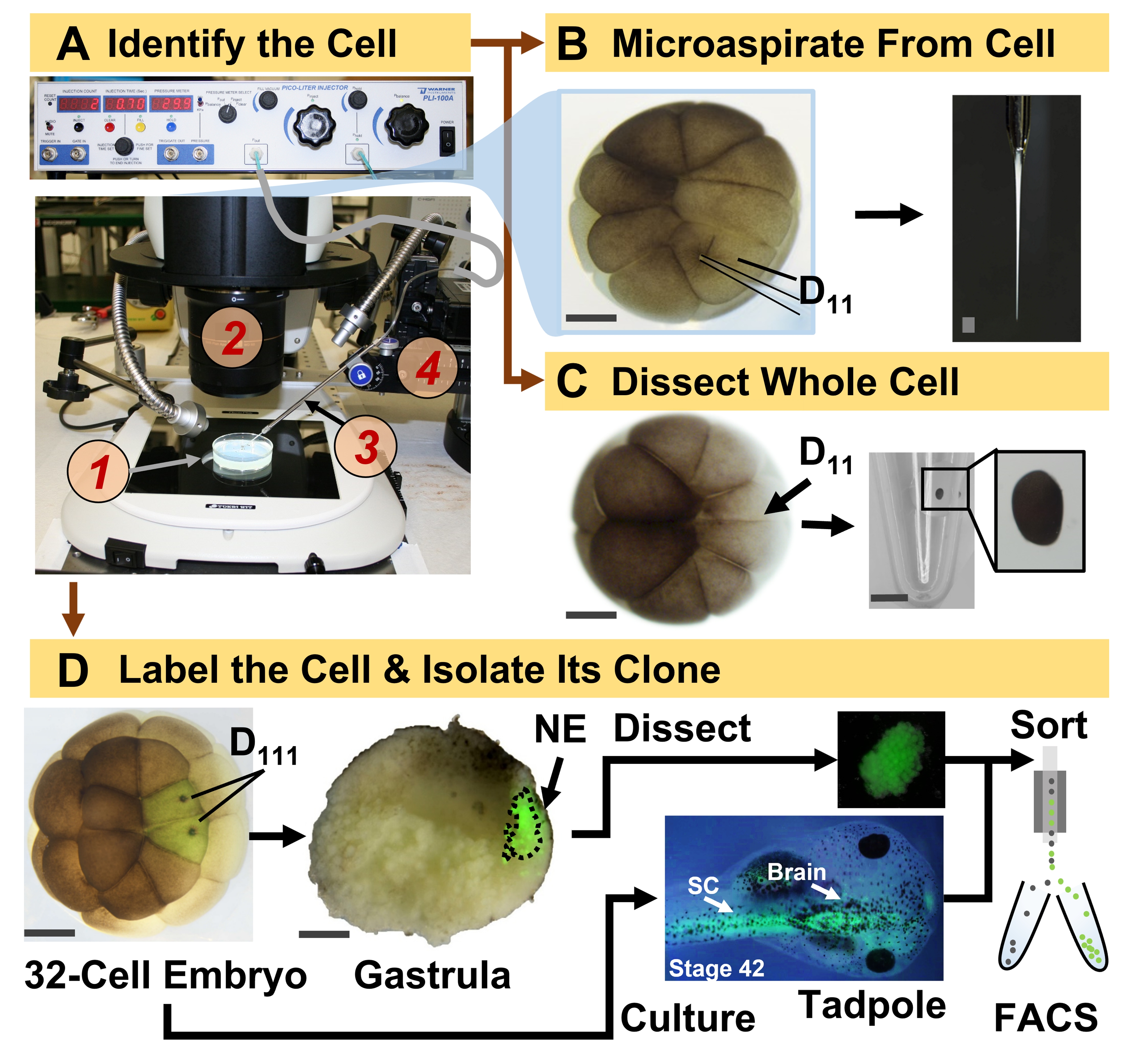
Figura 1: Proteómica escalable espaciotemporalmente que permite la proteómica HRMS guiada por linaje celular en el embrión en desarrollo (rana). (A) Visualización de la muestra (1) utilizando un microscopio estereoscópico (2) para la inyección de una célula identificada (recuadro), utilizando una micropipeta fabricada (3) bajo control por una etapa de traslación (4). (B) Muestreo subcelular de la célula D11 izquierda identificada en un embrión de 16 células. (C) Disección de una célula D11 completa de un embrión de 16 células. (D) Trazado fluorescente (verde) de las progenies D111 izquierda y derecha de un embrión de 32 células para guiar la disección del ectodermo neural (NE) en la gástrula (etapa 10) y el aislamiento del tejido descendente del renacuajo utilizando FACS. Barras de escala: 200 μm para embriones, 1,25 mm para el vial. Las figuras fueron adaptadas con permiso de las referencias 15,19,21,59. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
El protocolo presentado aquí permite la cuantificación basada en HRMS de un gran número de proteínas en células / tejidos identificados en embriones de X. laevis en desarrollo. El enfoque se basa en la identificación precisa de células, mapas de destino celular reproducibles y metodologías establecidas para rastrear linajes celulares en este modelo biológico 6,7,8. Como se muestra en la Figura 1, estudiamos proteomas de células individuales empleando disección de células enteras o micromuestreo capilar para aspirar el contenido celular. El monitoreo del linaje de una célula nos permite estudiar la evolución espacio-temporal del proteoma a medida que las células forman tejidos durante la gastrulación. La progenie celular se marca fluorescentemente inyectando un fluoróforo conjugado con dextrano inerte o ARNm para la proteína fluorescente (por ejemplo, proteína fluorescente verde o GFP). La progenie etiquetada se aísla en los puntos de tiempo de desarrollo deseados. Durante la gastrulación, los clones celulares que están estrechamente agrupados pueden aislarse por disección. Después de la gastrulación, los clones celulares pueden distribuirse dentro del embrión debido a movimientos migratorios y pueden aislarse de tejidos disociados mediante clasificación celular activada por fluorescencia (FACS). Las proteínas en estas células y tejidos se miden a través de la proteómica ascendente empleando HPLC o CE para la separación y ESI tándem HRMS para la identificación. La proteómica HRMS guiada por linaje celular es escalable a diferentes tamaños y linajes celulares dentro del embrión y es específica, sensible y cuantitativa. A través de ejemplos seleccionados que se muestran aquí, también demostramos que este protocolo es escalable y ampliamente adaptable a diferentes tipos de células y linajes celulares.

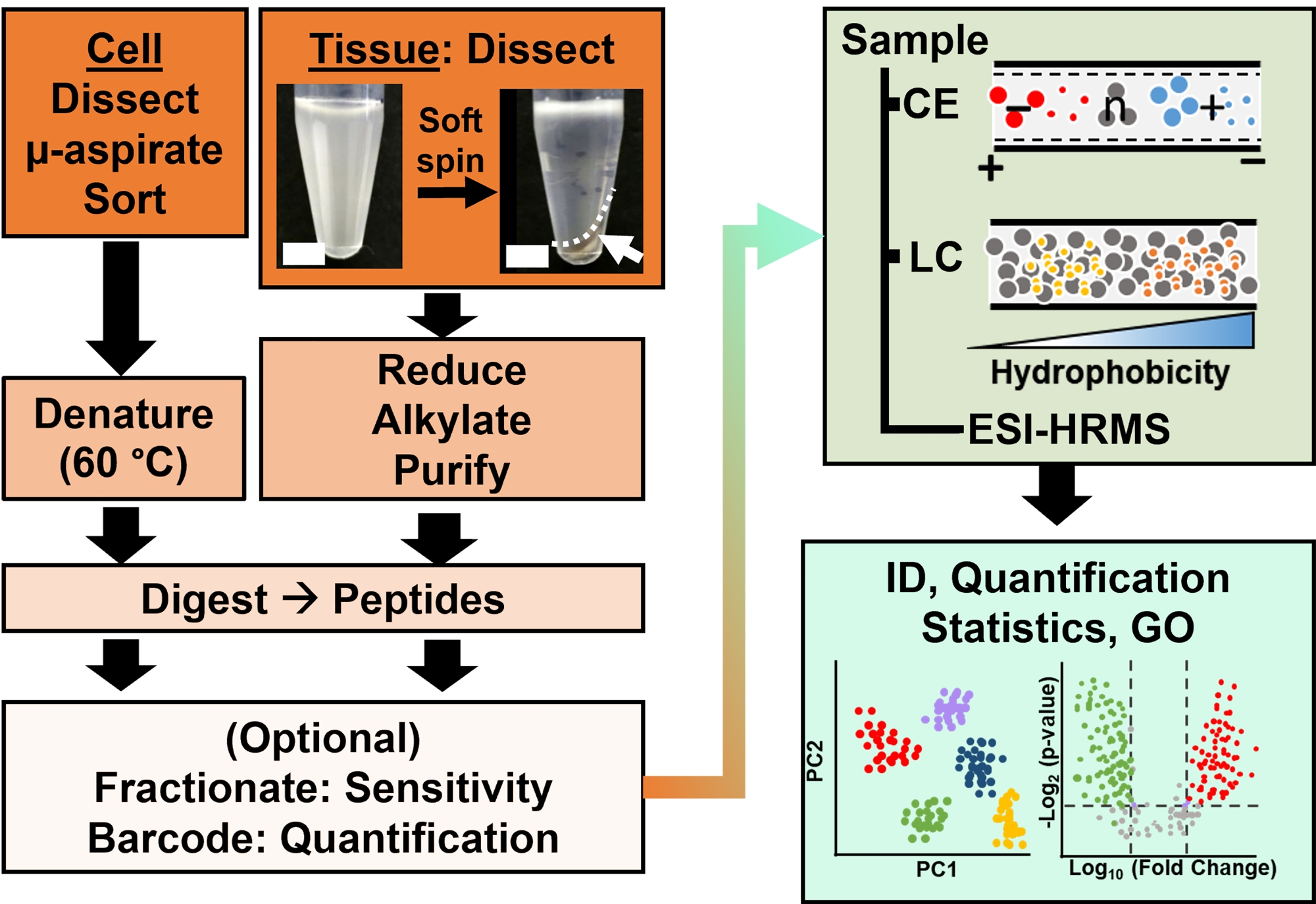
Figura 2: El flujo de trabajo bioanalítico. La microdisección y la aspiración capilar, o FACS, facilitaron el muestreo del contenido de proteínas celulares y clonales. Agotamiento de abundantes proteínas vitelinas y separación por electroforesis capilar (CE) o cromatografía líquida de nanoflujo (LC) mejora la sensibilidad de identificación (ID) utilizando espectrometría de masas de alta resolución (HRMS) por ionización por electrospray (ESI). La cuantificación reveló desregulación, proporcionando nueva información para estudios basados en hipótesis junto con la información disponible de la ontología génica (GO). Las figuras fueron adaptadas con permiso de la referencia15. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocolo
Todos los protocolos que garantizan el mantenimiento y manejo humanitario de las ranas adultas de Xenopus laevis fueron aprobados por el Comité Institucional de Cuidado y Uso de Animales de la Universidad de Maryland, College Park (números de aprobación R-DEC-17-57 y R-FEB-21-07).
1. Preparar las soluciones
- Para embriología
- Preparar 1x, 0.5x y 0.2x solución de Steinberg (SS), luego autoclavelos (120 °C durante 20 min) para esterilidad siguiendo los protocolos estándar12.
- Preparar Ficoll al 3% (p/v) en 1x SS esterilizado siguiendo los protocolos estándar12.
- Para el desejeo, prepare una solución de cisteína al 2% (p/v) y ajuste su pH a 8 agregando una solución de hidróxido de sodio 10 M gota a gota.
PRECAUCIÓN: La exposición a la cisteína puede causar daño cutáneo y respiratorio. El hidróxido de sodio es un corrosivo que puede causar daños graves a la piel y los ojos tras la exposición directa. Use el equipo de protección personal (EPP) adecuado cuando manipule estos productos químicos, como guantes y una bata de laboratorio. - Para el trazador de linaje, prepare 0.5% (v / v) de un dextrano fluorescente en agua desionizada estéril. Alternativamente, prepare una solución de 0,2 μg/μL de ARNm para proteínas fluorescentes en agua desionizada estéril (p. ej., GFP).
- Para disociar las células, prepare el tampón Newport 2.0 que contenga 0.1 M de isetionato de sodio, 20 mM de pirofosfato de sodio y 10 mM de CAPS, luego lleve su pH a 10.513.
PRECAUCIÓN: La exposición al pirofosfato de sodio puede causar irritación de la piel y los ojos. Use el EPP adecuado cuando manipule estos productos químicos.
- Para proteómica ascendente
- Prepare el tampón de lisis celular para incluir: 250 mM de sacarosa, sustituto nonidet P-40 al 1% (p/v), Tris-HCl 20 mM, EDTA 5 mM, citocalasina D de 10 μM y combretastatina 4A de 10 μM. Preparar una reserva de dodecil sulfato de sodio al 10% (p/v)14,15.
NOTA: Tris-HCl se eligió para minimizar la contaminación por HEPES durante el nanoflujo LC (nanoLC)-HRMS.
PRECAUCIÓN: La exposición al sustituto nonidet P-40 puede causar irritación de la piel. La citocalasina D es teratogénica si se consume y la combretastatina es agudamente tóxica tras la exposición directa. Use el EPP adecuado cuando manipule estos productos químicos. - Para separar los péptidos por CE, prepare los siguientes disolventes (v/v): Disolvente de muestra, acetonitrilo al 75% (ACN) que contiene 0,05% de ácido acético (AcOH) en agua; solución de vaina, 10% ACN que contiene 0,05% de AcOH en agua; electrolito de fondo (BGE), 25% ACN que contiene 1 M de ácido fórmico (FA) en agua.
PRECAUCIÓN: AcOH y FA son tóxicos cuando se inhalan o consumen y pueden causar daños graves en la piel y los ojos tras la exposición directa. Use el EPP adecuado cuando manipule estos productos químicos. - Para separar péptidos por nanoLC de fase inversa, prepare (v/v): Fase móvil A (acuosa), agua que contiene 0,1% de FA; fase móvil B (orgánica), 0,1% FA en ACN.
NOTA: Todas las mezclas deben prepararse utilizando disolventes de grado LC-MS para minimizar las interferencias químicas durante la detección de HRMS.
- Prepare el tampón de lisis celular para incluir: 250 mM de sacarosa, sustituto nonidet P-40 al 1% (p/v), Tris-HCl 20 mM, EDTA 5 mM, citocalasina D de 10 μM y combretastatina 4A de 10 μM. Preparar una reserva de dodecil sulfato de sodio al 10% (p/v)14,15.
2. Preparar las herramientas para microinyección y disección
- Para mover y orientar suavemente los embriones, haga bucles capilares fijando el cabello limpio en una pipeta Pasteur como se describe en otra parte16.
- Para la microinyección, fabricar agujas tirando de capilares de borosilicato (1 mm/500 μm de diámetro exterior/interior) utilizando un extractor de pipetas como se describe en otra parte16.
NOTA: Aquí, se utilizó un extractor de pipetas P-1000 con los siguientes ajustes para fabricar agujas: calor, 495; tirón, 30, velocidad, 60; tiempo, 150; presión, 200. - Con la observación bajo un microscopio estereoscópico, corte la punta del capilar usando un par de pinzas afiladas para fabricar esencialmente el capilar en una (micro) aguja (por ejemplo, Dumont # 5)16.
PRECAUCIÓN: Los capilares tirados son muy afilados y deben manejarse con cuidado.
NOTA: La punta de la aguja debe ser lo suficientemente afilada (diámetro exterior 10-15 μm) para poder perforar la célula con un daño mínimo a la membrana celular para que el contenido intracelular no se escape y la célula pueda sanar y continuar siendo viable. - Para mantener los embriones durante la microinyección, prepare pozos en un plato lleno de arcilla. En una placa de Petri de 15 mm, imprima pozos de ~1 mm de diámetro x ~0,5 mm de profundidad en arcilla de plastilina no tóxica como se describe en otra parte16.
- Para la microdisección, prepare platos recubiertos de agarosa. Hacer agarosa al 2% en 1x SS y autoclave para esterilizar la solución (120 °C durante 20 min). Llene las placas de Petri de 60 mm hasta la mitad y deje que las placas se solidifiquen. Haga pozos de ~1 mm de diámetro x ~0,5 mm de profundidad utilizando una herramienta de pipeta Pasteur en bola como se describió anteriormente16.
3. Aislar el linaje celular
NOTA: Los siguientes pasos se realizan para aislar células individuales identificadas y/o sus linajes celulares descendientes. Por lo general, el embrión se cultiva hasta la etapa de 16 o 32 células, donde los destinos tisulares de cada célula se mapean de manera reproducible 6,7,17. Las células embrionarias se identifican en función de la morfología, la ubicación y en referencia a sus mapas de destino. Para el análisis unicelular, las células identificadas se aíslan por disección manual, o su contenido intracelular se recoge en una pipeta capilar y se deposita en 5 μL de bicarbonato de amonio 0,5 mM. La muestra resultante se almacena a -80 °C hasta el análisis (Figura 1)18,19,20,21. Para el análisis del linaje celular, las células identificadas se inyectan con un trazador de linaje, y sus clones posteriores se aíslan en etapas clave de desarrollo (por ejemplo, durante la gastrulación para estudiar la inducción tisular, después de la neurulación para estudiar el compromiso tisular). En lo que sigue, se describen los pasos para etiquetar fluorescentemente el linaje de las células identificadas para el aislamiento por disección o FACS.
- Cultivar los embriones
- Obtener embriones mediante apareamiento natural o fecundación in vitro (FIV) siguiendo los protocolos establecidos12.
NOTA: El apareamiento natural es logísticamente más simple, evita a las ranas macho adultas y produce embriones en diferentes etapas de desarrollo, mientras que la FIV proporciona embriones sincronizados con el desarrollo para experimentos que requieren una estadificación precisa. - Dejelly los embriones. Retirar la capa gelatinosa que rodea los embriones mediante tratamiento con la solución desarticulada como se describe en otra parte12,16.
NOTA: Las microinyecciones y la disección requieren acceso a las células y tejidos, lo que requiere la eliminación en embriones de X. laevis. - Seleccionar embriones de 2 células con pigmentación estereotipada16,22.
NOTA: Este paso es importante para garantizar la precisión y reproducibilidad en la identificación de la célula y su linaje. - Cultive embriones a la etapa de desarrollo deseada. Transfiera los embriones gelatinosos a una placa de Petri que contenga 1x SS e incubarlos entre 14-25 °C para controlar la velocidad de desarrollo.
NOTA: La dependencia de la temperatura del desarrollo es reproducible y se representa en gráficos para X. laevis, disponible en Xenbase23 (www.xenbase.org). El cultivo de lotes de embriones a diferentes temperaturas permite etapas de desarrollo asombrosas. Hacerlo ayuda a distribuir el número de embriones disponibles en un momento dado para la experimentación. - Monitorear el patrón de escisión de los embriones y seleccionar embriones con pigmentación estereotipada y patrones de escisión para microinyección16.
NOTA: Al seleccionar embriones de 16 y 32 células, asegúrese de que las divisiones celulares sean simétricas para el trazado reproducible del linaje.
- Obtener embriones mediante apareamiento natural o fecundación in vitro (FIV) siguiendo los protocolos establecidos12.
- Etiquetar la(s) celda(s) de interés(es)
- Coloque la aguja de inyección que contiene la solución trazadora de linaje. Monte la aguja de microinyección en un soporte de micropipeta controlado por un micromanipulador multieje.
- Conecte el soporte de micropipeta a un microinyector. Llene la aguja con el trazador de linaje aplicando presión negativa como se describe en otra parte16. La figura 1A ejemplifica la configuración.
- Calibre la aguja. Ajuste el tamaño de la punta de la aguja y el tiempo de inyección para administrar ~ 1 nL de la solución trazadora de linaje, medida en aceite (mineral) siguiendo un protocolo disponible en otros lugares16.
NOTA: Los capilares con una punta más ancha tienden a dañar la membrana celular, causando que el contenido subcelular y el trazador de linaje inyectado se filtren, mientras que los capilares con puntas más pequeñas son propensos a obstruirse. Los capilares con ~ 10 μm de diámetro exterior de la punta son ideales, ya que requieren un pulso de presión de 40 psi sobre ~ 300 ms para entregar ~ 1 nL. - Inunde el plato de arcilla de microinyección con la solución Ficoll al 3% y transfiera ~ 10 embriones al plato de arcilla usando una pipeta de transferencia. Use un lazo de cabello para guiar cada embrión hacia un pozo y colóquelos suavemente para que la célula de interés objetivo esté en ángulo recto con la microaguja.
- Identificar la célula precursora del linaje de interés siguiendo los mapas de destino tisular de X. laevis. Por ejemplo, la Figura 1 demuestra el etiquetado de clones ectodérmicos neurales basados en la inyección de sus células precursoras en embriones de 32 células (células D111 izquierda y derecha).
NOTA: Los mapas detallados del destino de los embriones de 16-6 y 32 células de 7,8 están disponibles en una plataforma interactiva a través de Xenbase23. Es importante garantizar la pigmentación y las divisiones estereotipadas en los embriones cuando se utilizan para experimentos de rastreo de linaje. - Inyecte la(s) célula(s) de interés con ~1 nL del dextrano fluorescente o ~200 pg de ARNm como se describió anteriormente16.
NOTA: Utilice conjugados dextranos que sean 10.000-40.000 MW. Los conjugados de dextrano más pequeños podrían pasar a través de uniones de brecha, mientras que los conjugados de dextrano más grandes pueden no difundirse uniformemente en la célula inyectada. Planee inyectar células en ~ 10 embriones para tener suficientes tejidos para análisis proteómicos. - Confirmar el éxito del etiquetado celular bajo un microscopio estereoscópico. Asegúrese de que solo se inyecta la celda deseada. Deseche los embriones que contengan células lesionadas o etiquetadas incorrectamente siguiendo las políticas institucionales.
NOTA: Debido a que X. laevis es invasivo en muchos ambientes no naturales, los embriones pueden congelarse para garantizar la letalidad antes de descartar los embriones.
- Aislar la progenie celular marcada
- Transfiera los embriones inyectados a 0,5x SS en una placa de Petri y críelos entre 14-25 °C hasta que se alcance la etapa de desarrollo deseada.
NOTA: Consulte los protocolos establecidos para estadificar los embriones reportados en Xenbase. - Transfiera 3-5 embriones a una placa de agar con solución SS 0.2x para microdiciones.
NOTA: La reducción de la concentración de sal de la solución SS de 0.5x a 0.2x ayuda a separar las células durante la disección. - Use dos pinzas afiladas para extraer suavemente la membrana vitelina que rodea el embrión.
NOTA: Para evitar que el clon de interés sufra daños, despegue la membrana del lado opuesto del clon marcado con fluorescencia. - Aísle el clon etiquetado mediante disección manual (pasos 3.3.5-3.3.6) o FACS (pasos 3.3.7-3.3.8) de la siguiente manera.
- Use fórceps para diseccionar el clon marcado del embrión.
NOTA: Otras herramientas como tijeras microquirúrgicas, agujas de tungsteno o cuchillos para el cabello de cejas se pueden usar para la disección del clon etiquetado, como se detalla en otra parte16. - Recoger el tejido disecado con una pipeta de 0,5-10 μL y depositarlos en un vial de microcentrífuga. Usando una pipeta de transferencia, aspire los medios que rodean el tejido recolectado para limitar las sales en la muestra, que interfieren con el análisis HRMS en pasos posteriores.
NOTA: Utilice viales que minimicen la adsorción de proteínas en superficies de plástico para minimizar las pérdidas de proteínas en las superficies de los viales durante los pasos posteriores del flujo de trabajo. - Para aislar mediante FACS, transfiera ~5-8 embriones desvitellizados a cada pocillo de una placa de 12 pocillos que contenga ~5 ml de tampón Newport 2.0. Disociar los embriones nutrando la placa a 80 rpm durante 20-30 min a temperatura ambiente13.
NOTA: Los embriones/larvas mayores de la etapa 22 tienen abundantes proteínas de la matriz extracelular, lo que dificulta la disociación en células separadas. Se pueden adaptar enfoques enzimáticos adicionales para disociar tejidos de embriones más viejos, como se describe en otra parte24. - Purificar las células marcadas con fluorescencia de la suspensión utilizando FACS como se describe en otra parte24.
- Granular las células por centrifugación y desechar el sobrenadante.
NOTA: Utilice baja velocidad de centrifugación (400 × g) y temperatura (4 °C) para prevenir la lisis celular. Si utiliza albúmina sérica bovina (BSA) para el FACS, lave el pellet celular para reducir la interferencia de BSA durante la detección de HRMS. Resuspenda suavemente las células en solución salina tamponada con fosfato 1x (PBS) y centrifugar nuevamente a las células enjuagadas con pellets. Retire el líquido sobrenadante PBS. - Congele rápidamente las células aisladas colocando el vial de muestra sobre hielo seco o nitrógeno líquido.
NOTA: Mantenga las muestras (tejidos o células) enfriadas (por ejemplo, en hielo) durante los pasos de procesamiento. Congele las células con la menor cantidad posible de medios alrededor de la muestra para facilitar el procesamiento posterior. - Almacenar las muestras a -80 °C hasta el análisis HRMS.
- Transfiera los embriones inyectados a 0,5x SS en una placa de Petri y críelos entre 14-25 °C hasta que se alcance la etapa de desarrollo deseada.
4. Analizar las proteínas por espectrometría de masas
La caracterización proteómica de los tejidos o células aisladas se basa en una serie de pasos establecidos en HRMS. La figura 2 ilustra los pasos del flujo de trabajo bioanalítico. El protocolo de recolección de muestras utilizado aquí es compatible con los flujos de trabajo de proteómicabottom-up 11, middle-down25 otop-down 26 . En lo que sigue, se describe la estrategia ascendente utilizada en este estudio, que ha demostrado ser sensible, cuantitativa y adaptable a diversos tipos de espectrómetros de masas. Después de extraer y digerir enzimáticamente las proteínas, los péptidos resultantes se separan, seguidos de un análisis HRMS.
- Procesar los tejidos/células individuales
- Para el análisis de una sola célula por CE, caliente la muestra a 60 °C durante ~15 min para desnaturalizar las proteínas, luego equilibre la muestra a temperatura ambiente (RT, ~5 min)18,21.
NOTA: A diferencia del trabajo con tejidos, los pasos de reducción y alquilación se omiten para limitar las pérdidas de proteínas durante la preparación de la muestra de células individuales. Se pueden adoptar métodos de preparación de muestras asistidos por filtración (FASP)27,28, otras estrategias de recipiente único 29 y enfoques microfluídicos30 para minimizar las pérdidas de proteínas durante la preparación de la muestra. - Para el análisis por nanoLC, lisar hasta 5 tejidos disecados en 50 μL de tampón de lisis (~100 μg de proteína total). Facilite el proceso pipeteando la muestra hacia arriba y hacia abajo varias veces.
- Incubar el lisado a 4 °C durante 10 min, luego granular los restos celulares y las plaquetas de yema por centrifugación a 4.500 × g a 4 °C. Transfiera el sobrenadante a un vial de microcentrífuga limpio y agregue SDS al 10% para obtener una concentración final de SDS al 1% en el lisado (v / v).
- Para los tejidos, siga los pasos 4.1.5-4.1.7.
- Añadir 0,5 M de ditiothreitol al lisado para obtener una concentración final de ~25 mM (por ejemplo, 2,5 μL de 0,5 M de ditiothretol a 50 μL de lisado) e incubar el lisado durante 30 min a 60 °C para reducir químicamente los enlaces disulfuro en las proteínas.
- Añadir 0,5 M de yodoacetamida para obtener una concentración final de ~75 mM en el lisado e incubar la mezcla durante 15 min a RT en la oscuridad (Figura 2).
- Agregue 0,5 M de ditiotritol, igual que el volumen inicial (por ejemplo, 2,5 μL de 0,5 M de ditiothretol a 50 μL de lisado) para apagar los reactivos restantes de la reacción de alquilación.
PRECAUCIÓN: La yodoacetamida y el ditiothreitol pueden causar daños graves en la piel y los ojos tras la exposición directa. Use el EPP adecuado cuando manipule estos productos químicos. - Purificar las proteínas a través de la precipitación. La precipitación a base de cloroformo-metanol funciona bien31. Este protocolo también es adaptable a otros tipos de enfoques de precipitación32.
NOTA: Para el análisis de una sola célula, donde las pérdidas de proteínas son motivo de preocupación, omita el paso de precipitación para CE-HRMS. - Secar el precipitado proteico en un concentrador de vacío (4-37 °C), luego resuspender el proteoma extraído en 50 μL de bicarbonato de amonio de 50 mM. Estimar la concentración de proteína usando un ensayo colorimétrico de proteína total para determinar la cantidad de enzima requerida para la digestión (por ejemplo, ensayo de proteína de ácido bicinchonínico).
- Digerir las proteínas a péptidos. Añadir tripsina (1 μg/μL de cepa) para obtener una relación proteasa:proteína de 1:50 e incubar la mezcla a 37 °C durante un máximo de 5 h para muestras unicelulares y hasta 14 h para muestras de tejido. Consulte las recomendaciones específicas del proveedor para la reacción.
NOTA: La digestión con tripsina por concentraciones superiores a 14 h o superiores puede introducir escisiones que no son específicas de la secuencia de la proteína, desafiando así las identificaciones proteicas33. - Cuantificar la concentración total de péptidos mediante un ensayo colorimétrico.
- OPCIONAL: Para la cuantificación de multiplexación, etiquete los péptidos de cada muestra con una etiqueta de masa isobárica diferente siguiendo las instrucciones específicas del proveedor. Mezclar los péptidos con código de barras en proporciones iguales por muestra de péptido.
NOTA: Asegure un etiquetado y una mezcla precisos para evitar sesgos cuantitativos. Para muestras de cantidad limitada o muestras unicelulares, se puede incluir un canal portador basado en TMT compuesto de tejidos/células agrupados para minimizar las pérdidas de muestras durante los pasos de separación posteriores y para aumentar la sensibilidad de proteínas menos abundantes34. - OPCIONAL: Péptidos de desalinización para eliminar sales y contaminantes (por ejemplo, reactivos de etiqueta de masa isobárica sin reaccionar) en una columna/punta de giro de fase inversa C18 para proteger el sistema LC-MS.
- OPCIONAL: Fraccionar (por ejemplo, fraccionamiento de fase inversa de pH medio o alto) la mezcla peptídica para una detección más profunda del proteoma a través de plataformas manuales o automáticas. Use puntas estacionarias de fase C18 para fraccionar cantidades bajas (1-10 μg) de digeridos peptídicos.
- Secar la mezcla peptídica a 60 °C en un concentrador de vacío.
- Conservar la mezcla peptídica a -80 °C hasta la medición.
- Para el análisis de una sola célula por CE, caliente la muestra a 60 °C durante ~15 min para desnaturalizar las proteínas, luego equilibre la muestra a temperatura ambiente (RT, ~5 min)18,21.
- Separar los péptidos
NOTA: Después de extraer y digerir enzimáticamente las proteínas, los péptidos resultantes se separan por nanoLC o CE y se ionizan por ESI para su secuenciación por HRMS en tándem. La separación de nanoLC de fase inversa es ideal para péptidos que acumulan ~ 150 ng a ~ 1 μg por análisis. CE proporciona sensibilidad complementaria para péptidos que van desde femtogramas hasta <100 ng. Various custom-built and commercial CE-ESI interfaces allow for ready coupling of CE to HRMS with robust performance35 y se utilizan cada vez más para el análisis de una sola célula18,36,37.- Para separar usando CE, siga los pasos 4.2.2-4.2.7.
NOTA: A continuación, se describe el uso de la plataforma CE personalizada para medir los péptidos. Los protocolos para construir y usar este instrumento CE se proporcionaron anteriormente38, junto con un experimento visualizado sobre el uso de moléculas pequeñas20. Alternativamente, estas mediciones se pueden realizar en un sistema CE comercial, como el AB SCIEX CESI, Agilent 7100 o equivalente. - Reconstituir la digestión de proteínas en 1-2 μL del disolvente de muestra, vórtice para mezclar la muestra y centrifugarla a 10.000 x g durante 2 min para granular los restos celulares.
NOTA: La eliminación de los restos celulares minimiza la probabilidad de obstruir el capilar CE, prolongando así la vida útil del sistema de separación y aumentando el rendimiento de medición. - Inicialice el instrumento CE-ESI enjuagando el capilar CE con el BGE.
- Validar el rendimiento instrumental utilizando un estándar conocido (por ejemplo, citocromo C o digestión BSA, péptidos de angiotensina).
NOTA: Se recomienda evaluar el instrumento en términos de precisión de masa, sensibilidad de detección, reproducibilidad y rango dinámico lineal de cuantificación antes de medir muestras preciosas. Las notas adicionales sobre la validación y la resolución de problemas del rendimiento de CE-ESI-MS se enumeran en otra parte18,20,38. - Inyecte ~1-10 nL de la muestra en el capilar de separación CE.
NOTA: Este estudio utiliza capilar de sílice fundida de ~ 1 m de largo (40/110 μm de diámetro interno / externo) con la configuración de flujo de vaina bombeada electrocinéticamente. Los instrumentos comerciales de CE generalmente requieren la presentación de 5-10 μL de muestra en un microvial para inyección. La plataforma CE18,38 hecha a medida es compatible con ~250 nL a 1 μL de muestra depositada en un microvial de carga de muestras. - Transfiera el extremo de entrada del capilar de separación CE al BGE.
- Comience la separación electroforética aumentando gradualmente el voltaje de separación CE desde tierra (por ejemplo, paso a paso durante 1 minuto). Los potenciales de 20-28 kV con corriente por debajo de ~10 μA aseguran un rendimiento instrumental estable y reproducible para el análisis.
- Para separar usando nanoLC, siga los pasos 4.2.9-4.2.12.
- Resuspender la muestra de péptido en la fase móvil A. La concentración de la muestra y su volumen para inyección dependen del sistema LC y la columna disponibles. En este estudio, se inyectan ~ 250 ng-1 μg de digestión de proteínas en 1-20 μL de volumen de muestra en una columna de lecho empacado C18 (diámetro interno de 75 μm, tamaño de partícula de 2 μm con poros de 100 Å, columna de separación de 25 cm de longitud).
- Transfiera la muestra a un vial de LC.
NOTA: Asegúrese de que no haya burbujas de aire en el vial, que pueden dañar la columna analítica. Los viales con inserciones podrían usarse para muestras de tejido de bajo volumen o de una sola célula. - Cargue ~200 ng a 2 μg de muestra de péptido en la columna analítica C18.
NOTA: Opcionalmente, los péptidos se pueden cargar en una columna de trampa para desalinizar antes de la separación analítica. Por ejemplo, una columna de trampa C18 con 0,1 mm de diámetro interior, tamaño de partícula de 5 μm, tamaño de poro de 100 Å, longitud de 20 mm. Péptidos desalívoros con tampón A al 100% a un caudal de 5 μL/min durante 5 min antes de que comience el gradiente de separación. - Separar los péptidos mediante elución de gradiente. A una tasa de flujo de 300 nL/min, el gradiente de 120 min utilizado en este estudio es el siguiente: 0-5 min 2% B, 5-85 min 2-35% B, 86-90 min 70% B, 91-120 min 2% B.
- Para separar usando CE, siga los pasos 4.2.2-4.2.7.
- Ionizar los péptidos por ESI
NOTA: El capilar CE o nanoLC se acopla más típicamente a una fuente ESI para la ionización. Las interfaces CE-ESI de microflujo (punta roma) y nanoflujo(diseño de flujo de vaina bombeadaelectrocinética 39 y punta cónica 36) para la detección ultrasensible se han desarrollado previamente.- Suministre los péptidos de separación en una fuente de iones de electropulverización para la ionización utilizando una interfaz ESI comercial o personalizada. Para el análisis CE-ESI-MS de una sola célula en embriones de Xenopus , utilice una interfaz de bajo flujo bombeada electrocinéticamente en la que la salida capilar CE está encerrada en un emisor de borosilicato tirado.
- Compruebe el flujo de líquido a través del emisor de electrospray con una cámara e inspeccione visualmente la configuración para detectar posibles fugas.
- Ajuste el voltaje de electrospray a ~ 2.5 kV para iniciar la fuente ESI (frente a tierra a tierra).
- Asegure un nanospray estable para el análisis HRMS mediante el monitoreo de la corriente total de iones. Ajuste el voltaje de electrospray y la distancia del emisor a la entrada HRMS para lograr una pulverización estable (<15% de desviación estándar relativa en intensidad total).
- Detectar los péptidos
NOTA: La detección de péptidos sigue diferentes consideraciones instrumentales para péptidos isobáricos marcados en masa y no marcados y depende del tipo de espectrómetro de masas disponible. Este estudio utiliza un espectrómetro de masas tríbrido orbitrap de acuerdo con los siguientes pasos.- Adquiera eventos MS1 con la configuración: Analizador, orbitrap; Resolución espectral, 120.000 de ancho completo a la mitad del máximo (FWHM); tiempo máximo de inyección (IT), 50 ms; control automático de ganancia (AGC), 4 x 105 cuentas; microescaneos, 1.
- Para secuenciar péptidos, fragmente iones precursores para su detección en el analizador de trampas de iones utilizando la configuración: modo de fragmentación, disociación de colisión de mayor energía (HCD); gas de colisión, nitrógeno; energía de colisión, 32% de energía de colisión normalizada (NCE); TI máxima, 70 ms; AGC, 1 x 104 cuentas; microescaneos, 1.
- OPCIONAL: Cuantificar péptidos marcados con TMT utilizando HRMS en tándem/multietapa (MS2/MS3). Para MS3 que emplea selección síncrona de precursores, los ajustes instrumentales típicos son los siguientes. Los escaneos de una sola etapa (MS1) que estudian los iones más abundantes se disocian a través de la adquisición dependiente de los datos utilizando los parámetros: modo de fragmentación MS2 , disociación inducida por colisión (CID); gas de colisión, helio; energía de colisión, 35% NCE; analizador para iones de fragmento, trampa de iones de iones siguiente configuración: TI máxima, 50 ms; AGC, 5 x 104 cuentas; microescaneos, 1. Seleccione 10 iones de fragmentos MS2 y fragmentarlos con HCD en nitrógeno (65% NCE). Detecte iones de fragmentos MS3 utilizando los siguientes ajustes: resolución Orbitrap 15.000 FWHM, TI máxima, 120 ms; AGC, 1 × 105 cuentas; microescaneos, 1.
NOTA: Se pueden utilizar diferentes métodos y parámetros de adquisición de MS para muestras etiquetadas siguiendo las recomendaciones del proveedor como se describe en otra parte11,40.
- Analizar los datos
NOTA: Las proteínas se identifican y cuantifican utilizando paquetes bioinformáticos avanzados. La fidelidad de las identificaciones se calcula utilizando una base de datos señuelo, expresada como la tasa de descubrimiento falso (FDR) a nivel de péptidos y proteínas.- Procesar los datos utilizando paquetes de software comerciales o de código abierto (revisado en la referencia41). Compare los datos sin procesar con una base de datos preparada concatenando el proteoma Xenopus 9.2 con la base de datos PHROG derivada del ARNm42.
NOTA: Los parámetros de búsqueda son: enzima digestiva, tripsina; escotes perdidos, hasta 2; modificación variable, oxidación de metionina; modificación estática, carbamidometilación de cisteína; tolerancia de masa del precursor, 10 ppm; tolerancia de masa de fragmento, 0,6 Da; longitud mínima del péptido, 5; fidelidad de identificación, <1% FDR para péptidos y proteínas. Sin alquilación a péptidos, la carbamidometilación como modificación estática se excluye durante la búsqueda en la base de datos (por ejemplo, para el análisis de una sola célula). - Cuantificar las abundancias de proteínas a través de estrategias sin etiquetas43 o basadas en etiquetas44,45.
- OPCIONAL: Anotar proteínas para ontología génica. Se pueden utilizar PantherDB46, Reactome47 o Xenbase23 .
- OPCIONAL: Cuantifique la abundancia de proteínas y las diferencias en la abundancia de proteínas en los tipos de células / tejidos utilizando paquetes de software / herramientas web, como Trans-Proteomic Pipeline48, Perseus49 y Orange50.
NOTA: Las consideraciones adicionales sobre el diseño experimental y las opciones de software se revisaron en otra parte41,51. - OPCIONAL: Evalúe los resultados más a fondo utilizando bases de conocimiento, como STRING52 y BioPlex Display 53 para interacciones proteína-proteína conocidas y PhosphoSiteplus54 para fosforilaciones. Para analizar motivos y dominios que están representados en el proteoma, utilice herramientas web como Simple Modular Architecture Research (SMART)55.
- Procesar los datos utilizando paquetes de software comerciales o de código abierto (revisado en la referencia41). Compare los datos sin procesar con una base de datos preparada concatenando el proteoma Xenopus 9.2 con la base de datos PHROG derivada del ARNm42.
Access restricted. Please log in or start a trial to view this content.
Resultados
Este protocolo permitió el estudio de proteínas en células individuales y sus linajes a medida que establecen tejidos en embriones de X. laevis. La Figura 1 ilustra una de esas aplicaciones del enfoque para estudiar proteínas en células predestinadas al tejido neural y el ectodermo neural recién inducido en el embrión. Como se muestra en la Figura 1A, el flujo de trabajo bioanalítico integró herramientas tradicionales de biología celular y del...
Access restricted. Please log in or start a trial to view this content.
Discusión
Este protocolo permite la caracterización de la expresión de proteínas en linajes celulares identificados en embriones de la especie Xenopus. Derivada de HRMS, la metodología combina una especificidad exquisita en la identificación molecular, la capacidad de detección de múltiples proteínas sin sondas moleculares (generalmente cientos a miles de proteínas diferentes) y la capacidad de cuantificación. La adaptabilidad a las herramientas y flujos de trabajo clásicos en biología celular y del desarrollo...
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
Los autores declaran que no hay intereses contrapuestos.
Agradecimientos
Agradecemos a Jie Li (Universidad de Maryland, College Park) por sus valiosos debates sobre la disociación embrionaria y el sistema de control de los bienes sobre el terreno. Agradecemos a Vi M. Quach y Camille Lombard-Banek por su ayuda con la preparación de muestras y la recopilación de datos en estudios previos que ejemplifican las aplicaciones proteómicas que se destacan en este protocolo. Partes de este trabajo fueron apoyadas por la National Science Foundation bajo el número de premio IOS-1832968 CAREER (a P.N.), los Institutos Nacionales de Salud bajo el número de premio R35GM124755 (a P.N.), el Programa de Asociación de la Universidad de Maryland-Instituto Nacional del Cáncer (a P.N.) y los premios de investigación de la Fundación COSMOS Club (a A.B.B. y L.R.P.).
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| Acetonitrile (LC-MS-grade) | Fisher Scientific | A955 | |
| Agarose | ThermoFisher Scientific | R0492 | |
| Ammonium bicarbonate | Fisher Scientific | A643-500 | |
| Analytical Column | Thermo Scientific | 164941 | |
| Analytical microbalance | Mettler-Toledo | XSE105DU | |
| Automatic peptide fractionation platform | Agilent | 1260 Infinity II | |
| Borosilicate Capillaries | Sutter Instruments Co. | B100-50-10 | |
| Borosilicate Capillaries (for making Emmitters) | Sutter Instruments | B100-75-10 | |
| C18 spin columns (for desalting) | ThermoFisher Scientific | 89870 | |
| Camera ro monitor electrospray | Edmund Optics Inc. | EO-2018C | |
| Combretastatin A4 | Millipore Sigma | C7744 | |
| Commercial CESI system | AB SCIEX | CESI | |
| (Cyclohexylamino)-1-propanesulfonic acid (CAPS) | VWR | 97061-492 | |
| Cytochalasin D | Millipore Sigma | C8273 | |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable | ThermoFisher Scientific | D22910 | |
| Diothiothreitol | Fisher Scientific | FERR0861 | |
| Dumont #5 Forceps | Fine Science Tools | 11252-30 | |
| EDTA | Fisher Scientific | AAJ62786AP | |
| Epifluorescence light source | Lumencore | AURA III | |
| Eppendorf LoBing microcentrifuge tubes: protein | Fisher Scientific | 13-698-793 | |
| Formic acid (LC-MS-grade) | Fisher Scientific | A117-50 | |
| Freezer (-20 °C) | Fisher Scientific | 97-926-1 | |
| Freezer (-80 °C) | Thermo Scientific | TSX40086A | |
| Fused silica capillary | Molex | 1088150596 | |
| Heat Block | Benchmark | BSH300 | |
| High pressure liquid Chromatography System | ThermoFisher Scientific | Dionex Ultimate 3000 RSLC nanosystem | |
| High voltage power supply | Spellman | CZE1000R | |
| High-resolution Mass Spectrometer | ThermoFisher Scientific | Orbitrap Fusion Lumos Tribrid Mass Spectrometer | |
| HPLC caps | Thermo Scientific | C4013-40A | |
| HPLC Vials | Thermo Scientific | C4013-11 | |
| Illuminator e.g. Goosenecks | Nikon | C-FLED2 | |
| Ingenuity Pathway Analysis | Qiagen | ||
| Iodoacetamide | Fisher Scientific | AC122275000 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456-4 | |
| Microcapillary puller | Suttor Instruments | P-2000 | |
| Microinjector | Warner Instrument, Handem, CT | PLI-100A | |
| Micropippette puller | Sutter Instruments Co. | P-1000 | |
| MS data analysis software, commercial | ProteomeDiscoverer | ||
| MS data analysis software, opensource | MaxQuant | ||
| non-idet 40 substitute | Millipore Sigma | 11754599001 | |
| Petri dish 60 mm and 80 mm | Fisher Scientific | S08184 | |
| Pierce 10 µL bed Zip-tips (for desalting) | ThermoFisher Scientific | 87782 | |
| Pierce bicinchoninic acid protein assay kit | ThermoFisher Scientific | 23225 | |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 | |
| Pierce Trypsin Protease (MS Grade) | Fisher Scientific | PI90058 | |
| Protein LoBind vials | Eppendorf | 0030108434 , 0030108442 | |
| Refrigerated Centrifuge | Eppendorf | 5430R | |
| Refrigerated Incubator | Thermo Scientific | PR505755R/3721 | |
| sodium isethionate | Millipore Sigma | 220078 | |
| sodium pyrophosphate | Sigma Aldrich | 221368-100G | |
| Stainless steel BGE vial | Custom-Built | ||
| Stainless steel sample vials | Custom-Built | ||
| Stereomicroscope (objective 10x) | Nikon | SMZ 1270, SZX18 | |
| Sucrose | VWR | 97063-790 | |
| Syringe pumps (2) | Harvard Apparatus | 704506 | |
| Syringes (gas-tight): 500–1000 µL | Hamilton | 1750TTL | |
| Transfer pipettes (Plastic, disposable) | Fisher Scientific | 13-711-7M | |
| Trap Column | Thermo Scientific | 164750 | |
| Tris-HCl (1 M solution) | Fisher Scientific | AAJ22638AP | |
| Vacuum concentrator capable of operation at 4–10 °C | Labconco | 7310022 | |
| Vortex-mixer | Benchmark | BS-VM-1000 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| XYZ translation stage | Thorlabs | PT3 | |
| XYZ translation stage | Custom-Built |
Referencias
- Shoemaker, L. D., Kornblum, H. I. Neural Stem Cells (NSCs) and Proteomics. Molecular & Cellular Proteomics. 15 (2), 344-354 (2016).
- Cervenka, J., et al. Proteomic characterization of human neural stem cells and their secretome during in vitro differentiation. Frontiers in Cellular Neuroscience. 14, 612560(2021).
- Christian, J. L. Morphogen gradients in development: From form to function. Wiley Interdisciplinary Reviews. Developmental Biology. 1 (1), 3-15 (2012).
- Gurdon, J. B., Elsdale, T. R., M, F. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 182, 64-65 (1958).
- Harland, R. M., Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 27 (12), 507-515 (2011).
- Moody, S. A. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 119 (2), 560-578 (1987).
- Moody, S. A. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Developmental Biology. 122 (2), 300-319 (1987).
- Dale, L., Slack, J. M. W. Fate map for the 32-cell stage of Xenopus laevis. Development. 99 (4), 527-551 (1987).
- Sun, L. L., et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Analytical Chemistry. 88 (13), 6653-6657 (2016).
- Lombard-Banek, C., Moody, S. A., Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angewandte Chemie-International Edition. 55 (7), 2454-2458 (2016).
- Zhang, Y. Y., Fonslow, B. R., Shan, B., Baek, M. C., Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113 (4), 2343-2394 (2013).
- Sive, H. L., Grainger, R. M., Harland, R. M. Early development of Xenopus laevis: A laboratory manual. , Cold Spring Harbor Laboratory Press. New York. (2000).
- Briggs, J. A., et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 360 (6392), (2018).
- Gupta, M., Sonnett, M., Ryazanova, L., Presler, M., Wuhr, M. Quantitative proteomics of xenopus embryos I, sample preparation. Xenopus. Methods in Molecular Biology. Vleminckx, K. 1865, Humana Press Inc. NY. 175-194 (2018).
- Baxi, A. B., Lombard-Banek, C., Moody, S. A., Nemes, P. Proteomic characterization of the neural ectoderm fated cell clones in the Xenopus laevis embryo by high-resolution mass spectrometry. ACS Chemical Neuroscience. 9 (8), 2064-2073 (2018).
- Moody, S. A. Cell lineage analysis in Xenopus embryos. Methods in Molecular Biology. 135, 331-347 (2000).
- Sater, A. K., Moody, S. A. Using Xenopus to understand human diseases and developmental disorders. Genesis. 55 (1-2), 1-14 (2017).
- Lombard-Banek, C., Choi, S. B., Nemes, P. Enzyme Activity in Single Cells. Methods in Enzymology. Allbritton, N. L., Kovarik, M. L. 628, 263-292 (2019).
- Lombard-Banek, C., Moody, S. A., Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Frontiers in Cell and Developmental Biology. 4, 11(2016).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. Microprobe capillary electrophoresis mass spectrometry for single-cell metabolomics in live frog (Xenopus laevis) embryos. Journal of Visualized Experiments: JoVE. (130), e56956(2017).
- Lombard-Banek, C., Moody, S. A., Manzin, M. C., Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and zebrafish embryos. Analytical Chemistry. 91 (7), 4797-4805 (2019).
- Klein, S. L. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Developmental Biology. 120 (1), 299-304 (1987).
- Karimi, K., et al. Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Research. 46 (1), 861-868 (2018).
- Kakebeen, A. D., Chitsazan, A. D., Wills, A. E. Tissue disaggregation and isolation of specific cell types from transgenic Xenopus appendages for transcriptional analysis by FACS. Developmental Dynamics. 250 (9), 1381-1392 (2021).
- Garcia, B. A. What does the future hold for top down mass spectrometry. Journal of the American Society for Mass Spectrometry. 21 (2), 193-202 (2010).
- Toby, T. K., Fornelli, L., Kelleher, N. L. Progress in top-down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry. (Palo Alto Calif). 9 (1), 499-519 (2016).
- Zhang, Z. B., Dubiak, K. M., Huber, P. W., Dovichi, N. J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis Embryos. Analytical Chemistry. 92 (7), 5554-5560 (2020).
- Wisniewski, J. R. Microbial Proteomics: Methods and Protocols.Methods in Molecular Biology. Becher, D. 1841, Humana Press Inc. NY. 3-10 (2018).
- Hughes, C. S., et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Zhu, Y., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nature Communications. 9, 882(2018).
- Wessel, D., Flugge, U. I. A method for the quantitative recovery of protein in dilute-solution in the presence of detergents and lipids. Analytical Biochemistry. 138 (1), 141-143 (1984).
- Jiang, L., He, L., Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of Chromatography A. 1023 (2), 317-320 (2004).
- Hildonen, S., Halvorsen, T. G., Reubsaet, L. Why less is more when generating tryptic peptides in bottom-up proteomics. Proteomics. 14 (17-18), 2031-2041 (2014).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19, 161(2018).
- Drouin, N., et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: effective electrophoretic mobility for reproducible and robust compound annotation. Analytical Chemistry. 92 (20), 14103-14112 (2020).
- Sun, L. L., Zhu, G. J., Zhang, Z. B., Mou, S., Dovichi, N. J. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. Journal of Proteome Research. 14 (5), 2312-2321 (2015).
- DeLaney, K., Sauer, C. S., Vu, N. Q., Li, L. J. Recent advances and new perspectives in capillary electrophoresis-mass spectrometry for single cell "omics". Molecules. 24 (1), 21(2019).
- Nemes, P., Rubakhin, S. S., Aerts, J. T., Sweedler, J. V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nature Protocols. 8 (4), 783-799 (2013).
- Choi, S. B., Zamarbide, M., Manzini, M. C., Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. Journal of the American Society for Mass Spectrometry. 28 (4), 597-607 (2017).
- Pino, L. K., Rose, J., O'Broin, A., Shah, S., Schilling, B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions. 48 (5), 1953-1966 (2020).
- Chen, C., Hou, J., Tanner, J. J., Cheng, J. L. Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences. 21 (8), 25(2020).
- Peshkin, L., et al. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Developmental Cell. 35 (3), 383-394 (2015).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 13 (9), 2513-2526 (2014).
- Gygi, S. P., et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 17 (10), 994-999 (1999).
- Thompson, A., et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 75 (8), 1895-1904 (2003).
- Mi, H. Y., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive api. Nucleic Acids Research. 49, 394-403 (2021).
- Schmidt, E., et al. On the Move Federated Workshops. , Springer, Verlag. Berlin. 710-719 (2006).
- Deutsch, E. W., et al. Trans-Proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 9 (7-8), 745-754 (2015).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Demsar, J., et al. Orange: Data mining toolbox in Python. Journal of Machine Learning Research. 14, 2349-2353 (2013).
- Oberg, A. L., Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. Journal of Proteome Research. 8 (5), 2144-2156 (2009).
- Jensen, L. J., et al. STRING 8 - a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, 412-416 (2009).
- Schweppe, D. K., Huttlin, E. L., Harper, J. W., Gygi, S. P. BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data. Journal of Proteome Research. 17 (1), 722-726 (2018).
- Hornbeck, P. V., et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research. 43, 512-520 (2015).
- Letunic, I., Khedkar, S., Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Research. 49, 458-460 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie-International Edition. 60 (23), 12852-12858 (2021).
- Lombard-Banek, C., Reddy, S., Moody, S. A., Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Molecular & Cellular Proteomics. 15 (8), 2756-2768 (2016).
- Saha-Shah, A., et al. Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Analytical Chemistry. 91 (14), 8891-8899 (2019).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Analytical Chemistry. 89 (13), 7069-7076 (2017).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47, 442-450 (2019).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados