
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Cell-Lineage Guided Mass Spectrometry Proteomics in the Developing (Frog) Embryo
In This Article
Summary
Here we describe a mass spectrometry-based proteomic characterization of cell lineages with known tissue fates in the vertebrate Xenopus laevis embryo.
Abstract
Characterization of molecular events as cells give rise to tissues and organs raises a potential to better understand normal development and design efficient remedies for diseases. Technologies enabling accurate identification and quantification of diverse types and large numbers of proteins would provide still missing information on molecular mechanisms orchestrating tissue and organism development in space and time. Here, we present a mass spectrometry-based protocol that enables the measurement of thousands of proteins in identified cell lineages in Xenopus laevis (frog) embryos. The approach builds on reproducible cell-fate maps and established methods to identify, fluorescently label, track, and sample cells and their progeny (clones) from this model of vertebrate development. After collecting cellular contents using microsampling or isolating cells by dissection or fluorescence-activated cell sorting, proteins are extracted and processed for bottom-up proteomic analysis. Liquid chromatography and capillary electrophoresis are used to provide scalable separation for protein detection and quantification with high-resolution mass spectrometry (HRMS). Representative examples are provided for the proteomic characterization of neural-tissue fated cells. Cell-lineage-guided HRMS proteomics is adaptable to different tissues and organisms. It is sufficiently sensitive, specific, and quantitative to peer into the spatio-temporal dynamics of the proteome during vertebrate development.
Introduction
Our understanding of cell differentiation and the genesis of tissues and organs is the result of decades of elaborate targeted screens of genes and their products. Increasing our knowledge of all the biomolecules and their quantities during important cellular events would help unravel molecular mechanisms that control the spatial and temporal patterning of the vertebrate body plan. Technologies enabling molecular amplification and sequencing are now able to routinely report on large numbers of genes and transcripts, supporting hypothesis-driven studies in basic biological and translational research. To understand developing systems, a complex relationship between transcription and translation advocates for direct analysis of multiple proteins and their post-translational modifications. Global proteomics using in vitro biological systems, such as induced pluripotent stem cells, began to delineate mechanisms of tissue induction1,2. In complex organisms, such as the vertebrate embryo, development relies on morphogen gradients in the context of space and time3. It follows that gaining knowledge of proteomic changes as cells differentiate to form specialized tissues, such as neural tissues, offers a key to unlock molecular programs controlling normal and defective development and guide next-generation therapeutics.
The vertebrate South African clawed frog (Xenopus laevis) is a well-established model in cell and developmental, neuro-, and regenerative biology. Sir John Gurdon's 2012 Nobel Prize in Physiology or Medicine4,5 for the discovery of pluripotency of the somatic nucleus highlighted the importance of this model for discoveries in basic and translational studies. Xenopus embryos develop externally to the mother, thus facilitating direct manipulation of cells, cell clones, and gene expression over various stages of development. Asymmetrical pigmentation and stereotypical cell divisions enabled the charting of reproducible fate maps from the 16-6 and 32-cell7,8 stage embryo. For high-resolution mass spectrometry (HRMS) based proteomics, additional advantages of the model include relatively large size (~1 mm in diameter), which yields abundant protein content for analysis (~130 µg in early cleavage-stage embryos, ~10 µg of protein content in single cells of the 16-cell embryo)9,10.
At present, HRMS is the leading technology of choice for detecting proteins. This technology enables direct, sensitive, and specific detection and quantification of multiple, usually hundreds-to-thousands of different proteins11. Bottom-up proteomics by HRMS involves a series of interconnected steps. Following extraction from the cell/tissue sample, proteins are digested with a proteolytic enzyme, such as trypsin (bottom-up proteomics). The resulting peptides are separated based on their different physicochemical properties, including hydrophobicity (reversed-phase liquid chromatography, LC), net charge (ion-exchange chromatography), size (size exclusion chromatography), or electrophoretic mobility (capillary electrophoresis, CE). Peptides are then charged (ionized), typically using electrospray ionization (ESI), and peptide ions are detected and sequenced via gas-phase fragmentation by tandem HRMS. The resulting peptide data are mapped to the proteome of the organism being studied. With protein-specific (proteotypic) peptide ion signal intensity correlating with concentration, protein quantification can be performed label-free or label-based (multiplexing quantitation). HRMS proteomics yields a rich resource of information on the molecular state of the system under study, allowing for the generation of hypotheses and follow-up functional studies.

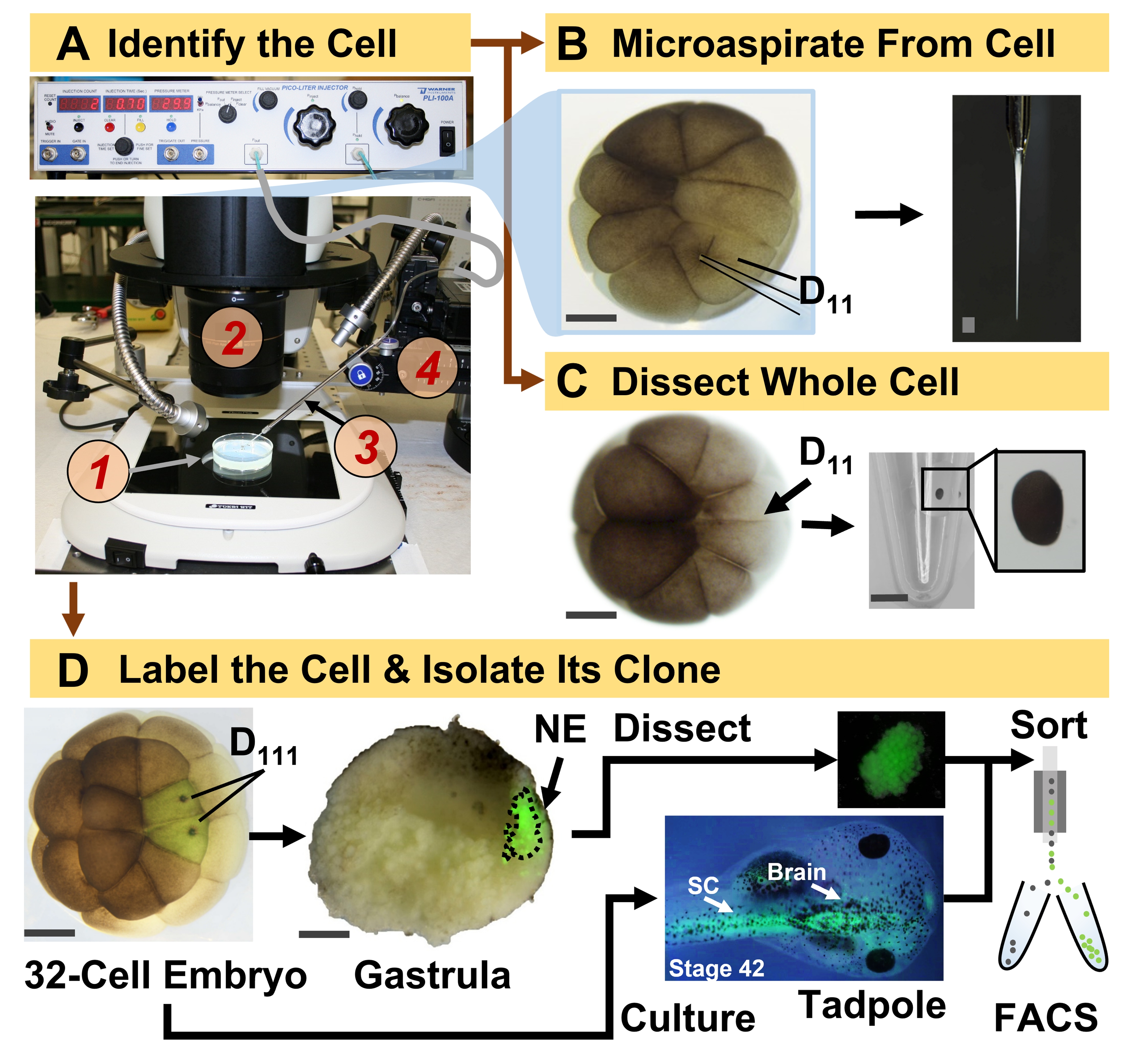
Figure 1: Spatiotemporally scalable proteomics enabling cell-lineage guided HRMS proteomics in the developing (frog) embryo. (A) Visualization of the specimen (1) using a stereomicroscope (2) for injection of an identified cell (inset), using a fabricated micropipette (3) under control by a translation-stage (4). (B) Subcellular sampling of the identified left D11 cell in a 16-cell embryo. (C) Dissection of a whole D11 cell from a 16-cell embryo. (D) Fluorescent (green) tracing of the left and right D111 progenies from a 32-cell embryo to guide dissection of the neural ectoderm (NE) in the gastrula (stage 10) and isolation of the descendent tissue from the tadpole using FACS. Scale bars: 200 µm for embryos, 1.25 mm for the vial. Figures were adapted with permission from references15,19,21,59. Please click here to view a larger version of this figure.
{kind=link}
The protocol presented here enables HRMS-based quantification of large numbers of proteins in identified cells/tissues in developing X. laevis embryos. The approach builds on accurate cell identification, reproducible cell fate maps, and established methodologies to track cell lineages in this biological model6,7,8. As shown in Figure 1, we study proteomes from single cells by employing whole-cell dissection or capillary microsampling to aspirate cellular content. Monitoring the lineage of a cell permits us to study the spatiotemporal evolution of the proteome as cells form tissues during gastrulation. The cell progeny is fluorescently marked by injecting a fluorophore conjugated to inert dextran or mRNA for fluorescent protein (e.g., green fluorescent protein, or GFP). The labeled progeny is isolated at desired developmental time points. During gastrulation, cell clones that are tightly clustered may be isolated by dissection. After gastrulation, cell clones may be distributed within the embryo owing to migratory movements and can be isolated from dissociated tissues by fluorescence-activated cell sorting (FACS). Proteins in these cells and tissues are measured via bottom-up proteomics employing HPLC or CE for separation and ESI tandem HRMS for identification. Cell-lineage-guided HRMS proteomics is scalable to different cell sizes and lineages within the embryo and is specific, sensitive, and quantitative. Through select examples shown here, we also demonstrate this protocol to be scalable and broadly adaptable to different types of cells and cell lineages.

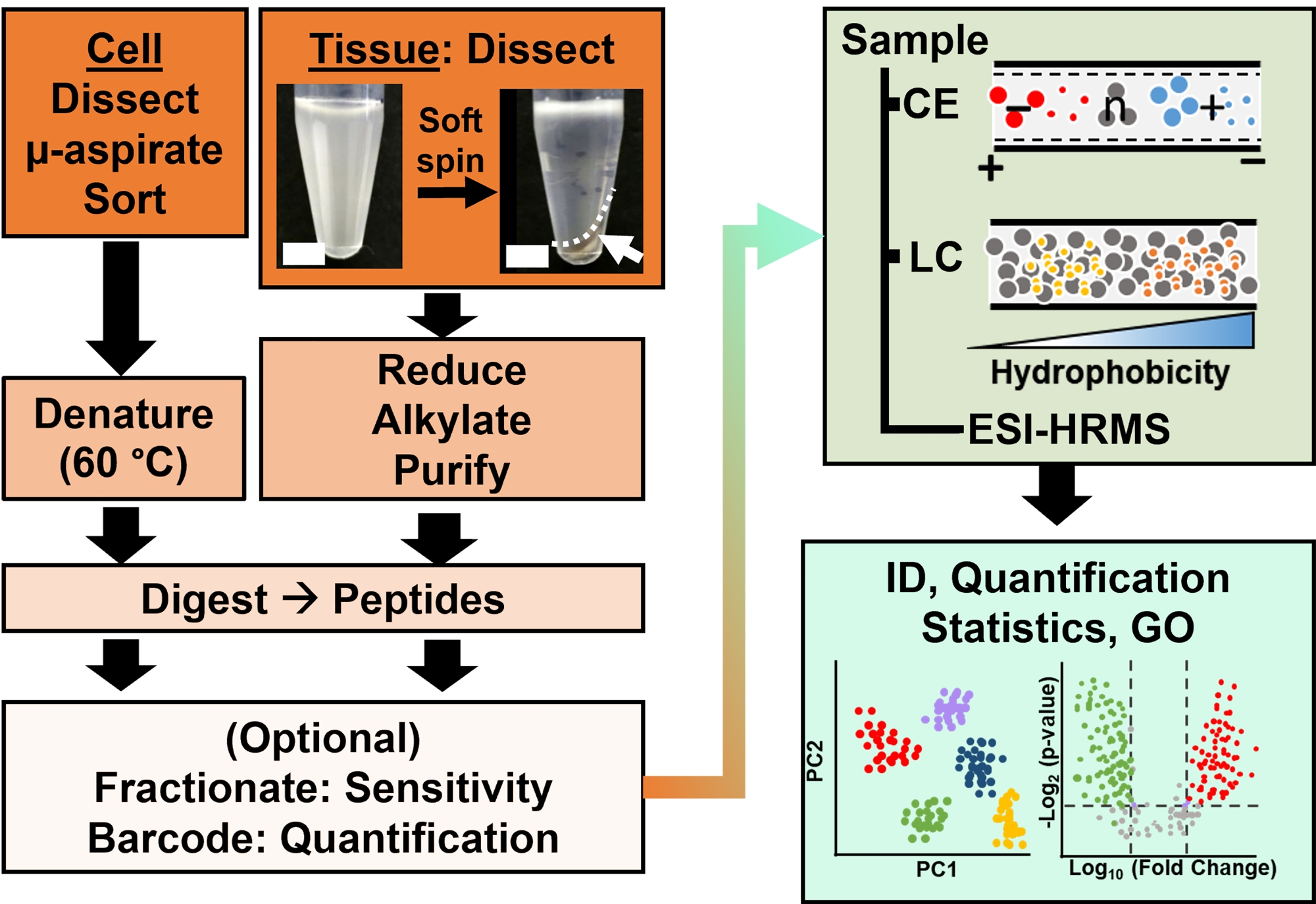
Figure 2: The bioanalytical workflow. Micro-dissection and capillary aspiration, or FACS facilitated sampling of cellular and clonal protein content. Depletion of abundant yolk proteins and separation by capillary electrophoresis (CE) or nano-flow liquid chromatography (LC) enhanced identification (ID) sensitivity using electrospray ionization (ESI) high-resolution mass spectrometry (HRMS). Quantification revealed dysregulation, supplying new information for hypothesis-driven studies in conjunction with information available from gene ontology (GO). Figures were adapted with permission from reference15. Please click here to view a larger version of this figure.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocol
All protocols ensuring the humane maintenance and handling of Xenopus laevis adult frogs were approved by the Institutional Animal Care and Use Committee at the University of Maryland, College Park (Approval numbers R-DEC-17-57 and R-FEB-21-07).
1. Prepare the solutions
- For embryology
- Prepare 1x, 0.5x, and 0.2x Steinberg's solution (SS), then autoclave them (120 °C for 20 min) to sterility following standard protocols12.
- Prepare 3% (w/v) Ficoll in sterilized 1x SS following standard protocols12.
- For dejellying, freshly prepare 2% (w/v) cysteine solution and adjust its pH to 8 by adding 10 M sodium hydroxide solution dropwise.
CAUTION: Exposure to cysteine can cause skin and respiratory damage. Sodium hydroxide is a corrosive that can cause serious damage to skin and eyes upon direct exposure. Use appropriate personal protective equipment (PPE) when handling these chemicals, such as gloves and a laboratory coat. - For the lineage tracer, prepare 0.5% (v/v) of a fluorescent dextran in sterile deionized water. Alternatively, prepare a solution of 0.2 µg/µL of mRNA for fluorescent proteins in sterile deionized water (e.g., GFP).
- To dissociate cells, prepare Newport 2.0 buffer containing 0.1 M sodium isethionate, 20 mM sodium pyrophosphate, and 10 mM CAPS, then bring its pH to 10.513.
CAUTION: Exposure to sodium pyrophosphate can cause skin and eye irritation. Use appropriate PPE when handling these chemicals.
- For bottom-up proteomics
- Prepare the cell lysis buffer to include: 250 mM sucrose, 1% nonidet P-40 substitute (w/v), 20 mM Tris-HCl, 5 mM EDTA, 10 µM cytochalasin D, and 10 µM combretastatin 4A. Prepare a stock of 10% (w/v) sodium dodecyl sulfate14,15.
NOTE: Tris-HCl was chosen to minimize HEPES contamination during nano-flow LC (nanoLC)-HRMS.
CAUTION: Exposure to nonidet P-40 substitute can cause skin irritation. Cytochalasin D is teratogenic if consumed and combretastatin is acutely toxic upon direct exposure. Use appropriate PPE when handling these chemicals. - To separate peptides by CE, prepare the following solvents (v/v): Sample solvent, 75% acetonitrile (ACN) containing 0.05% acetic acid (AcOH) in water; sheath solution, 10% ACN containing 0.05% AcOH in water; background electrolyte (BGE), 25% ACN containing 1 M formic acid (FA) in water.
CAUTION: AcOH and FA are toxic when inhaled or consumed and can cause serious skin and eye damage upon direct exposure. Use appropriate PPE when handling these chemicals. - To separate peptides by reversed-phase nanoLC, prepare (v/v): Mobile phase A (aqueous), water containing 0.1% FA; mobile phase B (organic), 0.1% FA in ACN.
NOTE: All mixtures should be prepared using LC-MS grade solvents to minimize chemical interferences during HRMS detection.
- Prepare the cell lysis buffer to include: 250 mM sucrose, 1% nonidet P-40 substitute (w/v), 20 mM Tris-HCl, 5 mM EDTA, 10 µM cytochalasin D, and 10 µM combretastatin 4A. Prepare a stock of 10% (w/v) sodium dodecyl sulfate14,15.
2. Prepare the tools for microinjection and dissection
- To gently move and orient embryos, make hair loops by fixing clean hair into a Pasteur pipette as described elsewhere16.
- For microinjection, fabricate needles by pulling borosilicate capillaries (1 mm/500 µm outer/inner diameter) using a pipette puller as described elsewhere16.
NOTE: Here, a P-1000 Pipette Puller with the following settings was used for fabricating needles: heat, 495; pull, 30, velocity, 60; time, 150; pressure, 200. - With observation under a stereomicroscope, cut the tip of the capillary using a pair of sharp forceps to essentially fabricate the capillary into a (micro)needle (e.g., Dumont #5)16.
CAUTION: Pulled capillaries are very sharp and should be handled with care.
NOTE: The tip of the needle should be sharp enough (outer diameter 10-15 µm) to be able to pierce the cell with minimal damage to the cell membrane so that the intracellular content does not leak out and the cell can heal and continue to be viable. - To hold embryos during microinjection, prepare wells in a clay-filled dish. In a 15 mm Petri dish, imprint ~1 mm diameter x ~0.5 mm deep wells into non-toxic plasticine clay as described elsewhere16.
- For microdissection, prepare agarose-coated dishes. Make 2% agarose in 1x SS and autoclave it to sterilize the solution (120 °C for 20 min). Fill 60 mm Petri dishes halfway, and let the plates solidify. Make ~1 mm diameter x ~0.5 mm deep wells using a balled Pasteur pipette tool as described previously16.
3. Isolate the cell lineage
NOTE: The following steps are performed to isolate identified single cells and/or their descendent cell lineages. Usually, the embryo is cultured to the 16- or 32-cell stage, where the tissue fates of each cell are reproducibly mapped6,7,17. The embryonic cells are identified based on morphology, location, and in reference to their fate maps. For single-cell analysis, identified cells are isolated by manual dissection, or their intracellular contents are collected into a capillary pipette and deposited in 5 μL of 0.5 mM ammonium bicarbonate. The resulting sample is stored at -80 °C until analysis (Figure 1)18,19,20,21. For cell lineage analysis, identified cells are injected with a lineage tracer, and their subsequent clones are isolated at key stages of development (e.g., during gastrulation to study tissue induction, following neurulation to study tissue commitment). In what follows, steps are outlined to fluorescently label the lineage of identified cells for isolation by dissection or FACS.
- Culture the embryos
- Obtain embryos via natural mating or in vitro fertilization (IVF) following established protocols12.
NOTE: Natural mating is logistically simpler, spares the adult male frogs, and yields embryos at different developmental stages, whereas IVF provides developmentally synchronized embryos for experiments that require accurate staging. - Dejelly the embryos. Remove the jelly coat surrounding the embryos via treatment with the dejellying solution as described elsewhere12,16.
NOTE: Microinjections and dissection require access to the cells and tissues, necessitating dejellying in X. laevis embryos. - Select 2-cell embryos with stereotypical pigmentation16,22.
NOTE: This step is important to ensure accuracy and reproducibility in the identification of the cell and its lineage. - Culture embryos to the desired developmental stage. Transfer the dejellied embryos to a Petri dish containing 1x SS and incubate them between 14-25 °C to control the speed of development.
NOTE: The temperature dependence of development is reproducible and charted for X. laevis, available on Xenbase23 (www.xenbase.org). Culturing batches of embryos at different temperatures allows for staggering developmental stages. Doing so helps distribute the number of embryos available at a given time for experimentation. - Monitor the cleavage pattern of embryos and select embryos with stereotypical pigmentation and cleavage patterns for microinjection16.
NOTE: When selecting 16- and 32-cell embryos, ensure that the cell cleavages are symmetrical for reproducible lineage tracing.
- Obtain embryos via natural mating or in vitro fertilization (IVF) following established protocols12.
- Label the cell(s) of interest
- Set up the injection needle containing the lineage tracer solution. Mount the microinjection needle into a micropipette holder controlled by a multi-axis micromanipulator.
- Connect the micropipette holder to a microinjector. Fill the needle with the lineage tracer by applying negative pressure as described elsewhere16. Figure 1A exemplifies the setup.
- Calibrate the needle. Adjust the size of the needle tip and injection time to deliver ~1 nL of the lineage tracer solution, measured in (mineral) oil following a protocol available elsewhere16.
NOTE: Capillaries with a wider tip tend to damage the cell membrane, causing subcellular contents and the injected lineage tracer to leak out, whereas capillaries with smaller tips are prone to clogging. Capillaries with ~10 µm tip outer diameter are ideal, requiring a 40 psi pressure pulse over ~300 ms to deliver ~1 nL. - Flood the microinjection clay dish with the 3% Ficoll solution and transfer ~10 embryos to the clay dish using a transfer pipette. Use a hair loop to guide each embryo into a well and gently position them so that the targeted cell of interest is at a right angle to the microneedle.
- Identify the precursor cell of the lineage of interest following the X. laevis tissue fate maps. For example, Figure 1 demonstrates the labeling of neural ectodermal clones based on the injection of its precursor cells in 32-cell embryos (left and right D111 cells).
NOTE: Detailed fate maps for the 16-6 and 32-cell7,8 embryos are available in an interactive platform via Xenbase23. It is important to ensure stereotypical pigmentation and cleavages on embryos when using them for lineage-tracing experiments. - Inject the cell(s) of interest with ~1 nL of the fluorescent dextran or ~200 pg of mRNA as described earlier16.
NOTE: Use dextran conjugates that are 10,000-40,000 MW. Smaller dextran conjugates could pass through gap junctions, whereas larger dextran conjugates may not diffuse evenly into the injected cell. Plan to inject cells in ~10 embryos to have sufficient tissues for proteomic analyses. - Confirm the success of cell labeling under a stereomicroscope. Ensure that only the intended cell is injected. Discard embryos containing injured or incorrectly labeled cells following institutional policies.
NOTE: Because X. laevis is invasive in many non-natural environments, embryos may be frozen to ensure lethality before discarding the embryos.
- Isolate the labeled cell progeny
- Transfer the injected embryos to 0.5x SS in a Petri dish and culture them between 14-25 °C until the desired developmental stage is reached.
NOTE: Consult established protocols to stage embryos reported on Xenbase. - Transfer 3-5 embryos to an agar dish with 0.2x SS solution for microdissections.
NOTE: Reducing the salt concentration of SS solution from 0.5x to 0.2x helps separate cells during dissection. - Use two sharpened forceps to gently remove the vitelline membrane surrounding the embryo.
NOTE: To spare the clone of interest from damage, peel the membrane from the opposite side of the fluorescently labeled clone. - Isolate the labeled clone by manual dissection (steps 3.3.5-3.3.6) or FACS (steps 3.3.7-3.3.8) as follows.
- Use forceps to dissect the labeled clone from the embryo.
NOTE: Other tools such as microsurgical scissors, tungsten needles, or eyebrow hair knives can be used for dissection of the labeled clone, as detailed elsewhere16. - Collect the dissected tissue with a 0.5-10 µL pipette and deposit them into a microcentrifuge vial. Using a transfer pipette, aspirate the media surrounding the collected tissue to limit salts in the sample, which interfere with HRMS analysis in later steps.
NOTE: Use vials that minimize protein adsorption onto plastic surfaces to minimize protein losses on vial surfaces during later steps of the workflow. - To isolate by FACS, transfer ~5-8 devitellized embryos into each well of a 12-well plate containing ~5 mL of Newport 2.0 buffer. Dissociate the embryos by nutating the plate at 80 rpm for 20-30 min at room temperature13.
NOTE: Embryos/larvae older than stage 22 have abundant extracellular matrix proteins, making dissociation into separate cells difficult. Additional enzymatic approaches can be adapted for dissociating tissues from older embryos as described elsewhere24. - Purify the fluorescently labeled cells from the suspension using FACS as described elsewhere24.
- Pellet cells by centrifugation and discard the supernatant.
NOTE: Use low centrifugation speed (400 × g) and temperature (4 °C) to prevent cell lysis. If using bovine serum albumin (BSA) for FACS, wash the cell pellet to reduce BSA interference during HRMS detection. Gently resuspend the cells in 1x phosphate buffered saline (PBS) and centrifuge again to pellet rinsed cells. Remove the supernatant PBS liquid. - Swiftly freeze the isolated cells by placing the sample vial on dry ice or liquid nitrogen.
NOTE: Keep the samples (tissues or cells) cooled (e.g., on ice) during processing steps. Freeze the cells with as little media around the sample as possible to facilitate downstream processing. - Store the samples at −80 °C until HRMS analysis.
- Transfer the injected embryos to 0.5x SS in a Petri dish and culture them between 14-25 °C until the desired developmental stage is reached.
4. Analyze the proteins by mass spectrometry
Proteomic characterization of the isolated tissues or cells is based on a series of established steps in HRMS. Figure 2 illustrates the steps of the bioanalytical workflow. The sample collection protocol used here is compatible with bottom-up11, middle-down25, or top-down26 workflows of proteomics. In what follows, the bottom-up strategy used in this study is described, which has proved to be sensitive, quantitative, and adaptable to diverse types of mass spectrometers. After extracting and enzymatically digesting proteins, the resulting peptides are separated, followed by HRMS analysis.
- Process the tissues/single cells
- For single-cell analysis by CE, heat the sample to 60 °C for ~15 min to denature proteins, then equilibrate the sample to room temperature (RT, ~5 min)18,21.
NOTE: Unlike during working with tissues, the reduction and alkylation steps are skipped to limit protein losses during sample preparation from single cells. Filter-aided sample preparation (FASP)27,28, other single pot strategies29, and microfluidic approaches30 can be adopted to minimize protein losses during sample preparation. - For analysis by nanoLC, lyse up to 5 dissected tissues in 50 µL of lysis buffer (~100 µg of total protein). Facilitate the process by pipetting the sample up and down a few times.
- Incubate the lysate at 4 °C for 10 min, then pellet the cell debris and yolk platelets by centrifugation at 4,500 × g at 4 °C. Transfer the supernatant into a clean microcentrifuge vial and add 10% SDS to obtain a final concentration of 1% SDS in the lysate (v/v).
- For tissues, follow steps 4.1.5-4.1.7.
- Add 0.5 M dithiothreitol to the lysate to obtain a final concentration of ~25 mM (e.g., 2.5 µL of 0.5 M dithiothretol to 50 µL of lysate) and incubate the lysate for 30 min at 60 °C to chemically reduce disulfide bonds in proteins.
- Add 0.5 M iodoacetamide to obtain a final concentration of ~75 mM in the lysate and incubate the mixture for 15 min at RT in the dark (Figure 2).
- Add 0.5 M dithiothreitol, same as the initial volume (e.g., 2.5 µL of 0.5 M dithiothretol to 50 µL of lysate) to quench reactants remaining from the alkylation reaction.
CAUTION: Iodoacetamide and dithiothreitol can cause serious skin and eye damage upon direct exposure. Use appropriate PPE when handling these chemicals. - Purify proteins via precipitation. Chloroform-methanol-based precipitation performs well31. This protocol is also adaptable to other types of precipitation approaches32.
NOTE: For single-cell analysis, where protein losses are of concern, skip the precipitation step for CE-HRMS. - Dry the protein precipitate in a vacuum concentrator (4-37 °C), then resuspend the extracted proteome in 50 µL of 50 mM ammonium bicarbonate. Estimate the protein concentration using a total-protein colorimetric assay to determine the amount of enzyme required for digestion (e.g., bicinchoninic acid protein assay).
- Digest the proteins to peptides. Add trypsin (1 µg/µL stock) to obtain a protease:protein ratio of 1:50 and incubate the mixture at 37 °C for up to 5 h for single-cell samples and up to 14 h for tissue samples. Consult vendor-specific recommendations for the reaction.
NOTE: Digestion with trypsin for longer than 14 h or higher concentrations may introduce cleavages that are nonspecific to the sequence of the protein, thus challenging protein identifications33. - Quantify the total peptide concentration using a colorimetric assay.
- OPTIONAL: For multiplexing quantification, tag the peptides from each sample with a different isobaric mass tag following vendor-specific instructions. Mix the barcoded peptides in equal proportions per peptide sample.
NOTE: Ensure accurate labeling and mixing to avoid quantitative biases. For quantity-limited samples or single-cell samples, a TMT-based carrier channel composed of pooled tissues/cells can be included to minimize sample losses during subsequent separation steps and to boost the sensitivity of lower abundant proteins34. - OPTIONAL: Desalt peptides to remove salts and contaminants (e.g., unreacted isobaric mass tag reagents) on a C18 reversed-phase spin column/tip to protect the LC-MS system.
- OPTIONAL: Fractionate (e.g., medium- or high-pH reversed-phase fractionation) the peptide mixture for deeper detection of the proteome via manual or automatic platforms. Use C18 stationary phase containing tips to fractionate low amounts (1-10 µg) of peptide digests.
- Dry the peptide mixture at 60 °C in a vacuum concentrator.
- Store the peptide mixture at −80 °C until measurement.
- For single-cell analysis by CE, heat the sample to 60 °C for ~15 min to denature proteins, then equilibrate the sample to room temperature (RT, ~5 min)18,21.
- Separate the Peptides
NOTE: After extracting and enzymatically digesting proteins, the resulting peptides are separated by nanoLC or CE and ionized by ESI for sequencing by tandem HRMS. Reversed-phase nanoLC separation is ideal for peptides amassing ~150 ng to ~1 µg per analysis. CE provides complementary sensitivity for peptides ranging from femtograms to <100 ng. Various custom-built and commercial CE-ESI interfaces allow for ready coupling of CE to HRMS with robust performance35 and are increasingly used for single-cell analysis18,36,37.- To separate using CE, follow steps 4.2.2-4.2.7.
NOTE: In what follows, the use of the custom-built CE platform for measuring the peptides is described. Protocols to build and use this CE instrument were provided earlier38, along with a visualized experiment on usage for small molecules20. Alternatively, these measurements can be performed on a commercial CE system, such as the AB SCIEX CESI, Agilent 7100, or equivalent. - Reconstitute the protein digest in 1-2 µL of the sample solvent, vortex to mix the sample, and centrifuge it at 10,000 x g for 2 min to pellet cell debris.
NOTE: Removal of the cell debris minimizes the likelihood of clogging the CE capillary, thus prolonging the lifetime of the separation system and boosting measurement throughput. - Initialize the CE-ESI instrument by flushing the CE capillary with the BGE.
- Validate instrumental performance using a known standard (e.g., cytochrome C or BSA digest, angiotensin peptides).
NOTE: Evaluating the instrument in terms of mass accuracy, detection sensitivity, reproducibility, and linear dynamic range of quantification is recommended before measuring precious samples. Additional notes on validation and troubleshooting of CE-ESI-MS performance are listed elsewhere18,20,38. - Inject ~1-10 nL of the sample into the CE separation capillary.
NOTE: This study uses ~1 m long fused silica capillary (40/110 µm inner/outer diameter) with the electrokinetically pumped sheath-flow setup. Commercial CE instruments usually require the presentation of 5-10 µL of sample in a microvial for injection. The custom-built CE platform18,38 is compatible with ~250 nL to 1 µL of sample deposited into a sample-loading microvial. - Transfer the inlet end of the CE separation capillary into the BGE.
- Start electrophoretic separation by gradually ramping the CE separation voltage from Earth ground (e.g., stepwise over 1 min). Potentials of 20-28 kV with current below ~10 µA ensure stable and reproducible instrumental performance for analysis.
- To separate using nanoLC, follow steps 4.2.9-4.2.12.
- Resuspend the peptide sample in Mobile Phase A. The concentration of the sample and its volume for injection depends on the available LC system and column. In this study, ~250 ng-1 µg of protein digest is injected in 1-20 µL of sample volume on a C18 packed-bed column (75 µm inner diameter, 2 µm particle size with 100 Å pores, 25 cm length separation column).
- Transfer the sample into an LC vial.
NOTE: Ensure that there are no air bubbles in the vial, which may damage the analytical column. Vials with inserts could be used for low-volume tissue or single-cell samples. - Load ~200 ng to 2 µg of peptide sample onto the C18 analytical column.
NOTE: Optionally, the peptides can be loaded on a trap column for desalting prior to analytical separation. For example, a C18 trap column with 0.1 mm inner diameter, 5 µm particle size, 100 Å pore size, 20 mm length. Desalt peptides with 100% buffer A at a flow rate of 5 µL/min for 5 min before the separation gradient begins. - Separate the peptides using gradient elution. At a 300 nL/min flow rate, the 120-min gradient used in this study is as follows: 0-5 min 2% B, 5-85 min 2-35% B, 86-90 min 70% B, 91-120 min 2% B.
- To separate using CE, follow steps 4.2.2-4.2.7.
- Ionize the peptides by ESI
NOTE: The CE or nanoLC capillary is most typically coupled into an ESI source for ionization. Micro-flow (blunt-tip) and nano-flow (tapered-tip39 and electrokinetic pumped sheath-flow36 design) CE-ESI interfaces for ultrasensitive detection have been developed previously.- Supply the separating peptides into an electrospray ion source for ionization using a commercial or custom-built ESI interface. For single-cell CE-ESI-MS analysis in Xenopus embryos, use an electrokinetically pumped low-flow interface wherein the CE capillary outlet is enclosed in a pulled borosilicate emitter.
- Check the liquid flow through the electrospray emitter using a camera and visually inspect the setup for possible leaks.
- Set the electrospray voltage to ~2.5 kV to start the ESI source (vs. Earth ground).
- Ensure a stable nanospray for HRMS analysis by monitoring the total ion current. Adjust the electrospray voltage and distance of the emitter to the HRMS inlet to achieve a stable spray (<15% relative standard deviation in total intensity).
- Detect the peptides
NOTE: Detection of peptides follows different instrumental considerations for isobaric mass tagged and untagged peptides and depends on the type of available mass spectrometer. This study uses an orbitrap tribrid mass spectrometer according to the following steps.- Acquire MS1 events with the settings: Analyzer, orbitrap; Spectral resolution, 120,000 full width at half maximum (FWHM); maximum injection time (IT), 50 ms; automatic gain control (AGC), 4 x 105 counts; microscans, 1.
- To sequence peptides, fragment precursor ions for detection in the ion trap analyzer using the settings: fragmentation mode, higher energy collision dissociation (HCD); collision gas, nitrogen; collision energy, 32% normalized collision energy (NCE); maximum IT, 70 ms; AGC, 1 x 104 counts; microscans, 1.
- OPTIONAL: Quantify TMT tagged peptides using tandem/multistage HRMS (MS2/MS3). For MS3 employing synchronous precursor selection, the typical instrumental settings are as follows. Single-stage (MS1) scans surveying the most abundant ions are dissociated via data-dependent acquisition using the parameters: MS2 fragmentation mode, collision-induced dissociation (CID); collision gas, helium; collision energy, 35% NCE; analyzer for fragment ions, ion trap following settings: maximum IT, 50 ms; AGC, 5 x 104 counts; microscans, 1. Select 10 MS2 fragment ions and fragment them with HCD in nitrogen (65% NCE). Detect MS3 fragment ions using the following settings: Orbitrap resolution 15,000 FWHM, maximum IT, 120 ms; AGC, 1 × 105 counts; microscans, 1.
NOTE: Different MS acquisition methods and parameters can be used for tagged samples following vendor recommendations as described elsewhere11,40.
- Analyze the data
NOTE: Proteins are identified and quantified using advanced bioinformatic packages. The fidelity of identifications is calculated using a decoy database, expressed as the false discovery rate (FDR) at the level of peptides and proteins.- Process the data using commercial or open-source software packages (reviewed in reference41). Match the raw data against a database that was prepared by concatenating the Xenopus proteome 9.2 with the mRNA-derived PHROG database42.
NOTE: The search parameters are: digestion enzyme, trypsin; missed cleavages, up to 2; variable modification, methionine oxidation; static modification, cysteine carbamidomethylation; precursor mass tolerance, 10 ppm; fragment mass tolerance, 0.6 Da; minimum peptide length, 5; identification fidelity, <1% FDR for peptides and proteins. Without alkylation to peptides, carbamidomethylation as a static modification is excluded during database search (e.g., for single-cell analysis). - Quantify protein abundances via label-free43 or label-based strategies44,45.
- OPTIONAL: Annotate proteins for gene ontology. PantherDB46, Reactome47, or Xenbase23 can be used.
- OPTIONAL: Quantify protein abundance and differences in protein abundances across cell/tissue types using software packages/webtools, such as the Trans-Proteomic Pipeline48, Perseus49, and Orange50.
NOTE: Additional considerations on experimental design and software options were reviewed elsewhere41,51. - OPTIONAL: Evaluate the results further using knowledge bases, such as STRING52 and BioPlex Display53 for known protein-protein interactions and PhosphoSiteplus54 for phosphorylations. For analyzing motifs and domains that are represented in the proteome, use webtools such as Simple Modular Architecture Research (SMART)55.
- Process the data using commercial or open-source software packages (reviewed in reference41). Match the raw data against a database that was prepared by concatenating the Xenopus proteome 9.2 with the mRNA-derived PHROG database42.
Access restricted. Please log in or start a trial to view this content.
Results
This protocol enabled the study of proteins in single cells and their lineages as they establish tissues in X. laevis embryos. Figure 1 illustrates one such application of the approach to study proteins in neural-tissue-fated cells and the newly induced neural ectoderm in the embryo. As shown in Figure 1A, the bioanalytical workflow integrated traditional tools of cell and developmental biology to identify, inject/aspirate cells, and collect specimens. ...
Access restricted. Please log in or start a trial to view this content.
Discussion
This protocol enables the characterization of protein expression in identified cell lineages in embryos of the Xenopus species. Stemming from HRMS, the methodology combines exquisite specificity in molecular identification, capability for multi-protein detection without molecular probes (usually hundreds to thousands of different proteins), and a capability for quantification. Adaptability to classical tools and workflows in cell and developmental (neuro)biology expand HRMS proteomics to exciting applications, i...
Access restricted. Please log in or start a trial to view this content.
Disclosures
The authors declare no competing interests.
Acknowledgements
We are grateful to Jie Li (University of Maryland, College Park) for valuable discussions on embryonic dissociation and FACS. We thank Vi M. Quach and Camille Lombard-Banek for assistance with sample preparation and data collection in previous studies exemplifying the proteomic applications that are highlighted in this protocol. Parts of this work were supported by the National Science Foundation under award number IOS-1832968 CAREER (to P.N.), the National Institutes of Health under award number R35GM124755 (to P.N.), the University of Maryland-National Cancer Institute Partnership Program (to P.N.), and COSMOS Club Foundation research awards (to A.B.B. and L.R.P.).
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| Acetonitrile (LC-MS-grade) | Fisher Scientific | A955 | |
| Agarose | ThermoFisher Scientific | R0492 | |
| Ammonium bicarbonate | Fisher Scientific | A643-500 | |
| Analytical Column | Thermo Scientific | 164941 | |
| Analytical microbalance | Mettler-Toledo | XSE105DU | |
| Automatic peptide fractionation platform | Agilent | 1260 Infinity II | |
| Borosilicate Capillaries | Sutter Instruments Co. | B100-50-10 | |
| Borosilicate Capillaries (for making Emmitters) | Sutter Instruments | B100-75-10 | |
| C18 spin columns (for desalting) | ThermoFisher Scientific | 89870 | |
| Camera ro monitor electrospray | Edmund Optics Inc. | EO-2018C | |
| Combretastatin A4 | Millipore Sigma | C7744 | |
| Commercial CESI system | AB SCIEX | CESI | |
| (Cyclohexylamino)-1-propanesulfonic acid (CAPS) | VWR | 97061-492 | |
| Cytochalasin D | Millipore Sigma | C8273 | |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable | ThermoFisher Scientific | D22910 | |
| Diothiothreitol | Fisher Scientific | FERR0861 | |
| Dumont #5 Forceps | Fine Science Tools | 11252-30 | |
| EDTA | Fisher Scientific | AAJ62786AP | |
| Epifluorescence light source | Lumencore | AURA III | |
| Eppendorf LoBing microcentrifuge tubes: protein | Fisher Scientific | 13-698-793 | |
| Formic acid (LC-MS-grade) | Fisher Scientific | A117-50 | |
| Freezer (-20 °C) | Fisher Scientific | 97-926-1 | |
| Freezer (-80 °C) | Thermo Scientific | TSX40086A | |
| Fused silica capillary | Molex | 1088150596 | |
| Heat Block | Benchmark | BSH300 | |
| High pressure liquid Chromatography System | ThermoFisher Scientific | Dionex Ultimate 3000 RSLC nanosystem | |
| High voltage power supply | Spellman | CZE1000R | |
| High-resolution Mass Spectrometer | ThermoFisher Scientific | Orbitrap Fusion Lumos Tribrid Mass Spectrometer | |
| HPLC caps | Thermo Scientific | C4013-40A | |
| HPLC Vials | Thermo Scientific | C4013-11 | |
| Illuminator e.g. Goosenecks | Nikon | C-FLED2 | |
| Ingenuity Pathway Analysis | Qiagen | ||
| Iodoacetamide | Fisher Scientific | AC122275000 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456-4 | |
| Microcapillary puller | Suttor Instruments | P-2000 | |
| Microinjector | Warner Instrument, Handem, CT | PLI-100A | |
| Micropippette puller | Sutter Instruments Co. | P-1000 | |
| MS data analysis software, commercial | ProteomeDiscoverer | ||
| MS data analysis software, opensource | MaxQuant | ||
| non-idet 40 substitute | Millipore Sigma | 11754599001 | |
| Petri dish 60 mm and 80 mm | Fisher Scientific | S08184 | |
| Pierce 10 µL bed Zip-tips (for desalting) | ThermoFisher Scientific | 87782 | |
| Pierce bicinchoninic acid protein assay kit | ThermoFisher Scientific | 23225 | |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 | |
| Pierce Trypsin Protease (MS Grade) | Fisher Scientific | PI90058 | |
| Protein LoBind vials | Eppendorf | 0030108434 , 0030108442 | |
| Refrigerated Centrifuge | Eppendorf | 5430R | |
| Refrigerated Incubator | Thermo Scientific | PR505755R/3721 | |
| sodium isethionate | Millipore Sigma | 220078 | |
| sodium pyrophosphate | Sigma Aldrich | 221368-100G | |
| Stainless steel BGE vial | Custom-Built | ||
| Stainless steel sample vials | Custom-Built | ||
| Stereomicroscope (objective 10x) | Nikon | SMZ 1270, SZX18 | |
| Sucrose | VWR | 97063-790 | |
| Syringe pumps (2) | Harvard Apparatus | 704506 | |
| Syringes (gas-tight): 500–1000 µL | Hamilton | 1750TTL | |
| Transfer pipettes (Plastic, disposable) | Fisher Scientific | 13-711-7M | |
| Trap Column | Thermo Scientific | 164750 | |
| Tris-HCl (1 M solution) | Fisher Scientific | AAJ22638AP | |
| Vacuum concentrator capable of operation at 4–10 °C | Labconco | 7310022 | |
| Vortex-mixer | Benchmark | BS-VM-1000 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| XYZ translation stage | Thorlabs | PT3 | |
| XYZ translation stage | Custom-Built |
References
- Shoemaker, L. D., Kornblum, H. I. Neural Stem Cells (NSCs) and Proteomics. Molecular & Cellular Proteomics. 15 (2), 344-354 (2016).
- Cervenka, J., et al. Proteomic characterization of human neural stem cells and their secretome during in vitro differentiation. Frontiers in Cellular Neuroscience. 14, 612560(2021).
- Christian, J. L. Morphogen gradients in development: From form to function. Wiley Interdisciplinary Reviews. Developmental Biology. 1 (1), 3-15 (2012).
- Gurdon, J. B., Elsdale, T. R., M, F. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 182, 64-65 (1958).
- Harland, R. M., Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 27 (12), 507-515 (2011).
- Moody, S. A. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 119 (2), 560-578 (1987).
- Moody, S. A. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Developmental Biology. 122 (2), 300-319 (1987).
- Dale, L., Slack, J. M. W. Fate map for the 32-cell stage of Xenopus laevis. Development. 99 (4), 527-551 (1987).
- Sun, L. L., et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Analytical Chemistry. 88 (13), 6653-6657 (2016).
- Lombard-Banek, C., Moody, S. A., Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angewandte Chemie-International Edition. 55 (7), 2454-2458 (2016).
- Zhang, Y. Y., Fonslow, B. R., Shan, B., Baek, M. C., Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113 (4), 2343-2394 (2013).
- Sive, H. L., Grainger, R. M., Harland, R. M. Early development of Xenopus laevis: A laboratory manual. , Cold Spring Harbor Laboratory Press. New York. (2000).
- Briggs, J. A., et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 360 (6392), (2018).
- Gupta, M., Sonnett, M., Ryazanova, L., Presler, M., Wuhr, M. Quantitative proteomics of xenopus embryos I, sample preparation. Xenopus. Methods in Molecular Biology. Vleminckx, K. 1865, Humana Press Inc. NY. 175-194 (2018).
- Baxi, A. B., Lombard-Banek, C., Moody, S. A., Nemes, P. Proteomic characterization of the neural ectoderm fated cell clones in the Xenopus laevis embryo by high-resolution mass spectrometry. ACS Chemical Neuroscience. 9 (8), 2064-2073 (2018).
- Moody, S. A. Cell lineage analysis in Xenopus embryos. Methods in Molecular Biology. 135, 331-347 (2000).
- Sater, A. K., Moody, S. A. Using Xenopus to understand human diseases and developmental disorders. Genesis. 55 (1-2), 1-14 (2017).
- Lombard-Banek, C., Choi, S. B., Nemes, P. Enzyme Activity in Single Cells. Methods in Enzymology. Allbritton, N. L., Kovarik, M. L. 628, 263-292 (2019).
- Lombard-Banek, C., Moody, S. A., Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Frontiers in Cell and Developmental Biology. 4, 11(2016).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. Microprobe capillary electrophoresis mass spectrometry for single-cell metabolomics in live frog (Xenopus laevis) embryos. Journal of Visualized Experiments: JoVE. (130), e56956(2017).
- Lombard-Banek, C., Moody, S. A., Manzin, M. C., Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and zebrafish embryos. Analytical Chemistry. 91 (7), 4797-4805 (2019).
- Klein, S. L. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Developmental Biology. 120 (1), 299-304 (1987).
- Karimi, K., et al. Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Research. 46 (1), 861-868 (2018).
- Kakebeen, A. D., Chitsazan, A. D., Wills, A. E. Tissue disaggregation and isolation of specific cell types from transgenic Xenopus appendages for transcriptional analysis by FACS. Developmental Dynamics. 250 (9), 1381-1392 (2021).
- Garcia, B. A. What does the future hold for top down mass spectrometry. Journal of the American Society for Mass Spectrometry. 21 (2), 193-202 (2010).
- Toby, T. K., Fornelli, L., Kelleher, N. L. Progress in top-down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry. (Palo Alto Calif). 9 (1), 499-519 (2016).
- Zhang, Z. B., Dubiak, K. M., Huber, P. W., Dovichi, N. J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis Embryos. Analytical Chemistry. 92 (7), 5554-5560 (2020).
- Wisniewski, J. R. Microbial Proteomics: Methods and Protocols.Methods in Molecular Biology. Becher, D. 1841, Humana Press Inc. NY. 3-10 (2018).
- Hughes, C. S., et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Zhu, Y., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nature Communications. 9, 882(2018).
- Wessel, D., Flugge, U. I. A method for the quantitative recovery of protein in dilute-solution in the presence of detergents and lipids. Analytical Biochemistry. 138 (1), 141-143 (1984).
- Jiang, L., He, L., Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of Chromatography A. 1023 (2), 317-320 (2004).
- Hildonen, S., Halvorsen, T. G., Reubsaet, L. Why less is more when generating tryptic peptides in bottom-up proteomics. Proteomics. 14 (17-18), 2031-2041 (2014).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19, 161(2018).
- Drouin, N., et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: effective electrophoretic mobility for reproducible and robust compound annotation. Analytical Chemistry. 92 (20), 14103-14112 (2020).
- Sun, L. L., Zhu, G. J., Zhang, Z. B., Mou, S., Dovichi, N. J. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. Journal of Proteome Research. 14 (5), 2312-2321 (2015).
- DeLaney, K., Sauer, C. S., Vu, N. Q., Li, L. J. Recent advances and new perspectives in capillary electrophoresis-mass spectrometry for single cell "omics". Molecules. 24 (1), 21(2019).
- Nemes, P., Rubakhin, S. S., Aerts, J. T., Sweedler, J. V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nature Protocols. 8 (4), 783-799 (2013).
- Choi, S. B., Zamarbide, M., Manzini, M. C., Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. Journal of the American Society for Mass Spectrometry. 28 (4), 597-607 (2017).
- Pino, L. K., Rose, J., O'Broin, A., Shah, S., Schilling, B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions. 48 (5), 1953-1966 (2020).
- Chen, C., Hou, J., Tanner, J. J., Cheng, J. L. Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences. 21 (8), 25(2020).
- Peshkin, L., et al. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Developmental Cell. 35 (3), 383-394 (2015).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 13 (9), 2513-2526 (2014).
- Gygi, S. P., et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 17 (10), 994-999 (1999).
- Thompson, A., et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 75 (8), 1895-1904 (2003).
- Mi, H. Y., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive api. Nucleic Acids Research. 49, 394-403 (2021).
- Schmidt, E., et al. On the Move Federated Workshops. , Springer, Verlag. Berlin. 710-719 (2006).
- Deutsch, E. W., et al. Trans-Proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 9 (7-8), 745-754 (2015).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Demsar, J., et al. Orange: Data mining toolbox in Python. Journal of Machine Learning Research. 14, 2349-2353 (2013).
- Oberg, A. L., Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. Journal of Proteome Research. 8 (5), 2144-2156 (2009).
- Jensen, L. J., et al. STRING 8 - a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, 412-416 (2009).
- Schweppe, D. K., Huttlin, E. L., Harper, J. W., Gygi, S. P. BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data. Journal of Proteome Research. 17 (1), 722-726 (2018).
- Hornbeck, P. V., et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research. 43, 512-520 (2015).
- Letunic, I., Khedkar, S., Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Research. 49, 458-460 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie-International Edition. 60 (23), 12852-12858 (2021).
- Lombard-Banek, C., Reddy, S., Moody, S. A., Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Molecular & Cellular Proteomics. 15 (8), 2756-2768 (2016).
- Saha-Shah, A., et al. Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Analytical Chemistry. 91 (14), 8891-8899 (2019).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Analytical Chemistry. 89 (13), 7069-7076 (2017).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47, 442-450 (2019).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved