
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Протеомика масс-спектрометрии под контролем клеточных линий у развивающегося эмбриона (лягушки)
В этой статье
Резюме
Здесь мы описываем основанную на масс-спектрометрии протеомную характеристику клеточных линий с известными тканевыми судьбами эмбриона позвоночных Xenopus laevis .
Аннотация
Характеристика молекулярных событий, когда клетки дают начало тканям и органам, повышает потенциал для лучшего понимания нормального развития и разработки эффективных средств от болезней. Технологии, обеспечивающие точную идентификацию и количественную оценку различных типов и большого количества белков, предоставят до сих пор недостающую информацию о молекулярных механизмах, организующих развитие тканей и организмов в пространстве и времени. Здесь мы представляем протокол на основе масс-спектрометрии, который позволяет измерять тысячи белков в идентифицированных клеточных линиях эмбрионов Xenopus laevis (лягушек). Этот подход основан на воспроизводимых картах клеточной судьбы и установленных методах идентификации, флуоресцентной маркировки, отслеживания и отбора образцов клеток и их потомства (клонов) из этой модели развития позвоночных. После сбора клеточного содержимого с помощью микровыборки или выделения клеток путем диссекции или сортировки клеток, активированной флуоресценцией, белки извлекаются и обрабатываются для восходящего протеомного анализа. Жидкостная хроматография и капиллярный электрофорез используются для обеспечения масштабируемого разделения для обнаружения и количественного определения белка с помощью масс-спектрометрии высокого разрешения (HRMS). Приведены репрезентативные примеры протеомной характеристики клеток нервной ткани. Протеомика HRMS, управляемая клеточными линиями, адаптируется к различным тканям и организмам. Он достаточно чувствителен, специфичен и количественный, чтобы заглянуть в пространственно-временную динамику протеома во время развития позвоночных.
Введение
Наше понимание дифференцировки клеток и генезиса тканей и органов является результатом десятилетий сложных целевых скринингов генов и их продуктов. Расширение наших знаний обо всех биомолекулах и их количествах во время важных клеточных событий поможет разгадать молекулярные механизмы, которые контролируют пространственную и временную структуру плана тела позвоночных. Технологии, обеспечивающие молекулярную амплификацию и секвенирование, теперь могут регулярно сообщать о большом количестве генов и транскриптов, поддерживая исследования, основанные на гипотезах, в фундаментальных биологических и трансляционных исследованиях. Чтобы понять развивающиеся системы, сложная взаимосвязь между транскрипцией и трансляцией требует прямого анализа нескольких белков и их посттрансляционных модификаций. Глобальная протеомика с использованием биологических систем in vitro, таких как индуцированные плюрипотентные стволовые клетки, начала очерчивать механизмы тканевой индукции 1,2. У сложных организмов, таких как эмбрион позвоночных, развитие зависит от градиентов морфогена в контексте пространства и времени3. Из этого следует, что получение знаний о протеомных изменениях по мере того, как клетки дифференцируются с образованием специализированных тканей, таких как нервные ткани, дает ключ к разблокированию молекулярных программ, контролирующих нормальное и дефектное развитие, и направляет терапию следующего поколения.
Позвоночная южноафриканская когтистая лягушка (Xenopus laevis) является хорошо зарекомендовавшей себя моделью в клеточной и развивающейся, нейро- и регенеративной биологии. Нобелевская премия сэра Джона Гердона по физиологии и медицине 2012 года 4,5 за открытие плюрипотентности соматического ядра подчеркнула важность этой модели для открытий в фундаментальных и трансляционных исследованиях. Эмбрионы Xenopus развиваются внешне по отношению к матери, тем самым облегчая прямое манипулирование клетками, клеточными клонами и экспрессией генов на различных стадиях развития. Асимметричная пигментация и стереотипное деление клеток позволили составить карту воспроизводимых карт судьбы 16-6 и 32-клеточногоэмбриона на 7,8 стадии. Для протеомики на основе масс-спектрометрии высокого разрешения (HRMS) дополнительные преимущества модели включают относительно большой размер (~1 мм в диаметре), что дает обильное содержание белка для анализа (~130 мкг у эмбрионов на ранней стадии расщепления, ~10 мкг белка в одиночных клетках 16-клеточного эмбриона)9,10.
В настоящее время HRMS является ведущей технологией выбора для обнаружения белков. Эта технология позволяет прямо, чувствительно и специфически обнаруживать и количественно определять множественные, обычно от сотен до тысяч различныхбелков11. Восходящая протеомика HRMS включает в себя ряд взаимосвязанных этапов. После экстракции из образца клетки / ткани белки перевариваются протеолитическим ферментом, таким как трипсин (восходящая протеомика). Полученные пептиды разделяют на основе их различных физико-химических свойств, включая гидрофобность (обращенно-фазовая жидкостная хроматография, LC), суммарный заряд (ионообменная хроматография), размер (эксклюзионная хроматография) или электрофоретическую подвижность (капиллярный электрофорез, CE). Затем пептиды заряжаются (ионизируются), как правило, с помощью электрораспыления ионизации (ESI), а пептидные ионы обнаруживаются и секвенируются посредством газофазной фрагментации с помощью тандемной HRMS. Полученные пептидные данные сопоставляются с протеомом исследуемого организма. При том, что интенсивность сигнала пептид-ионов специфического белка (протеотипа) коррелирует с концентрацией, количественное определение белка может быть выполнено без меток или на основе меток (мультиплексирующее количественное определение). Протеомика HRMS дает богатый источник информации о молекулярном состоянии исследуемой системы, что позволяет генерировать гипотезы и проводить последующие функциональные исследования.

Рисунок 1: Пространственно-временная масштабируемая протеомика, обеспечивающая протеомику HRMS, управляемую клеточной линией, в развивающемся эмбрионе (лягушке). (A) Визуализация образца (1) с использованием стереомикроскопа (2) для инъекции идентифицированной клетки (врезка) с использованием изготовленной микропипетки (3) под контролем трансляционной стадии (4). (B) Субклеточный отбор идентифицированных левых клеток D11 в 16-клеточном эмбрионе. (C) Рассечение целой клетки D11 из 16-клеточного эмбриона. (D) Флуоресцентное (зеленое) отслеживание левого и правого потомства D111 от 32-клеточного эмбриона для направления рассечения невральной эктодермы (NE) в гаструле (стадия 10) и выделения ткани-потомка от головастика с использованием FACS. Масштабные линейки: 200 мкм для эмбрионов, 1,25 мм для флакона. Рисунки были адаптированы с разрешения ссылок 15,19,21,59. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
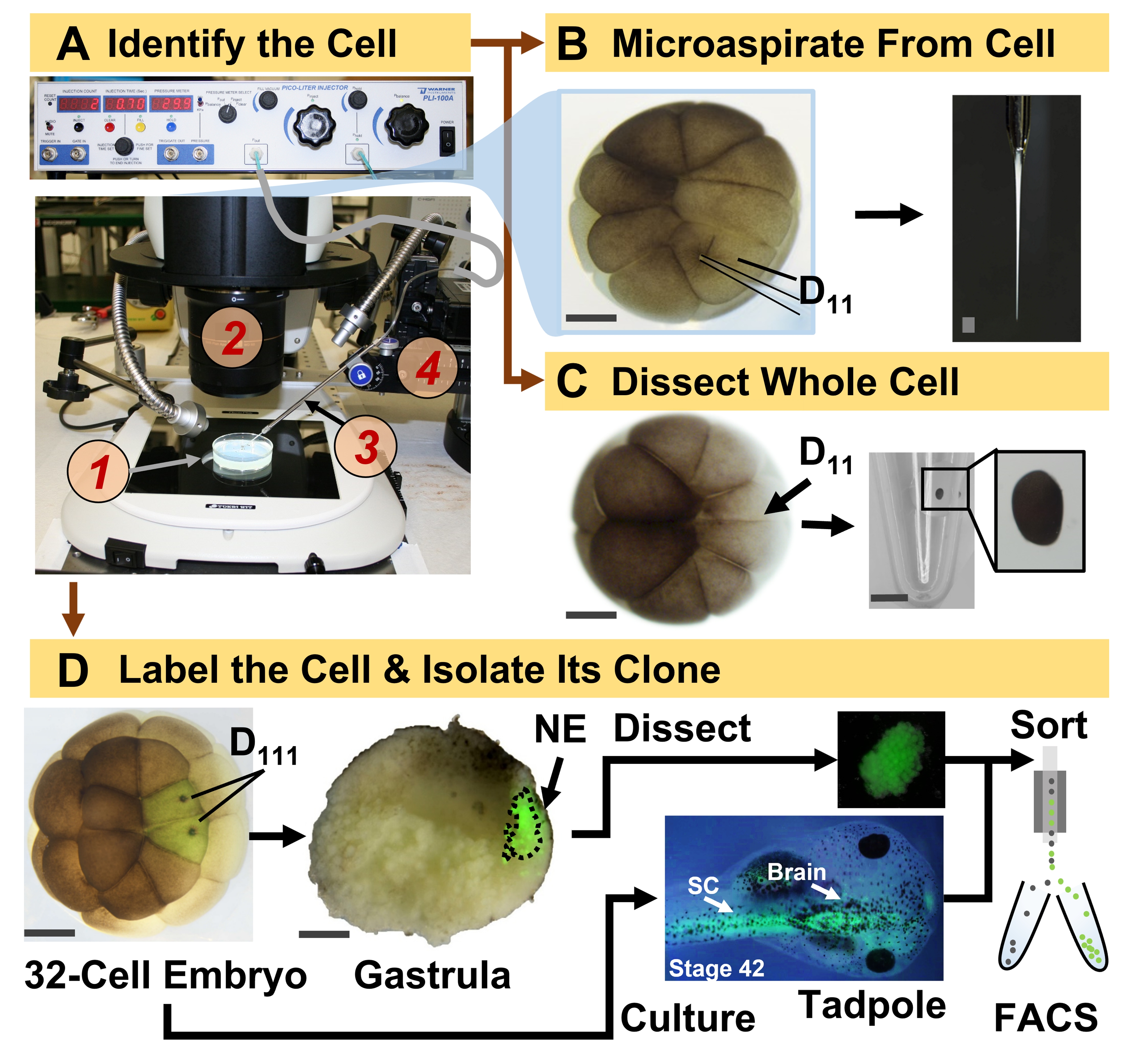{kind=link}
Представленный здесь протокол позволяет количественно оценить большое количество белков в идентифицированных клетках/тканях развивающихся эмбрионов X. laevis на основе HRMS. Подход основан на точной идентификации клеток, воспроизводимых картах клеточной судьбы и установленных методологиях отслеживания клеточных линий в этой биологической модели 6,7,8. Как показано на рисунке 1, мы изучаем протеомы из отдельных клеток, используя диссекцию целых клеток или капиллярный микропробоотбор для аспирации клеточного содержимого. Наблюдение за происхождением клетки позволяет нам изучать пространственно-временную эволюцию протеома по мере того, как клетки образуют ткани во время гаструляции. Клеточное потомство флуоресцентно маркируют путем введения флуорофора, конъюгированного с инертным декстраном или мРНК для флуоресцентного белка (например, зеленого флуоресцентного белка или GFP). Меченое потомство изолируют в желаемые моменты развития. Во время гаструляции клоны клеток, которые плотно сгруппированы, могут быть выделены путем рассечения. После гаструляции клеточные клоны могут быть распределены внутри эмбриона за счет миграционных движений и могут быть выделены из диссоциированных тканей с помощью флуоресцентно-активированной сортировки клеток (FACS). Белки в этих клетках и тканях измеряются с помощью восходящей протеомики с использованием ВЭЖХ или КЭ для разделения и тандемной HRMS ESI для идентификации. Протеомика HRMS, управляемая клеточными линиями, масштабируется для различных размеров клеток и линий внутри эмбриона и является специфической, чувствительной и количественной. На отдельных примерах, показанных здесь, мы также демонстрируем, что этот протокол является масштабируемым и широко адаптируемым к различным типам клеток и клеточных линий.

Рисунок 2: Биоаналитический рабочий процесс. Микродиссекция и капиллярная аспирация, или FACS, облегчают отбор проб клеточного и клонального белка. Истощение обильных белков желтка и разделение с помощью капиллярного электрофореза (CE) или нанопоточной жидкостной хроматографии (LC) с повышенной чувствительностью идентификации (ID) с использованием масс-спектрометрии высокого разрешения (HRMS) с помощью ионизации электрораспылением (ESI). Количественная оценка выявила дисрегуляцию, предоставив новую информацию для исследований, основанных на гипотезах, в сочетании с информацией, доступной из онтологии генов (GO). Рисунки были адаптированы с разрешения ссылки15. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
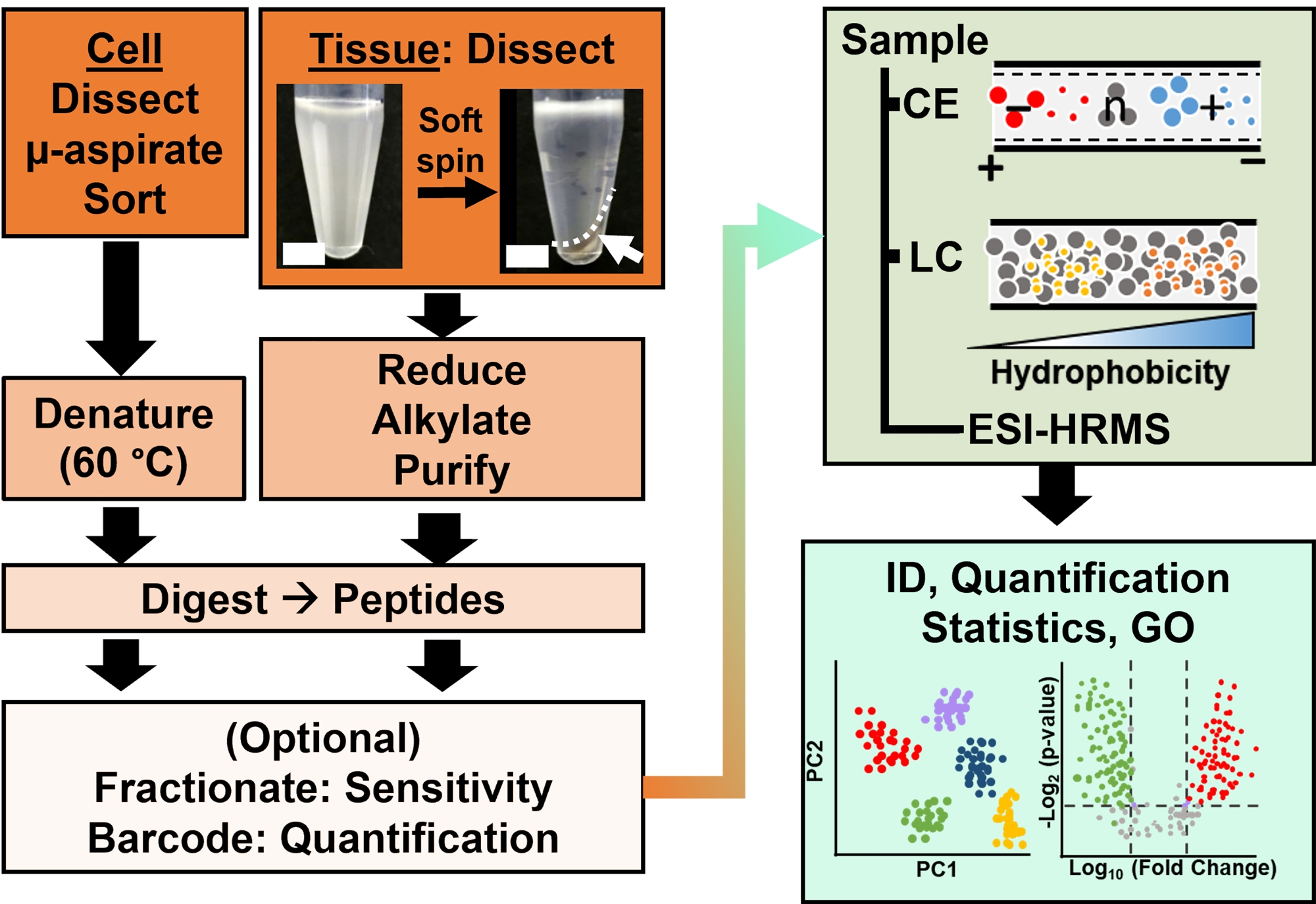{kind=link}
Access restricted. Please log in or start a trial to view this content.
протокол
Все протоколы, обеспечивающие гуманное содержание и обращение со взрослыми лягушками Xenopus laevis , были одобрены Институциональным комитетом по уходу за животными и их использованию в Университете Мэриленда, Колледж-Парк (номера одобрения R-DEC-17-57 и R-FEB-21-07).
1. Подготовьте растворы
- Для эмбриологии
- Приготовьте 1x, 0,5x и 0,2x раствор Штейнберга (SS), затем автоклавируйте их (120 ° C в течение 20 мин) до стерильности в соответствии со стандартными протоколами12.
- Приготовьте 3% (мас./об.) Ficoll в стерилизованном 1x SS в соответствии со стандартными протоколами12.
- Для удаления желе приготовьте 2% (мас./об.) раствор цистеина и отрегулируйте его рН до 8, добавив 10 М раствора гидроксида натрия по каплям.
ВНИМАНИЕ: Воздействие цистеина может вызвать повреждение кожи и дыхательных путей. Гидроксид натрия является коррозионным веществом, которое может нанести серьезный вред коже и глазам при прямом воздействии. При работе с этими химическими веществами используйте соответствующие средства индивидуальной защиты (СИЗ), такие как перчатки и лабораторный халат. - Для индикатора происхождения приготовьте 0,5% (об. / об.) флуоресцентного декстрана в стерильной деионизированной воде. В качестве альтернативы готовят раствор 0,2 мкг/мкл мРНК для флуоресцентных белков в стерильной деионизированной воде (например, GFP).
- Для диссоциации клеток приготовьте буфер Newport 2.0, содержащий 0,1 М изетионата натрия, 20 мМ пирофосфата натрия и 10 мМ CAPS, затем доведите его рН до 10,513.
ВНИМАНИЕ: Воздействие пирофосфата натрия может вызвать раздражение кожи и глаз. Используйте соответствующие СИЗ при работе с этими химическими веществами.
- Для восходящей протеомики
- Подготовьте буфер для лизиса клеток, включающий: 250 мМ сахарозы, 1% заменитель нонидета P-40 (мас./об.), 20 мМ трис-HCl, 5 мМ ЭДТА, 10 мкМ цитохалазин D и 10 мкМ комбретастатин 4А. Приготовьте бульон из 10% (мас./об.) додецилсульфата натрия14,15.
ПРИМЕЧАНИЕ: Tris-HCl был выбран для минимизации загрязнения HEPES во время нанопотока LC (nanoLC)-HRMS.
ВНИМАНИЕ: Воздействие заменителя нонидета P-40 может вызвать раздражение кожи. Цитохалазин D является тератогенным при употреблении, а комбретастатин остро токсичен при прямом воздействии. Используйте соответствующие СИЗ при работе с этими химическими веществами. - Для разделения пептидов с помощью CE приготовьте следующие растворители (v/v): растворитель образца, 75% ацетонитрил (ACN), содержащий 0,05% уксусной кислоты (AcOH) в воде; раствор оболочки, 10% ACN, содержащий 0,05% AcOH в воде; фоновый электролит (BGE), 25% ACN, содержащий 1 М муравьиную кислоту (FA) в воде.
ВНИМАНИЕ: AcOH и FA токсичны при вдыхании или употреблении и могут вызвать серьезное повреждение кожи и глаз при прямом воздействии. Используйте соответствующие СИЗ при работе с этими химическими веществами. - Для разделения пептидов с помощью обращенно-фазового наноЛК подготовьте (об./об.): подвижная фаза А (водная), вода, содержащая 0,1% ЖК; подвижная фаза B (органическая), 0,1% FA в ACN.
ПРИМЕЧАНИЕ: Все смеси должны быть приготовлены с использованием растворителей класса LC-MS, чтобы свести к минимуму химические помехи во время обнаружения HRMS.
- Подготовьте буфер для лизиса клеток, включающий: 250 мМ сахарозы, 1% заменитель нонидета P-40 (мас./об.), 20 мМ трис-HCl, 5 мМ ЭДТА, 10 мкМ цитохалазин D и 10 мкМ комбретастатин 4А. Приготовьте бульон из 10% (мас./об.) додецилсульфата натрия14,15.
2. Подготовьте инструменты для микроинъекций и вскрытия
- Чтобы мягко перемещать и ориентировать эмбрионы, сделайте волосяные петли, зафиксировав чистые волосы в пипетке Пастера, как описано в другом месте16.
- Для микроинъекций изготовляют иглы, вытягивая боросиликатные капилляры (1 мм/500 мкм внешний/внутренний диаметр) с помощью пипетки-съемницы, как описано в другом месте16.
ПРИМЕЧАНИЕ: Здесь для изготовления игл использовался съемник пипеток P-1000 со следующими настройками: тепловой, 495; тяга — 30, скорость — 60; время, 150; давление, 200. - При наблюдении под стереомикроскопом разрежьте кончик капилляра с помощью пары острых щипцов, чтобы по существу превратить капилляр в (микро)иглу (например, Dumont #5)16.
ВНИМАНИЕ: Вытянутые капилляры очень острые, и с ними следует обращаться осторожно.
ПРИМЕЧАНИЕ: Кончик иглы должен быть достаточно острым (наружный диаметр 10-15 мкм), чтобы иметь возможность проткнуть клетку с минимальным повреждением клеточной мембраны, чтобы внутриклеточное содержимое не вытекало наружу, и клетка могла зажить и продолжать быть жизнеспособной. - Чтобы удерживать эмбрионы во время микроинъекций, подготовьте лунки в глиняной посуде. В чашке Петри диаметром 15 мм отпечатайте отверстия диаметром ~1 мм и глубиной ~0,5 мм, как описано в другом месте16.
- Для микродиссекции приготовьте блюда, покрытые агарозой. Сделайте 2% агарозы в 1x SS и автоклавируйте его для стерилизации раствора (120 ° C в течение 20 мин). Заполните чашки Петри диаметром 60 мм наполовину и дайте тарелкам застыть. Сделайте лунки диаметром ~1 мм и глубиной ~0,5 мм с помощью шарикового пипетки Пастера, как описано ранее16.
3. Изолируйте клеточную линию
ПРИМЕЧАНИЕ: Следующие шаги выполняются для выделения идентифицированных одиночных клеток и/или их потомственных клеточных линий. Обычно эмбрион культивируют до 16- или 32-клеточной стадии, где воспроизводимо картируются тканевые судьбы каждой клетки 6,7,17. Эмбриональные клетки идентифицируются на основе морфологии, местоположения и со ссылкой на карты их судьбы. Для одноклеточного анализа идентифицированные клетки выделяют методом ручного вскрытия или собирают их внутриклеточное содержимое в капиллярную пипетку и депонируют в 5 мкл 0,5 мМ бикарбоната аммония. Полученный образец хранят при температуре -80 °C до анализа (рис. 1)18,19,20,21. Для анализа клеточного происхождения идентифицированным клеткам вводят индикатор происхождения, а их последующие клоны выделяют на ключевых стадиях развития (например, во время гаструляции для изучения индукции ткани, после нейруляции для изучения приверженности ткани). Далее изложены шаги по флуоресцентной маркировке происхождения идентифицированных клеток для выделения путем диссекции или FACS.
- Культивирование эмбрионов
- Получение эмбрионов путем естественного спаривания или экстракорпорального оплодотворения (ЭКО) в соответствии с установленными протоколами12.
ПРИМЕЧАНИЕ: Естественное спаривание логистически проще, щадит взрослых самцов лягушек и дает эмбрионы на разных стадиях развития, тогда как ЭКО обеспечивает синхронизированные эмбрионы с развитием для экспериментов, требующих точной стадии. - Дежелли эмбрионов. Удаляют желейную оболочку, окружающую эмбрионы, путем обработки раствором для удаления желелей, как описано в другом месте12,16.
ПРИМЕЧАНИЕ: Микроинъекции и диссекция требуют доступа к клеткам и тканям, что требует удаления желлинга у эмбрионов X. laevis. - Выберите 2-клеточные эмбрионы со стереотипной пигментацией16,22.
ПРИМЕЧАНИЕ: Этот шаг важен для обеспечения точности и воспроизводимости при идентификации клетки и ее происхождения. - Культивирование эмбрионов до желаемой стадии развития. Перенесите очищенные эмбрионы в чашку Петри, содержащую 1x SS, и инкубируйте их при температуре 14-25 ° C, чтобы контролировать скорость развития.
ПРИМЕЧАНИЕ: Температурная зависимость развития воспроизводима и отображена на диаграмме для X. laevis, доступной на Xenbase23 (www.xenbase.org). Культивирование партий эмбрионов при разных температурах позволяет получить ошеломляющие стадии развития. Это помогает распределить количество эмбрионов, доступных в данный момент времени для экспериментов. - Следите за характером расщепления эмбрионов и отбирайте эмбрионы со стереотипной пигментацией и паттернами расщепления для микроинъекции16.
ПРИМЕЧАНИЕ: При отборе 16- и 32-клеточных эмбрионов убедитесь, что клеточные расщепления симметричны для воспроизводимого отслеживания линии.
- Получение эмбрионов путем естественного спаривания или экстракорпорального оплодотворения (ЭКО) в соответствии с установленными протоколами12.
- Пометьте интересующие ячейки
- Установите инъекционную иглу, содержащую раствор индикатора происхождения. Установите иглу для микроинъекций в держатель микропипетки, управляемый многоосевым микроманипулятором.
- Подсоедините держатель микропипетки к микроинжектору. Заполните иглу индикатором происхождения, применяя отрицательное давление, как описано в другом месте16. Рисунок 1A иллюстрирует установку.
- Откалибруйте иглу. Отрегулируйте размер кончика иглы и время впрыска, чтобы доставить ~ 1 нл раствора индикатора происхождения, измеренного в (минеральном) масле в соответствии с протоколом, доступным в другом месте16.
ПРИМЕЧАНИЕ: Капилляры с более широким кончиком, как правило, повреждают клеточную мембрану, вызывая утечку субклеточного содержимого и введенного индикатора линии, тогда как капилляры с меньшими кончиками склонны к закупорке. Идеально подходят капилляры с наружным диаметром наконечника ~10 мкм, для доставки ~ 1 нл требуется импульс давления 40 фунтов на квадратный дюйм в течение ~ 300 мс. - Залейте глиняную чашку для микроинъекций 3% раствором Фиколла и перенесите ~ 10 эмбрионов в глиняную форму с помощью пипетки для переноса. Используйте волосяную петлю, чтобы направить каждый эмбрион в лунку и осторожно расположить их так, чтобы интересующая клетка-мишень находилась под прямым углом к микроигле.
- Идентификация клетки-предшественника интересующей линии по картам судьбы тканей X. laevis. Например, на рисунке 1 показана маркировка нейронных эктодермальных клонов на основе инъекции его клеток-предшественников в 32-клеточные эмбрионы (левая и правая клетки D111 ).
ПРИМЕЧАНИЕ: Подробные карты судьбы 16-6 и 32-клеточныхэмбрионов 7,8 доступны на интерактивной платформе через Xenbase23. Важно обеспечить стереотипную пигментацию и расщепление на эмбрионах при использовании их для экспериментов по отслеживанию родословной. - Вводят интересующей клетке (клеткам) ~ 1 нл флуоресцентного декстрана или ~ 200 пг мРНК, как описано ранее16.
ПРИМЕЧАНИЕ: Используйте декстрановые сопряжения мощностью 10 000-40 000 МВт. Меньшие конъюгаты декстрана могут проходить через щелевые соединения, тогда как более крупные конъюгаты декстрана могут не равномерно диффундировать в инъецируемую клетку. Планируйте ввести клетки ~ 10 эмбрионам, чтобы иметь достаточное количество тканей для протеомного анализа. - Подтвердите успешность маркировки клеток под стереомикроскопом. Убедитесь, что вводится только предполагаемая клетка. Откажитесь от эмбрионов, содержащих поврежденные или неправильно помеченные клетки, в соответствии с институциональной политикой.
ПРИМЕЧАНИЕ: Поскольку X. laevis является инвазивным во многих неестественных средах, эмбрионы могут быть заморожены, чтобы обеспечить летальность перед выбрасыванием эмбрионов.
- Изолировать меченое клеточное потомство
- Перенесите инъецированные эмбрионы в 0,5x SS в чашку Петри и культивируйте их при температуре 14-25 ° C до достижения желаемой стадии развития.
ПРИМЕЧАНИЕ: Ознакомьтесь с установленными протоколами для стадирования эмбрионов, о которых сообщается в Xenbase. - Перенесите 3-5 эмбрионов в чашку с агаром с 0,2x раствором SS для микродиссекций.
ПРИМЕЧАНИЕ: Снижение концентрации соли в растворе SS с 0,5x до 0,2x помогает разделить клетки во время вскрытия. - Используйте два заостренных щипца, чтобы аккуратно удалить вителлиновую мембрану, окружающую эмбрион.
ПРИМЕЧАНИЕ: Чтобы уберечь интересующий клон от повреждений, снимите мембрану с противоположной стороны флуоресцентно меченного клона. - Изолируйте помеченный клон путем ручного вскрытия (этапы 3.3.5-3.3.6) или FACS (этапы 3.3.7-3.3.8) следующим образом.
- Используйте щипцы, чтобы отделить меченый клон от эмбриона.
ПРИМЕЧАНИЕ: Другие инструменты, такие как микрохирургические ножницы, вольфрамовые иглы или ножи для волос бровей, могут быть использованы для вскрытия меченого клона, как подробно описано в другом месте16. - Рассеченную ткань соберите пипеткой 0,5-10 мкл и поместите в микроцентрифужный флакон. Используя трансферную пипетку, аспирируйте среду, окружающую собранную ткань, чтобы ограничить содержание солей в образце, которые мешают анализу HRMS на более поздних этапах.
ПРИМЕЧАНИЕ: Используйте флаконы, которые минимизируют адсорбцию белка на пластиковых поверхностях, чтобы свести к минимуму потери белка на поверхности флаконов на более поздних этапах рабочего процесса. - Чтобы изолировать с помощью FACS, перенесите ~ 5-8 девителлизованных эмбрионов в каждую лунку 12-луночного планшета, содержащего ~ 5 мл буфера Newport 2.0. Диссоциируют эмбрионы, нутируя пластину при 80 об/мин в течение 20-30 мин при комнатной температуре13.
ПРИМЕЧАНИЕ: Эмбрионы/личинки старше 22 стадии имеют обильные белки внеклеточного матрикса, что затрудняет диссоциацию на отдельные клетки. Дополнительные ферментативные подходы могут быть адаптированы для диссоциации тканей более старых эмбрионов, как описано в другом месте24. - Очистите флуоресцентно меченные клетки от суспензии с помощью FACS, как описано в другом месте24.
- Гранулированные ячейки центрифугируют и отбрасывают надосадочную жидкость.
ПРИМЕЧАНИЕ: Используйте низкую скорость центрифугирования (400 × г) и температуру (4 °C), чтобы предотвратить лизис клеток. Если вы используете бычий сывороточный альбумин (BSA) для FACS, промойте клеточную гранулу, чтобы уменьшить помехи BSA во время обнаружения HRMS. Осторожно ресуспендируйте клетки в 1x фосфатном буферном физиологическом растворе (PBS) и снова центрифугируйте до клеток, промытых гранулами. Удалите надосадочную жидкость PBS. - Быстро заморозьте изолированные клетки, поместив флакон с образцом на сухой лед или жидкий азот.
ПРИМЕЧАНИЕ: Храните образцы (ткани или клетки) охлажденными (например, на льду) во время этапов обработки. Заморозьте ячейки с как можно меньшим количеством среды вокруг образца, чтобы облегчить последующую обработку. - Храните образцы при температуре -80 °C до анализа HRMS.
- Перенесите инъецированные эмбрионы в 0,5x SS в чашку Петри и культивируйте их при температуре 14-25 ° C до достижения желаемой стадии развития.
4. Проанализируйте белки с помощью масс-спектрометрии
Протеомная характеристика изолированных тканей или клеток основана на серии установленных этапов в HRMS. На рисунке 2 показаны этапы биоаналитического рабочего процесса. Используемый здесь протокол сбора образцов совместим с рабочими процессами протеомики снизувверх 11, середина вниз25 илисверху вниз 26 . Далее описывается стратегия «снизу вверх», используемая в этом исследовании, которая оказалась чувствительной, количественной и адаптируемой к различным типам масс-спектрометров. После экстракции и ферментативного переваривания белков полученные пептиды разделяют с последующим анализом HRMS.
- Обработка тканей / отдельных клеток
- Для одноклеточного анализа методом CE нагрейте образец до 60 °C в течение ~15 мин, чтобы денатурировать белки, затем уравновесьте образец до комнатной температуры (RT, ~5 мин)18,21.
ПРИМЕЧАНИЕ: В отличие от работы с тканями, этапы восстановления и алкилирования пропускаются, чтобы ограничить потери белка во время подготовки образца из отдельных клеток. Для минимизации потерь белка во время пробоподготовки могут быть применены фильтроподготовка проб (FASP)27,28, другие стратегии 29 с одним горшком и микрофлюидные подходы30. - Для анализа с помощью nanoLC лизируют до 5 рассеченных тканей в 50 мкл лизисного буфера (~ 100 мкг общего белка). Облегчите процесс, пипетируя образец вверх и вниз несколько раз.
- Инкубируют лизат при 4 ° C в течение 10 мин, затем гранулируют клеточный мусор и желточные тромбоциты центрифугированием при 4 500 × г при 4 ° C. Перенесите надосадочную жидкость в чистый флакон микроцентрифуги и добавьте 10% SDS для получения конечной концентрации 1% SDS в лизате (v/v).
- Для салфеток выполните шаги 4.1.5-4.1.7.
- Добавьте 0,5 М дитиотреитол к лизату для получения конечной концентрации ~ 25 мМ (например, 2,5 мкл 0,5 М дитиотретола до 50 мкл лизата) и инкубируйте лизат в течение 30 мин при 60 ° C для химического восстановления дисульфидных связей в белках.
- Добавьте 0,5 М йодоацетамида для получения конечной концентрации ~ 75 мМ в лизате и инкубируйте смесь в течение 15 мин при RT в темноте (рис. 2).
- Добавьте 0,5 М дитиотреитол, такой же, как исходный объем (например, 2,5 мкл 0,5 М дитиотретола на 50 мкл лизата), чтобы погасить реагенты, оставшиеся от реакции алкилирования.
ВНИМАНИЕ: Йодоацетамид и дитиотреитол могут вызвать серьезное повреждение кожи и глаз при прямом воздействии. Используйте соответствующие СИЗ при работе с этими химическими веществами. - Очищайте белки с помощью осаждения. Осаждение на основе хлороформ-метанола хорошо работает31. Этот протокол также может быть адаптирован к другим типам подходов к осадкам32.
ПРИМЕЧАНИЕ: Для анализа отдельных клеток, где потери белка вызывают беспокойство, пропустите этап осаждения для CE-HRMS. - Высушите белковый осадок в вакуумном концентраторе (4-37 °C), затем ресуспендируйте экстрагированный протеом в 50 мкл 50 мМ бикарбоната аммония. Оцените концентрацию белка с помощью колориметрического анализа общего белка, чтобы определить количество фермента, необходимого для пищеварения (например, анализ белка бицинхониновой кислоты).
- Переваривают белки до пептидов. Добавьте трипсин (1 мкг/мкл) для получения соотношения протеаза/белок 1:50 и инкубируйте смесь при 37 °C до 5 ч для одноклеточных образцов и до 14 ч для тканевых образцов. Проконсультируйтесь с рекомендациями по реагированию на реакцию для конкретных поставщиков.
ПРИМЕЧАНИЕ: Переваривание трипсином в концентрациях более 14 ч или выше может привести к расщеплению, неспецифичному для последовательности белка, что затрудняет идентификацию белка33. - Количественно определите общую концентрацию пептидов с помощью колориметрического анализа.
- ДОПОЛНИТЕЛЬНО: Для количественного определения мультиплексирования пометьте пептиды из каждого образца другой изобарической меткой массы, следуя инструкциям конкретного поставщика. Смешайте пептиды со штрих-кодом в равных пропорциях на образец пептида.
ПРИМЕЧАНИЕ: Обеспечьте точную маркировку и смешивание, чтобы избежать количественных погрешностей. Для образцов с ограниченным количеством или одноклеточных образцов может быть включен несущий канал на основе ТМТ, состоящий из объединенных тканей/клеток, чтобы свести к минимуму потери образца на последующих этапах разделения и повысить чувствительность белков с меньшим количеством34. - ДОПОЛНИТЕЛЬНО: Обессолите пептиды для удаления солей и загрязняющих веществ (например, непрореагировавших изобарических реагентов с масс-меткой) на спиновой колонке/наконечнике с обращенной фазой C18 для защиты системы LC-MS.
- ДОПОЛНИТЕЛЬНО: Фракционирование (например, фракционирование в обратной фазе со средним или высоким pH) пептидной смеси для более глубокого обнаружения протеома с помощью ручных или автоматических платформ. Используйте неподвижную фазу C18, содержащую наконечники, для фракционирования небольших количеств (1-10 мкг) пептидных расщеплений.
- Высушите пептидную смесь при 60 °C в вакуумном концентраторе.
- Храните пептидную смесь при температуре -80 °C до измерения.
- Для одноклеточного анализа методом CE нагрейте образец до 60 °C в течение ~15 мин, чтобы денатурировать белки, затем уравновесьте образец до комнатной температуры (RT, ~5 мин)18,21.
- Разделение пептидов
ПРИМЕЧАНИЕ: После экстракции и ферментативного переваривания белков полученные пептиды разделяют nanoLC или CE и ионизируют ESI для секвенирования с помощью тандемных HRMS. Разделение наноЖКХ в обращенной фазе идеально подходит для пептидов, накапливающих от ~150 нг до ~1 мкг за анализ. CE обеспечивает дополнительную чувствительность для пептидов, начиная от фемтограмм и заканчивая <100 ng. Various custom-built and commercial CE-ESI interfaces allow for ready coupling of CE to HRMS with robust performance35 и все чаще используются для анализа отдельных клеток18,36,37.- Чтобы отделить с помощью CE, выполните шаги 4.2.2-4.2.7.
ПРИМЕЧАНИЕ: Далее описано использование специально созданной платформы CE для измерения пептидов. Протоколы для создания и использования этого инструмента CE были предоставлены ранее38 вместе с визуализированным экспериментом по использованию малых молекул20. Кроме того, эти измерения могут быть выполнены на коммерческой системе CE, такой как AB SCIEX CESI, Agilent 7100 или эквивалентной. - Восстановите расщепление белка в 1-2 мкл растворителя образца, встряхните для перемешивания образца и центрифугируйте его при 10 000 x g в течение 2 мин для гранулирования остатков клеток.
ПРИМЕЧАНИЕ: Удаление клеточного мусора сводит к минимуму вероятность засорения капилляра CE, тем самым продлевая срок службы системы разделения и повышая пропускную способность измерений. - Инициализируйте прибор CE-ESI, промыв капилляр CE с помощью BGE.
- Проверка инструментальной эффективности с использованием известного стандарта (например, дигест цитохрома С или БСА, пептиды ангиотензина).
ПРИМЕЧАНИЕ: Перед измерением драгоценных образцов рекомендуется оценить прибор с точки зрения точности массы, чувствительности обнаружения, воспроизводимости и линейного динамического диапазона количественного определения. Дополнительные примечания по проверке и устранению неисправностей характеристик CE-ESI-MS перечислены в других разделах18,20,38. - Введите ~ 1-10 нл образца в капилляр разделения CE.
ПРИМЕЧАНИЕ: В этом исследовании используется капилляр плавленого кварца длиной ~ 1 м (внутренний/внешний диаметр 40/110 мкм) с электрокинетически накачанной оболочкой. Коммерческие инструменты CE обычно требуют представления 5-10 мкл образца в микрофлаконе для инъекций. Изготовленная по индивидуальному заказу платформа CE18,38 совместима с ~ 250 нл до 1 мкл образца, осажденного в микрофлакон для загрузки образца. - Перенесите входной конец разделительного капилляра CE в BGE.
- Начните электрофоретическую сепарацию, постепенно увеличивая напряжение разделения CE от земли (например, ступенчато в течение 1 мин). Потенциалы 20-28 кВ с током ниже ~10 мкА обеспечивают стабильную и воспроизводимую инструментальную производительность для анализа.
- Чтобы отделить с помощью nanoLC, выполните шаги 4.2.9-4.2.12.
- Ресуспендировать образец пептида в мобильной фазе А. Концентрация образца и его объем для впрыска зависят от имеющейся системы LC и колонки. В этом исследовании ~ 250 нг-1 мкг белкового расщепления вводят в 1-20 мкл объема образца на насадочную колонку C18 (внутренний диаметр 75 мкм, размер частиц 2 мкм с порами 100 Å, разделительная колонка длиной 25 см).
- Переложите образец во флакон LC.
ПРИМЕЧАНИЕ: Убедитесь, что во флаконе нет пузырьков воздуха, которые могут повредить аналитическую колонку. Флаконы со вставками могут быть использованы для небольших образцов тканей или отдельных клеток. - Загрузите от ~200 нг до 2 мкг образца пептида на аналитическую колонку C18.
ПРИМЕЧАНИЕ: Опционально пептиды могут быть загружены в колонну-ловушку для обессоливания перед аналитической сепарацией. Например, колонна-ловушка C18 с внутренним диаметром 0,1 мм, размером частиц 5 мкм, размером пор 100 Å, длиной 20 мм. Обессолите пептиды 100% буфером А со скоростью потока 5 мкл / мин в течение 5 минут до начала градиента разделения. - Разделите пептиды с помощью градиентного элюирования. При скорости потока 300 нл / мин 120-минутный градиент, используемый в этом исследовании, выглядит следующим образом: 0-5 мин 2% B, 5-85 мин 2-35% B, 86-90 мин 70% B, 91-120 мин 2% B.
- Чтобы отделить с помощью CE, выполните шаги 4.2.2-4.2.7.
- Ионизируйте пептиды с помощью ESI
ПРИМЕЧАНИЕ: Капилляр CE или nanoLC чаще всего соединен с источником ESI для ионизации. Ранее были разработаны микропроточные (тупые наконечники) и нанопотоки (конические наконечники39 и электрокинетическая накачка с оболочкой36 ) интерфейсы CE-ESI для сверхчувствительного обнаружения.- Подайте разделительные пептиды в источник ионов электрораспыления для ионизации с помощью коммерческого или специально разработанного интерфейса ESI. Для одноклеточного анализа CE-ESI-MS у эмбрионов Xenopus используйте электрокинетически накачанный интерфейс с низким потоком, в котором выходное отверстие капилляра CE заключено в вытянутый боросиликатный излучатель.
- Проверьте поток жидкости через электрораспылитель с помощью камеры и визуально осмотрите установку на предмет возможных утечек.
- Установите напряжение электрораспыления на ~ 2.5 кВ, чтобы запустить источник ESI (по сравнению с землей).
- Обеспечьте стабильный наноспрей для анализа HRMS, контролируя общий ионный ток. Отрегулируйте напряжение электрораспыления и расстояние от излучателя до входа HRMS для достижения стабильного распыления (относительное стандартное отклонение общей интенсивности <15%).
- Обнаружение пептидов
ПРИМЕЧАНИЕ: Обнаружение пептидов следует различным инструментальным соображениям для изобарических масс-меченных и немеченых пептидов и зависит от типа доступного масс-спектрометра. В этом исследовании используется масс-спектрометр orbitrap tribrid в соответствии со следующими шагами.- Получение событий MS1 с настройками: Analyzer, orbitrap; Спектральное разрешение, полная ширина 120 000 при половинном максимуме (FWHM); максимальное время впрыска (IT), 50 мс; автоматическая регулировка усиления (АРУ), 4 x 105 отсчетов; микросканирование, 1.
- Для секвенирования пептидов фрагментировать ионы-предшественники для детектирования в анализаторе ионных ловушек с помощью настроек: режим фрагментации, диссоциация столкновения с более высокой энергией (HCD); ударный газ, азот; энергия столкновения, 32% нормированная энергия столкновения (NCE); максимальное ИТ, 70 мс; АРУ, 1 x 104 счета; микросканирование, 1.
- ДОПОЛНИТЕЛЬНО: Количественное определение пептидов, меченных ТМТ, с использованием тандемной/многоступенчатой HRMS (MS2 / MS3). Для MS3 , использующего синхронный выбор предшественника, типичные инструментальные настройки следующие. Одноступенчатое (MS1) сканирование, исследующее наиболее распространенные ионы, диссоциирует с помощью данных-зависимого сбора с использованием параметров: режим фрагментации MS2 , диссоциация, вызванная столкновением (CID); ударный газ, гелий; энергия столкновения, 35% NCE; анализатор осколочных ионов, ионных ловушек со следующими настройками: максимальный IT, 50 мс; АРУ, 5 х 104 счета; микросканирование, 1. Выберите 10 мс2 фрагментированных ионов и фрагментируйте их с HCD в азоте (65% NCE). Обнаружение ионов фрагментов MS3 с помощью следующих настроек: разрешение Orbitrap 15 000 FWHM, максимальное IT, 120 мс; СМЖЛ, 1 × 105 счетов; микросканирование, 1.
ПРИМЕЧАНИЕ: Для помеченных образцов можно использовать различные методы и параметры сбора данных MS в соответствии с рекомендациями поставщиков, как описано в другом месте11,40.
- Анализ данных
ПРИМЕЧАНИЕ: Белки идентифицируются и количественно оцениваются с использованием передовых биоинформационных пакетов. Точность идентификации рассчитывается с использованием базы данных-приманок, выраженной как частота ложных открытий (FDR) на уровне пептидов и белков.- Обрабатывать данные с помощью коммерческих пакетов программного обеспечения или пакетов программного обеспечения с открытым исходным кодом (см. ссылку41). Сопоставьте необработанные данные с базой данных, которая была подготовлена путем объединения протеома Xenopus 9.2 с базойданных 42 PHROG, полученной из мРНК.
ПРИМЕЧАНИЕ: Параметры поиска: фермент пищеварения, трипсин; пропущенные спайности, до 2; вариабельная модификация, окисление метионина; статическая модификация, цистеинкарбамидометилирование; допуск по массе прекурсора, 10 ppm; допуск по массе фрагмента, 0,6 Да; минимальная длина пептида, 5; точность идентификации, <1% FDR для пептидов и белков. Без алкилирования до пептидов карбамидометилирование как статическая модификация исключается при поиске в базе данных (например, для одноклеточного анализа). - Количественно оценивайте содержание белка с помощью стратегий без меток43 или на основе меток44,45.
- ДОПОЛНИТЕЛЬНО: Аннотирование белков для онтологии генов. Можно использовать PantherDB46, Reactome47 или Xenbase23 .
- НЕОБЯЗАТЕЛЬНО: Количественная оценка содержания белка и различий в количестве белка между типами клеток / тканей с помощью программных пакетов / веб-инструментов, таких как Trans-Proteomic Pipeline48, Perseus49 и Orange50.
ПРИМЕЧАНИЕ: Дополнительные соображения по экспериментальному дизайну и вариантам программного обеспечения были рассмотрены в другом месте41,51. - НЕОБЯЗАТЕЛЬНО: Дальнейшая оценка результатов с использованием баз знаний, таких как STRING52 и BioPlex Display 53 для известных белок-белковых взаимодействий и PhosphoSiteplus54 для фосфорилирования. Для анализа мотивов и областей, представленных в протеоме, используйте веб-инструменты, такие как Simple Modular Architecture Research (SMART)55.
- Обрабатывать данные с помощью коммерческих пакетов программного обеспечения или пакетов программного обеспечения с открытым исходным кодом (см. ссылку41). Сопоставьте необработанные данные с базой данных, которая была подготовлена путем объединения протеома Xenopus 9.2 с базойданных 42 PHROG, полученной из мРНК.
Access restricted. Please log in or start a trial to view this content.
Результаты
Этот протокол позволил изучить белки в отдельных клетках и их линии, поскольку они создают ткани в эмбрионах X. laevis. Рисунок 1 иллюстрирует одно из таких применений подхода к изучению белков в клетках нервной ткани и недавно индуцированной нервной эктодермы у эмбрио...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Этот протокол позволяет охарактеризовать экспрессию белка в идентифицированных клеточных линиях эмбрионов вида Xenopus . Методология, основанная на HRMS, сочетает в себе исключительную специфичность в молекулярной идентификации, способность к обнаружению нескольких белков без молек...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторы заявляют об отсутствии конкурирующих интересов.
Благодарности
Мы благодарны Цзе Ли (Университет Мэриленда, Колледж-Парк) за ценные дискуссии об эмбриональной диссоциации и FACS. Мы благодарим Ви М. Куача и Камиллу Ломбард-Банек за помощь в подготовке образцов и сборе данных в предыдущих исследованиях, иллюстрирующих протеомные приложения, выделенные в этом протоколе. Часть этой работы была поддержана Национальным научным фондом под номером награды IOS-1832968 CAREER (для P.N.), Национальными институтами здравоохранения под номером R35GM124755 (для P.N.), Партнерской программой Университета Мэриленда и Национального института рака (для P.N.) и исследовательскими наградами Фонда клуба COSMOS (для ABB и L.R.P.).
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| Acetonitrile (LC-MS-grade) | Fisher Scientific | A955 | |
| Agarose | ThermoFisher Scientific | R0492 | |
| Ammonium bicarbonate | Fisher Scientific | A643-500 | |
| Analytical Column | Thermo Scientific | 164941 | |
| Analytical microbalance | Mettler-Toledo | XSE105DU | |
| Automatic peptide fractionation platform | Agilent | 1260 Infinity II | |
| Borosilicate Capillaries | Sutter Instruments Co. | B100-50-10 | |
| Borosilicate Capillaries (for making Emmitters) | Sutter Instruments | B100-75-10 | |
| C18 spin columns (for desalting) | ThermoFisher Scientific | 89870 | |
| Camera ro monitor electrospray | Edmund Optics Inc. | EO-2018C | |
| Combretastatin A4 | Millipore Sigma | C7744 | |
| Commercial CESI system | AB SCIEX | CESI | |
| (Cyclohexylamino)-1-propanesulfonic acid (CAPS) | VWR | 97061-492 | |
| Cytochalasin D | Millipore Sigma | C8273 | |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable | ThermoFisher Scientific | D22910 | |
| Diothiothreitol | Fisher Scientific | FERR0861 | |
| Dumont #5 Forceps | Fine Science Tools | 11252-30 | |
| EDTA | Fisher Scientific | AAJ62786AP | |
| Epifluorescence light source | Lumencore | AURA III | |
| Eppendorf LoBing microcentrifuge tubes: protein | Fisher Scientific | 13-698-793 | |
| Formic acid (LC-MS-grade) | Fisher Scientific | A117-50 | |
| Freezer (-20 °C) | Fisher Scientific | 97-926-1 | |
| Freezer (-80 °C) | Thermo Scientific | TSX40086A | |
| Fused silica capillary | Molex | 1088150596 | |
| Heat Block | Benchmark | BSH300 | |
| High pressure liquid Chromatography System | ThermoFisher Scientific | Dionex Ultimate 3000 RSLC nanosystem | |
| High voltage power supply | Spellman | CZE1000R | |
| High-resolution Mass Spectrometer | ThermoFisher Scientific | Orbitrap Fusion Lumos Tribrid Mass Spectrometer | |
| HPLC caps | Thermo Scientific | C4013-40A | |
| HPLC Vials | Thermo Scientific | C4013-11 | |
| Illuminator e.g. Goosenecks | Nikon | C-FLED2 | |
| Ingenuity Pathway Analysis | Qiagen | ||
| Iodoacetamide | Fisher Scientific | AC122275000 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456-4 | |
| Microcapillary puller | Suttor Instruments | P-2000 | |
| Microinjector | Warner Instrument, Handem, CT | PLI-100A | |
| Micropippette puller | Sutter Instruments Co. | P-1000 | |
| MS data analysis software, commercial | ProteomeDiscoverer | ||
| MS data analysis software, opensource | MaxQuant | ||
| non-idet 40 substitute | Millipore Sigma | 11754599001 | |
| Petri dish 60 mm and 80 mm | Fisher Scientific | S08184 | |
| Pierce 10 µL bed Zip-tips (for desalting) | ThermoFisher Scientific | 87782 | |
| Pierce bicinchoninic acid protein assay kit | ThermoFisher Scientific | 23225 | |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 | |
| Pierce Trypsin Protease (MS Grade) | Fisher Scientific | PI90058 | |
| Protein LoBind vials | Eppendorf | 0030108434 , 0030108442 | |
| Refrigerated Centrifuge | Eppendorf | 5430R | |
| Refrigerated Incubator | Thermo Scientific | PR505755R/3721 | |
| sodium isethionate | Millipore Sigma | 220078 | |
| sodium pyrophosphate | Sigma Aldrich | 221368-100G | |
| Stainless steel BGE vial | Custom-Built | ||
| Stainless steel sample vials | Custom-Built | ||
| Stereomicroscope (objective 10x) | Nikon | SMZ 1270, SZX18 | |
| Sucrose | VWR | 97063-790 | |
| Syringe pumps (2) | Harvard Apparatus | 704506 | |
| Syringes (gas-tight): 500–1000 µL | Hamilton | 1750TTL | |
| Transfer pipettes (Plastic, disposable) | Fisher Scientific | 13-711-7M | |
| Trap Column | Thermo Scientific | 164750 | |
| Tris-HCl (1 M solution) | Fisher Scientific | AAJ22638AP | |
| Vacuum concentrator capable of operation at 4–10 °C | Labconco | 7310022 | |
| Vortex-mixer | Benchmark | BS-VM-1000 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| XYZ translation stage | Thorlabs | PT3 | |
| XYZ translation stage | Custom-Built |
Ссылки
- Shoemaker, L. D., Kornblum, H. I. Neural Stem Cells (NSCs) and Proteomics. Molecular & Cellular Proteomics. 15 (2), 344-354 (2016).
- Cervenka, J., et al. Proteomic characterization of human neural stem cells and their secretome during in vitro differentiation. Frontiers in Cellular Neuroscience. 14, 612560(2021).
- Christian, J. L. Morphogen gradients in development: From form to function. Wiley Interdisciplinary Reviews. Developmental Biology. 1 (1), 3-15 (2012).
- Gurdon, J. B., Elsdale, T. R., M, F. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 182, 64-65 (1958).
- Harland, R. M., Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 27 (12), 507-515 (2011).
- Moody, S. A. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 119 (2), 560-578 (1987).
- Moody, S. A. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Developmental Biology. 122 (2), 300-319 (1987).
- Dale, L., Slack, J. M. W. Fate map for the 32-cell stage of Xenopus laevis. Development. 99 (4), 527-551 (1987).
- Sun, L. L., et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Analytical Chemistry. 88 (13), 6653-6657 (2016).
- Lombard-Banek, C., Moody, S. A., Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angewandte Chemie-International Edition. 55 (7), 2454-2458 (2016).
- Zhang, Y. Y., Fonslow, B. R., Shan, B., Baek, M. C., Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113 (4), 2343-2394 (2013).
- Sive, H. L., Grainger, R. M., Harland, R. M. Early development of Xenopus laevis: A laboratory manual. , Cold Spring Harbor Laboratory Press. New York. (2000).
- Briggs, J. A., et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 360 (6392), (2018).
- Gupta, M., Sonnett, M., Ryazanova, L., Presler, M., Wuhr, M. Quantitative proteomics of xenopus embryos I, sample preparation. Xenopus. Methods in Molecular Biology. Vleminckx, K. 1865, Humana Press Inc. NY. 175-194 (2018).
- Baxi, A. B., Lombard-Banek, C., Moody, S. A., Nemes, P. Proteomic characterization of the neural ectoderm fated cell clones in the Xenopus laevis embryo by high-resolution mass spectrometry. ACS Chemical Neuroscience. 9 (8), 2064-2073 (2018).
- Moody, S. A. Cell lineage analysis in Xenopus embryos. Methods in Molecular Biology. 135, 331-347 (2000).
- Sater, A. K., Moody, S. A. Using Xenopus to understand human diseases and developmental disorders. Genesis. 55 (1-2), 1-14 (2017).
- Lombard-Banek, C., Choi, S. B., Nemes, P. Enzyme Activity in Single Cells. Methods in Enzymology. Allbritton, N. L., Kovarik, M. L. 628, 263-292 (2019).
- Lombard-Banek, C., Moody, S. A., Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Frontiers in Cell and Developmental Biology. 4, 11(2016).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. Microprobe capillary electrophoresis mass spectrometry for single-cell metabolomics in live frog (Xenopus laevis) embryos. Journal of Visualized Experiments: JoVE. (130), e56956(2017).
- Lombard-Banek, C., Moody, S. A., Manzin, M. C., Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and zebrafish embryos. Analytical Chemistry. 91 (7), 4797-4805 (2019).
- Klein, S. L. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Developmental Biology. 120 (1), 299-304 (1987).
- Karimi, K., et al. Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Research. 46 (1), 861-868 (2018).
- Kakebeen, A. D., Chitsazan, A. D., Wills, A. E. Tissue disaggregation and isolation of specific cell types from transgenic Xenopus appendages for transcriptional analysis by FACS. Developmental Dynamics. 250 (9), 1381-1392 (2021).
- Garcia, B. A. What does the future hold for top down mass spectrometry. Journal of the American Society for Mass Spectrometry. 21 (2), 193-202 (2010).
- Toby, T. K., Fornelli, L., Kelleher, N. L. Progress in top-down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry. (Palo Alto Calif). 9 (1), 499-519 (2016).
- Zhang, Z. B., Dubiak, K. M., Huber, P. W., Dovichi, N. J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis Embryos. Analytical Chemistry. 92 (7), 5554-5560 (2020).
- Wisniewski, J. R. Microbial Proteomics: Methods and Protocols.Methods in Molecular Biology. Becher, D. 1841, Humana Press Inc. NY. 3-10 (2018).
- Hughes, C. S., et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Zhu, Y., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nature Communications. 9, 882(2018).
- Wessel, D., Flugge, U. I. A method for the quantitative recovery of protein in dilute-solution in the presence of detergents and lipids. Analytical Biochemistry. 138 (1), 141-143 (1984).
- Jiang, L., He, L., Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of Chromatography A. 1023 (2), 317-320 (2004).
- Hildonen, S., Halvorsen, T. G., Reubsaet, L. Why less is more when generating tryptic peptides in bottom-up proteomics. Proteomics. 14 (17-18), 2031-2041 (2014).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19, 161(2018).
- Drouin, N., et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: effective electrophoretic mobility for reproducible and robust compound annotation. Analytical Chemistry. 92 (20), 14103-14112 (2020).
- Sun, L. L., Zhu, G. J., Zhang, Z. B., Mou, S., Dovichi, N. J. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. Journal of Proteome Research. 14 (5), 2312-2321 (2015).
- DeLaney, K., Sauer, C. S., Vu, N. Q., Li, L. J. Recent advances and new perspectives in capillary electrophoresis-mass spectrometry for single cell "omics". Molecules. 24 (1), 21(2019).
- Nemes, P., Rubakhin, S. S., Aerts, J. T., Sweedler, J. V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nature Protocols. 8 (4), 783-799 (2013).
- Choi, S. B., Zamarbide, M., Manzini, M. C., Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. Journal of the American Society for Mass Spectrometry. 28 (4), 597-607 (2017).
- Pino, L. K., Rose, J., O'Broin, A., Shah, S., Schilling, B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions. 48 (5), 1953-1966 (2020).
- Chen, C., Hou, J., Tanner, J. J., Cheng, J. L. Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences. 21 (8), 25(2020).
- Peshkin, L., et al. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Developmental Cell. 35 (3), 383-394 (2015).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 13 (9), 2513-2526 (2014).
- Gygi, S. P., et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 17 (10), 994-999 (1999).
- Thompson, A., et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 75 (8), 1895-1904 (2003).
- Mi, H. Y., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive api. Nucleic Acids Research. 49, 394-403 (2021).
- Schmidt, E., et al. On the Move Federated Workshops. , Springer, Verlag. Berlin. 710-719 (2006).
- Deutsch, E. W., et al. Trans-Proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 9 (7-8), 745-754 (2015).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Demsar, J., et al. Orange: Data mining toolbox in Python. Journal of Machine Learning Research. 14, 2349-2353 (2013).
- Oberg, A. L., Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. Journal of Proteome Research. 8 (5), 2144-2156 (2009).
- Jensen, L. J., et al. STRING 8 - a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, 412-416 (2009).
- Schweppe, D. K., Huttlin, E. L., Harper, J. W., Gygi, S. P. BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data. Journal of Proteome Research. 17 (1), 722-726 (2018).
- Hornbeck, P. V., et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research. 43, 512-520 (2015).
- Letunic, I., Khedkar, S., Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Research. 49, 458-460 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie-International Edition. 60 (23), 12852-12858 (2021).
- Lombard-Banek, C., Reddy, S., Moody, S. A., Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Molecular & Cellular Proteomics. 15 (8), 2756-2768 (2016).
- Saha-Shah, A., et al. Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Analytical Chemistry. 91 (14), 8891-8899 (2019).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Analytical Chemistry. 89 (13), 7069-7076 (2017).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47, 442-450 (2019).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены