
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Proteomica della spettrometria di massa guidata da livrea cellulare nell'embrione in via di sviluppo (rana)
In questo articolo
Riepilogo
Qui descriviamo una caratterizzazione proteomica basata sulla spettrometria di massa di linee cellulari con destini tissutali noti nell'embrione vertebrato di Xenopus laevis .
Abstract
La caratterizzazione degli eventi molecolari quando le cellule danno origine a tessuti e organi aumenta il potenziale per comprendere meglio lo sviluppo normale e progettare rimedi efficaci per le malattie. Le tecnologie che consentono l'identificazione e la quantificazione accurate di diversi tipi e di un gran numero di proteine fornirebbero informazioni ancora mancanti sui meccanismi molecolari che orchestrano lo sviluppo di tessuti e organismi nello spazio e nel tempo. Qui, presentiamo un protocollo basato sulla spettrometria di massa che consente la misurazione di migliaia di proteine in linee cellulari identificate in embrioni di Xenopus laevis (rana). L'approccio si basa su mappe riproducibili del destino cellulare e metodi consolidati per identificare, etichettare, tracciare e campionare in modo fluorescente le cellule e la loro progenie (cloni) da questo modello di sviluppo dei vertebrati. Dopo aver raccolto il contenuto cellulare utilizzando il microcampionamento o isolando le cellule mediante dissezione o selezione cellulare attivata dalla fluorescenza, le proteine vengono estratte e processate per l'analisi proteomica bottom-up. La cromatografia liquida e l'elettroforesi capillare vengono utilizzate per fornire una separazione scalabile per il rilevamento e la quantificazione delle proteine con spettrometria di massa ad alta risoluzione (HRMS). Vengono forniti esempi rappresentativi per la caratterizzazione proteomica delle cellule adipose del tessuto neurale. La proteomica HRMS guidata dalla linea cellulare è adattabile a diversi tessuti e organismi. È sufficientemente sensibile, specifico e quantitativo per scrutare le dinamiche spazio-temporali del proteoma durante lo sviluppo dei vertebrati.
Introduzione
La nostra comprensione della differenziazione cellulare e della genesi di tessuti e organi è il risultato di decenni di elaborati screening mirati di geni e dei loro prodotti. Aumentare la nostra conoscenza di tutte le biomolecole e delle loro quantità durante importanti eventi cellulari aiuterebbe a svelare i meccanismi molecolari che controllano il modello spaziale e temporale del piano corporeo dei vertebrati. Le tecnologie che consentono l'amplificazione molecolare e il sequenziamento sono ora in grado di riferire regolarmente su un gran numero di geni e trascrizioni, supportando studi basati su ipotesi nella ricerca biologica e traslazionale di base. Per comprendere i sistemi in via di sviluppo, una complessa relazione tra trascrizione e traduzione sostiene l'analisi diretta di più proteine e le loro modificazioni post-traduzionali. La proteomica globale che utilizza sistemi biologici in vitro, come le cellule staminali pluripotenti indotte, ha iniziato a delineare i meccanismi di induzione tissutale 1,2. Negli organismi complessi, come l'embrione vertebrato, lo sviluppo si basa su gradienti morfogeni nel contesto dello spazio e del tempo3. Ne consegue che acquisire conoscenze sui cambiamenti proteomici man mano che le cellule si differenziano per formare tessuti specializzati, come i tessuti neurali, offre una chiave per sbloccare programmi molecolari che controllano lo sviluppo normale e difettoso e guidare le terapie di prossima generazione.
La rana artigliata sudafricana vertebrata (Xenopus laevis) è un modello consolidato nella biologia cellulare e dello sviluppo, neuro e rigenerativa. Il Premio Nobel 2012 per la Fisiologia ola Medicina 4,5 di Sir John Gurdon per la scoperta della pluripotenza del nucleo somatico ha evidenziato l'importanza di questo modello per le scoperte negli studi di base e traslazionali. Gli embrioni di Xenopus si sviluppano esternamente alla madre, facilitando così la manipolazione diretta delle cellule, dei cloni cellulari e dell'espressione genica nelle varie fasi di sviluppo. La pigmentazione asimmetrica e le divisioni cellulari stereotipate hanno permesso di tracciare mappe del destino riproducibili dall'embrione a 7,8 e 16-6e 32 cellule. Per la proteomica basata sulla spettrometria di massa ad alta risoluzione (HRMS), ulteriori vantaggi del modello includono dimensioni relativamente grandi (~ 1 mm di diametro), che producono un abbondante contenuto proteico per l'analisi (~ 130 μg negli embrioni in fase di scissione precoce, ~ 10 μg di contenuto proteico in singole cellule dell'embrione a 16 cellule)9,10.
Attualmente, HRMS è la tecnologia leader di scelta per la rilevazione delle proteine. Questa tecnologia consente il rilevamento e la quantificazione diretti, sensibili e specifici di proteine multiple, di solito da centinaia a migliaia di proteine diverse11. La proteomica bottom-up di HRMS comporta una serie di passaggi interconnessi. Dopo l'estrazione dal campione di cellula/tessuto, le proteine vengono digerite con un enzima proteolitico, come la tripsina (proteomica bottom-up). I peptidi risultanti vengono separati in base alle loro diverse proprietà fisico-chimiche, tra cui idrofobicità (cromatografia liquida a fase inversa, LC), carica netta (cromatografia a scambio ionico), dimensioni (cromatografia di esclusione dimensionale) o mobilità elettroforetica (elettroforesi capillare, CE). I peptidi vengono quindi caricati (ionizzati), tipicamente utilizzando la ionizzazione elettrospray (ESI), e gli ioni peptide vengono rilevati e sequenziati tramite frammentazione in fase gassosa mediante HRMS tandem. I dati peptidici risultanti sono mappati sul proteoma dell'organismo studiato. Con l'intensità del segnale dello ione peptidico specifica per proteine (proteotipica) correlata alla concentrazione, la quantificazione delle proteine può essere eseguita senza etichetta o basata sull'etichetta (quantificazione multiplexing). La proteomica HRMS fornisce una ricca risorsa di informazioni sullo stato molecolare del sistema in studio, consentendo la generazione di ipotesi e studi funzionali di follow-up.

Figura 1: Proteomica spaziotemporalmente scalabile che consente la proteomica HRMS guidata dalla linea cellulare nell'embrione in via di sviluppo (rana). (A) Visualizzazione del campione (1) utilizzando uno stereomicroscopio (2) per l'iniezione di una cellula identificata (inserto), utilizzando una micropipetta fabbricata (3) sotto controllo da uno stadio di traslazione (4). (B) Campionamento subcellulare della cellula D11 sinistra identificata in un embrione a 16 cellule. (C) Dissezione di un'intera cellula D11 da un embrione a 16 cellule. (D) Tracciamento fluorescente (verde) delle progenie D111 sinistra e destra da un embrione a 32 cellule per guidare la dissezione dell'ectoderma neurale (NE) nella gastrula (stadio 10) e l'isolamento del tessuto discendente dal girino mediante FACS. Barre della scala: 200 μm per gli embrioni, 1,25 mm per la fiala. Le figure sono state adattate con il permesso dei riferimenti 15,19,21,59. Fare clic qui per visualizzare una versione ingrandita di questa figura.
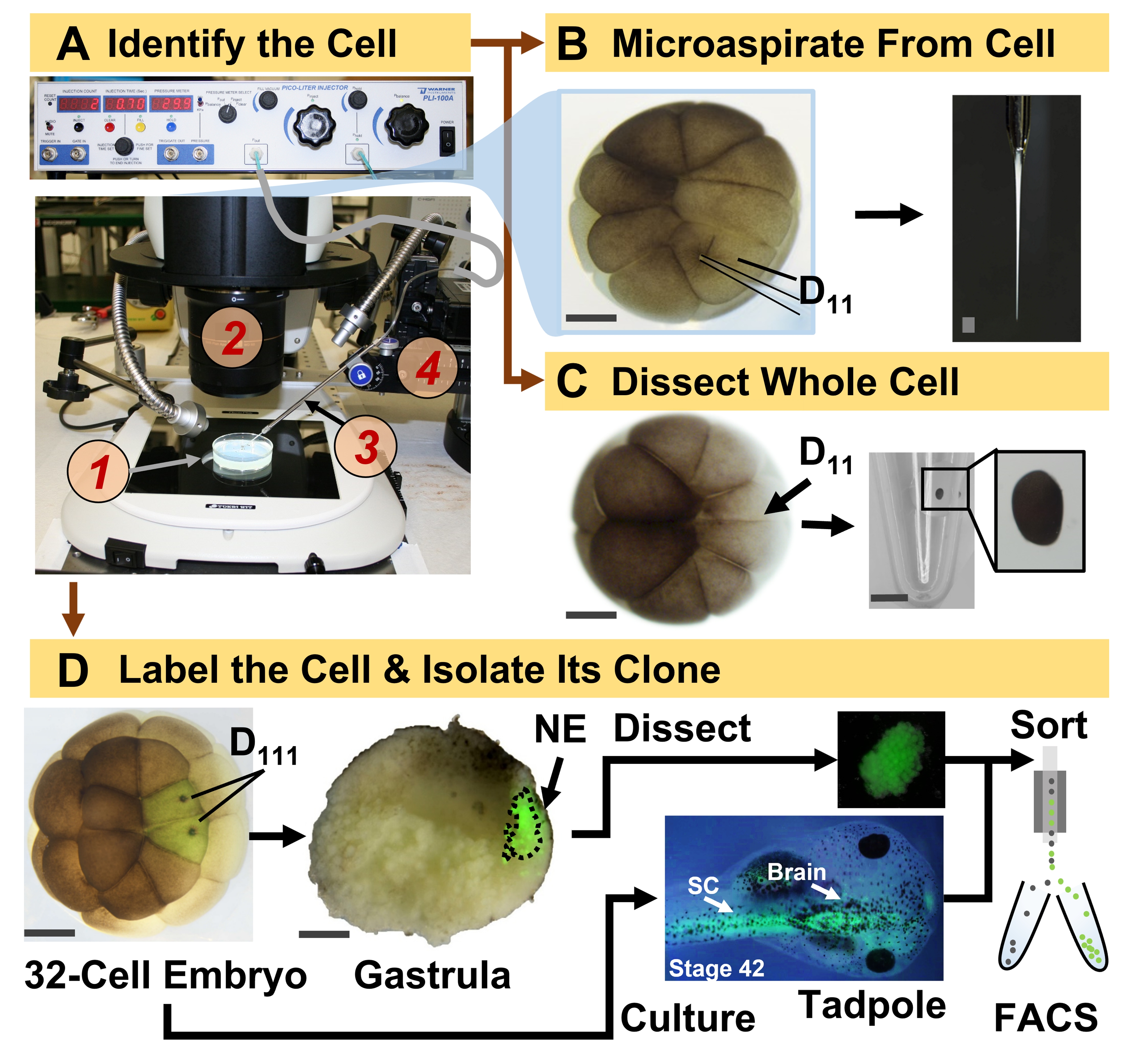{kind=link}
Il protocollo qui presentato consente la quantificazione basata su HRMS di un gran numero di proteine in cellule/tessuti identificati nello sviluppo di embrioni di X. laevis. L'approccio si basa su un'accurata identificazione cellulare, mappe del destino cellulare riproducibili e metodologie consolidate per tracciare le linee cellulari in questo modello biologico 6,7,8. Come mostrato nella Figura 1, studiamo i proteomi da singole cellule impiegando la dissezione dell'intera cellula o il microcampionamento capillare per aspirare il contenuto cellulare. Il monitoraggio del lignaggio di una cellula ci permette di studiare l'evoluzione spaziotemporale del proteoma quando le cellule formano i tessuti durante la gastrulazione. La progenie cellulare è marcata in modo fluorescente iniettando un fluoroforo coniugato a destrano inerte o mRNA per proteine fluorescenti (ad esempio, proteina fluorescente verde o GFP). La progenie marcata è isolata nei punti temporali di sviluppo desiderati. Durante la gastrulazione, i cloni cellulari che sono strettamente raggruppati possono essere isolati per dissezione. Dopo la gastrulazione, i cloni cellulari possono essere distribuiti all'interno dell'embrione a causa di movimenti migratori e possono essere isolati dai tessuti dissociati mediante selezione cellulare attivata dalla fluorescenza (FACS). Le proteine in queste cellule e tessuti sono misurate tramite proteomica bottom-up che impiega HPLC o CE per la separazione e HRMS tandem ESI per l'identificazione. La proteomica HRMS guidata dalla linea cellulare è scalabile a diverse dimensioni cellulari e linee cellulari all'interno dell'embrione ed è specifica, sensibile e quantitativa. Attraverso esempi selezionati mostrati qui, dimostriamo anche che questo protocollo è scalabile e ampiamente adattabile a diversi tipi di cellule e linee cellulari.

Figura 2: Il flusso di lavoro bioanalitico. Micro-dissezione e aspirazione capillare, o FACS ha facilitato il campionamento del contenuto proteico cellulare e clonale. Deplezione delle abbondanti proteine del tuorlo e separazione mediante elettroforesi capillare (CE) o cromatografia liquida a nanoflusso (LC) con sensibilità di identificazione migliorata (ID) mediante spettrometria di massa ad alta risoluzione (HRMS) a ionizzazione elettrospray (ESI). La quantificazione ha rivelato la disregolazione, fornendo nuove informazioni per studi basati su ipotesi in combinazione con le informazioni disponibili dall'ontologia genica (GO). Le cifre sono state adattate con il permesso del riferimento15. Fare clic qui per visualizzare una versione ingrandita di questa figura.
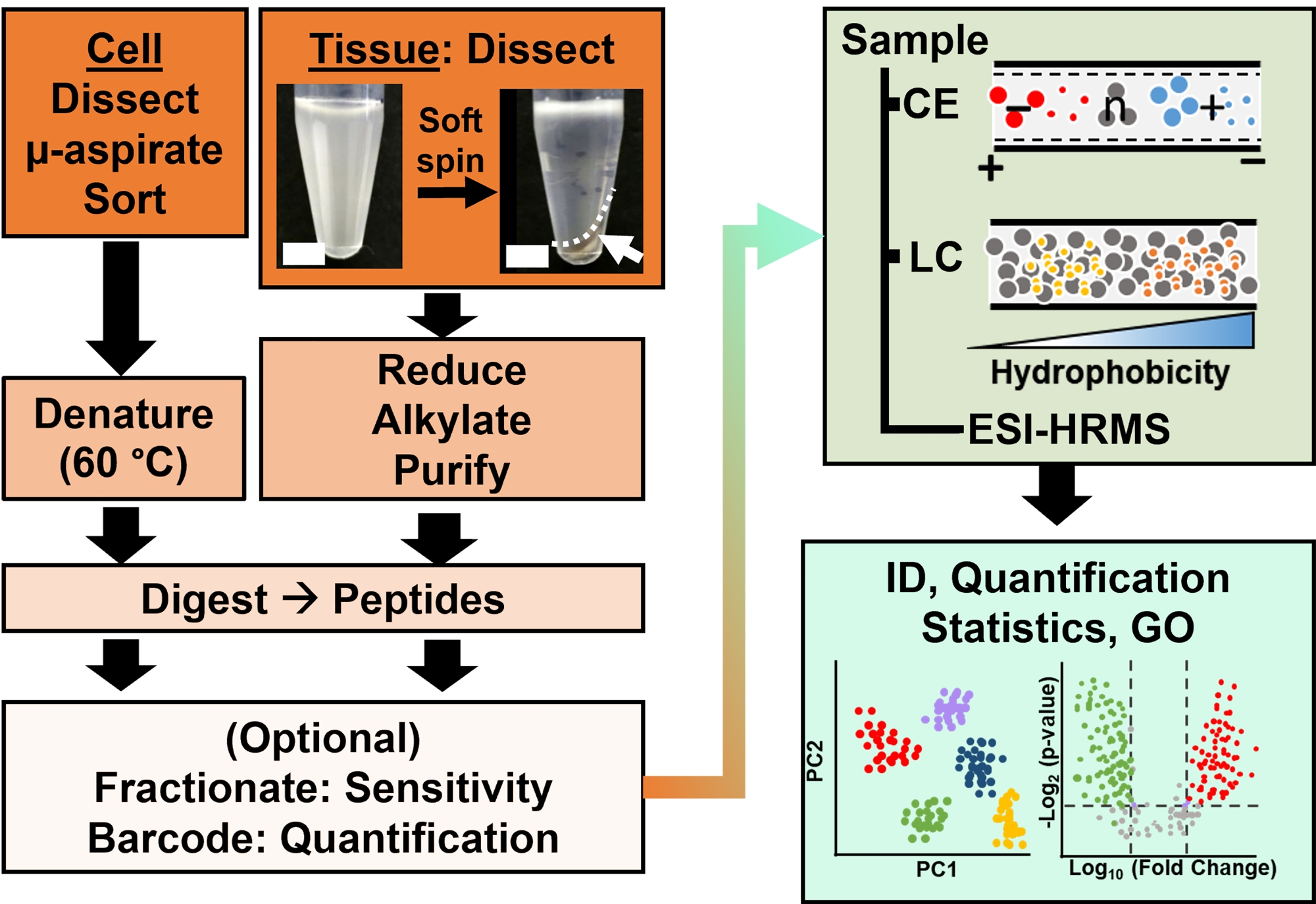{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocollo
Tutti i protocolli che garantiscono il mantenimento e la manipolazione umana delle rane adulte di Xenopus laevis sono stati approvati dal Comitato istituzionale per la cura e l'uso degli animali presso l'Università del Maryland, College Park (numeri di approvazione R-DEC-17-57 e R-FEB-21-07).
1. Preparare le soluzioni
- Per l'embriologia
- Preparare 1x, 0,5x e 0,2x la soluzione di Steinberg (SS), quindi sterilizzarli in autoclave (120 °C per 20 min) fino alla sterilità seguendo i protocolli standard12.
- Preparare il 3% (p/v) di Ficoll in 1x SS sterilizzato seguendo i protocolli standard12.
- Per la degelatina, preparare una soluzione di cisteina al 2% (p/v) e regolare il pH a 8 aggiungendo 10 M soluzione di idrossido di sodio goccia a goccia.
ATTENZIONE: L'esposizione alla cisteina può causare danni cutanei e respiratori. L'idrossido di sodio è un corrosivo che può causare gravi danni alla pelle e agli occhi in caso di esposizione diretta. Utilizzare dispositivi di protezione individuale (DPI) appropriati quando si maneggiano queste sostanze chimiche, come guanti e un camice da laboratorio. - Per il tracciante di lignaggio, preparare lo 0,5% (v / v) di un destrano fluorescente in acqua deionizzata sterile. In alternativa, preparare una soluzione di 0,2 μg/μL di mRNA per proteine fluorescenti in acqua deionizzata sterile (ad es. GFP).
- Per dissociare le cellule, preparare il tampone Newport 2.0 contenente 0,1 M isethionate di sodio, 20 mM di pirofosfato di sodio e 10 mM CAPS, quindi portare il suo pH a 10,513.
ATTENZIONE: L'esposizione al pirofosfato di sodio può causare irritazione della pelle e degli occhi. Utilizzare DPI appropriati quando si maneggiano queste sostanze chimiche.
- Per la proteomica bottom-up
- Preparare il tampone di lisi cellulare per includere: 250 mM di saccarosio, 1% di nonidet P-40 sostitutivo (w / v), 20 mM Tris-HCl, 5 mM EDTA, 10 μM citocalasina D e 10 μM combretastatina 4A. Preparare una scorta di sodio dodecilsolfato al 10% (p/v)14,15.
NOTA: Tris-HCl è stato scelto per ridurre al minimo la contaminazione HEPES durante il nano-flusso LC (nanoLC)-HRMS.
ATTENZIONE: L'esposizione al sostituto del nonidet P-40 può causare irritazione cutanea. La citocalasina D è teratogena se consumata e la combretastatina è acutamente tossica in caso di esposizione diretta. Utilizzare DPI appropriati quando si maneggiano queste sostanze chimiche. - Per separare i peptidi mediante CE, preparare i seguenti solventi (v/v): Campione di solvente, 75% acetonitrile (ACN) contenente 0,05% di acido acetico (AcOH) in acqua; soluzione di guaina, 10% ACN contenente 0,05% AcOH in acqua; elettrolita di fondo (BGE), 25% ACN contenente 1 M di acido formico (FA) in acqua.
ATTENZIONE: AcOH e FA sono tossici se inalati o consumati e possono causare gravi danni alla pelle e agli occhi in caso di esposizione diretta. Utilizzare DPI appropriati quando si maneggiano queste sostanze chimiche. - Per separare i peptidi mediante nanoLC in fase inversa, preparare (v / v): fase mobile A (acquosa), acqua contenente 0,1% FA; fase mobile B (organica), 0,1% FA in ACN.
NOTA: Tutte le miscele devono essere preparate utilizzando solventi di grado LC-MS per ridurre al minimo le interferenze chimiche durante il rilevamento HRMS.
- Preparare il tampone di lisi cellulare per includere: 250 mM di saccarosio, 1% di nonidet P-40 sostitutivo (w / v), 20 mM Tris-HCl, 5 mM EDTA, 10 μM citocalasina D e 10 μM combretastatina 4A. Preparare una scorta di sodio dodecilsolfato al 10% (p/v)14,15.
2. Preparare gli strumenti per la microiniezione e la dissezione
- Per spostare e orientare delicatamente gli embrioni, fare cappi fissando i capelli puliti in una pipetta Pasteur come descritto altrove16.
- Per la microiniezione, fabbricare gli aghi tirando capillari borosilicati (diametro esterno/interno 1 mm/500 μm) utilizzando un estrattore di pipette come descritto altrove16.
NOTA: Qui, per la fabbricazione di aghi è stato utilizzato un estrattore per pipette P-1000 con le seguenti impostazioni: calore, 495; tirare, 30, velocità, 60; tempo, 150; pressione, 200. - Con l'osservazione sotto uno stereomicroscopio, tagliare la punta del capillare usando un paio di pinze affilate per fabbricare essenzialmente il capillare in un (micro) ago (ad esempio, Dumont #5)16.
ATTENZIONE: I capillari tirati sono molto affilati e devono essere maneggiati con cura.
NOTA: La punta dell'ago deve essere abbastanza affilata (diametro esterno 10-15 μm) per poter perforare la cellula con danni minimi alla membrana cellulare in modo che il contenuto intracellulare non fuoriesca e la cellula possa guarire e continuare ad essere vitale. - Per trattenere gli embrioni durante la microiniezione, preparare i pozzetti in un piatto pieno di argilla. In una capsula di Petri di 15 mm, imprimere pozzetti di diametro ~ 1 mm x ~ 0,5 mm di profondità in argilla plastilina non tossica come descritto altrove16.
- Per la microdissezione, preparare piatti ricoperti di agarosio. Preparare il 2% di agarosio in 1x SS e sterilizzarlo in autoclave per sterilizzare la soluzione (120 °C per 20 min). Riempire le piastre di Petri da 60 mm a metà e lasciare solidificare le piastre. Realizzare pozzetti profondi ~1 mm di diametro x ~0,5 mm utilizzando uno strumento per pipette Pasteur a sfera come descritto in precedenza16.
3. Isolare la linea cellulare
NOTA: i seguenti passaggi vengono eseguiti per isolare singole cellule identificate e/o le loro linee cellulari discendenti. Di solito, l'embrione viene coltivato allo stadio di 16 o 32 cellule, dove i destini tissutali di ogni cellula sono mappati in modo riproducibile 6,7,17. Le cellule embrionali sono identificate in base alla morfologia, alla posizione e in riferimento alle loro mappe del destino. Per l'analisi di singole cellule, le cellule identificate vengono isolate mediante dissezione manuale o il loro contenuto intracellulare viene raccolto in una pipetta capillare e depositato in 5 μL di bicarbonato di ammonio 0,5 mM. Il campione risultante viene conservato a -80 °C fino all'analisi (Figura 1)18,19,20,21. Per l'analisi della linea cellulare, le cellule identificate vengono iniettate con un tracciante di linea e i loro cloni successivi vengono isolati nelle fasi chiave dello sviluppo (ad esempio, durante la gastrulazione per studiare l'induzione tissutale, dopo la neurulazione per studiare l'impegno tissutale). In quanto segue, vengono delineati i passaggi per etichettare in modo fluorescente il lignaggio delle cellule identificate per l'isolamento mediante dissezione o FACS.
- Coltura degli embrioni
- Ottenere embrioni tramite accoppiamento naturale o fecondazione in vitro (FIV) seguendo i protocolli stabiliti12.
NOTA: L'accoppiamento naturale è logisticamente più semplice, risparmia le rane maschi adulte e produce embrioni in diverse fasi di sviluppo, mentre la fecondazione in vitro fornisce embrioni sincronizzati con lo sviluppo per esperimenti che richiedono una stadiazione accurata. - Dejelly gli embrioni. Rimuovere il rivestimento gelatinoso che circonda gli embrioni mediante trattamento con la soluzione di dejellying come descritto altrove12,16.
NOTA: Le microiniezioni e la dissezione richiedono l'accesso alle cellule e ai tessuti, rendendo necessaria la dejellying negli embrioni di X. laevis. - Selezionare embrioni a 2 cellule con pigmentazione stereotipata16,22.
NOTA: Questo passaggio è importante per garantire l'accuratezza e la riproducibilità nell'identificazione della cellula e della sua discendenza. - Embrioni di coltura allo stadio di sviluppo desiderato. Trasferire gli embrioni degellati in una capsula di Petri contenente 1x SS e incubarli tra 14-25 °C per controllare la velocità di sviluppo.
NOTA: La dipendenza dalla temperatura dello sviluppo è riproducibile e tracciata per X. laevis, disponibile su Xenbase23 (www.xenbase.org). La coltura di lotti di embrioni a diverse temperature consente fasi di sviluppo sconcertanti. Ciò aiuta a distribuire il numero di embrioni disponibili in un dato momento per la sperimentazione. - Monitorare il modello di scissione degli embrioni e selezionare gli embrioni con pigmentazione stereotipata e modelli di scissione per microiniezione16.
NOTA: Quando si selezionano embrioni a 16 e 32 cellule, assicurarsi che le scissioni cellulari siano simmetriche per il tracciamento del lignaggio riproducibile.
- Ottenere embrioni tramite accoppiamento naturale o fecondazione in vitro (FIV) seguendo i protocolli stabiliti12.
- Etichettare le celle di interesse
- Impostare l'ago per iniezione contenente la soluzione tracciante della linea. Montare l'ago per microiniezione in un supporto per micropipette controllato da un micromanipolatore multiasse.
- Collegare il supporto della micropipetta a un microiniettore. Riempire l'ago con il tracciante di lignaggio applicando una pressione negativa come descritto altrove16. La Figura 1A illustra la configurazione.
- Calibrare l'ago. Regolare le dimensioni della punta dell'ago e il tempo di iniezione per fornire ~ 1 nL della soluzione tracciante della linea, misurata in olio (minerale) seguendo un protocollo disponibile altrove16.
NOTA: I capillari con una punta più larga tendono a danneggiare la membrana cellulare, causando la fuoriuscita del contenuto subcellulare e del tracciante della linea iniettato, mentre i capillari con punte più piccole sono inclini all'intasamento. I capillari con diametro esterno della punta di ~10 μm sono ideali, richiedendo un impulso di pressione di 40 psi oltre ~300 ms per fornire ~1 nL. - Inondare il piatto di argilla di microiniezione con la soluzione di Ficoll al 3% e trasferire ~ 10 embrioni nel piatto di argilla usando una pipetta di trasferimento. Utilizzare un cappio per guidare ogni embrione in un pozzo e posizionarli delicatamente in modo che la cellula di interesse mirata sia ad angolo retto rispetto al microago.
- Identificare la cellula precursore del lignaggio di interesse seguendo le mappe del destino tissutale di X. laevis. Ad esempio, la Figura 1 mostra la marcatura dei cloni ectodermici neurali basata sull'iniezione delle sue cellule precursori in embrioni a 32 cellule (cellule D111 sinistra e destra).
NOTA: Le mappe dettagliate del destino per gli embrioni a 16-6 e 32 cellule 7,8 sono disponibili in una piattaforma interattiva tramite Xenbase23. È importante garantire la pigmentazione stereotipata e le scissioni sugli embrioni quando li si utilizza per esperimenti di tracciamento del lignaggio. - Iniettare la/e cellula/e di interesse con ~1 nL del destrano fluorescente o ~200 pg di mRNA come descritto in precedenza16.
NOTA: utilizzare coniugati destrano da 10.000 a 40.000 MW. I coniugati destrani più piccoli potrebbero passare attraverso giunzioni gap, mentre i coniugati destrani più grandi potrebbero non diffondersi uniformemente nella cellula iniettata. Pianificare l'iniezione di cellule in ~ 10 embrioni per avere tessuti sufficienti per le analisi proteomiche. - Conferma il successo dell'etichettatura cellulare sotto uno stereomicroscopio. Assicurarsi che venga iniettata solo la cellula desiderata. Scartare gli embrioni contenenti cellule danneggiate o etichettate in modo errato seguendo le politiche istituzionali.
NOTA: Poiché X. laevis è invasivo in molti ambienti non naturali, gli embrioni possono essere congelati per garantire la letalità prima di scartare gli embrioni.
- Isolare la progenie cellulare marcata
- Trasferire gli embrioni iniettati a 0,5x SS in una capsula di Petri e coltivarli tra 14-25 °C fino al raggiungimento dello stadio di sviluppo desiderato.
NOTA: Consultare i protocolli stabiliti per mettere in scena gli embrioni riportati su Xenbase. - Trasferire 3-5 embrioni in un piatto di agar con soluzione 0,2x SS per microdissezioni.
NOTA: Ridurre la concentrazione salina della soluzione di SS da 0,5x a 0,2x aiuta a separare le cellule durante la dissezione. - Utilizzare due pinze affilate per rimuovere delicatamente la membrana vitellina che circonda l'embrione.
NOTA: Per evitare che il clone di interesse si danneggi, staccare la membrana dal lato opposto del clone marcato con fluorescenza. - Isolare il clone marcato mediante dissezione manuale (passaggi 3.3.5-3.3.6) o FACS (passaggi 3.3.7-3.3.8) come segue.
- Utilizzare una pinza per sezionare il clone etichettato dall'embrione.
NOTA: Altri strumenti come forbici microchirurgiche, aghi di tungsteno o coltelli per sopracciglia possono essere utilizzati per la dissezione del clone etichettato, come dettagliato altrove16. - Raccogliere il tessuto sezionato con una pipetta da 0,5-10 μL e depositarlo in un flaconcino di microcentrifuga. Utilizzando una pipetta di trasferimento, aspirare il mezzo che circonda il tessuto raccolto per limitare i sali nel campione, che interferiscono con l'analisi HRMS nelle fasi successive.
NOTA: utilizzare flaconcini che riducono al minimo l'adsorbimento delle proteine sulle superfici in plastica per ridurre al minimo le perdite di proteine sulle superfici dei flaconcini durante le fasi successive del flusso di lavoro. - Per isolare mediante FACS, trasferire ~ 5-8 embrioni devitellizzati in ciascun pozzetto di una piastra a 12 pozzetti contenente ~ 5 ml di tampone Newport 2.0. Dissociare gli embrioni nutando la piastra a 80 giri / min per 20-30 minuti a temperatura ambiente13.
NOTA: Gli embrioni/larve più vecchi dello stadio 22 hanno abbondanti proteine della matrice extracellulare, rendendo difficile la dissociazione in cellule separate. Ulteriori approcci enzimatici possono essere adattati per dissociare i tessuti dagli embrioni più vecchi, come descritto altrove24. - Purificare le cellule marcate con fluorescenza dalla sospensione utilizzando FACS come descritto altrove24.
- Celle a pellet mediante centrifugazione e scartare il surnatante.
NOTA: Utilizzare bassa velocità di centrifugazione (400 × g) e temperatura (4 °C) per prevenire la lisi cellulare. Se si utilizza l'albumina sierica bovina (BSA) per FACS, lavare il pellet cellulare per ridurre l'interferenza BSA durante il rilevamento della HRMS. Risospendere delicatamente le cellule in 1x soluzione salina tamponata fosfato (PBS) e centrifugare nuovamente in celle risciacquate a pellet. Rimuovere il liquido PBS surnatante . - Congelare rapidamente le cellule isolate ponendo il flaconcino del campione su ghiaccio secco o azoto liquido.
NOTA: Conservare i campioni (tessuti o cellule) raffreddati (ad esempio, su ghiaccio) durante le fasi di lavorazione. Congelare le celle con il minor supporto possibile attorno al campione per facilitare l'elaborazione a valle. - Conservare i campioni a -80 °C fino all'analisi HRMS.
- Trasferire gli embrioni iniettati a 0,5x SS in una capsula di Petri e coltivarli tra 14-25 °C fino al raggiungimento dello stadio di sviluppo desiderato.
4. Analizzare le proteine mediante spettrometria di massa
La caratterizzazione proteomica dei tessuti o delle cellule isolati si basa su una serie di passaggi stabiliti nella HRMS. Nella Figura 2 vengono illustrati i passaggi del flusso di lavoro bioanalitico. Il protocollo di raccolta dei campioni utilizzato qui è compatibile con i flussi di lavoro bottom-up11, middle-down25 o top-down26 di proteomica. In quanto segue, viene descritta la strategia bottom-up utilizzata in questo studio, che si è dimostrata sensibile, quantitativa e adattabile a diversi tipi di spettrometri di massa. Dopo aver estratto e digerito enzimaticamente le proteine, i peptidi risultanti vengono separati, seguiti dall'analisi HRMS.
- Elaborare i tessuti/singole cellule
- Per l'analisi a singola cellula mediante CE, riscaldare il campione a 60 °C per ~15 minuti per denaturare le proteine, quindi equilibrare il campione a temperatura ambiente (RT, ~5 min)18,21.
NOTA: A differenza del lavoro con i tessuti, le fasi di riduzione e alchilazione vengono saltate per limitare le perdite proteiche durante la preparazione del campione da singole cellule. La preparazione del campione assistita da filtro (FASP)27,28, altre strategie single pot 29 e approcci microfluidici30 possono essere adottati per ridurre al minimo le perdite proteiche durante la preparazione del campione. - Per l'analisi mediante nanoLC, lisare fino a 5 tessuti sezionati in 50 μL di tampone di lisi (~100 μg di proteine totali). Facilitare il processo pipettando il campione su e giù alcune volte.
- Incubare il lisato a 4 °C per 10 minuti, quindi pellettare i detriti cellulari e le piastrine del tuorlo mediante centrifugazione a 4.500 × g a 4 °C. Trasferire il surnatante in un flaconcino di microcentrifuga pulito e aggiungere il 10% di SDS per ottenere una concentrazione finale dell'1% di SDS nel lisato (v/v).
- Per i tessuti, seguire i passaggi 4.1.5-4.1.7.
- Aggiungere 0,5 M di ditiotreitolo al lisato per ottenere una concentrazione finale di ~25 mM (ad esempio, 2,5 μL di 0,5 M ditiotretolo a 50 μL di lisato) e incubare il lisato per 30 minuti a 60 °C per ridurre chimicamente i legami disolfuro nelle proteine.
- Aggiungere 0,5 M iodoacetamide per ottenere una concentrazione finale di ~75 mM nel lisato e incubare la miscela per 15 minuti a RT al buio (Figura 2).
- Aggiungere 0,5 M di ditiotreitolo, uguale al volume iniziale (ad esempio, 2,5 μL di 0,5 M di ditiotretolo a 50 μL di lisato) per estinguere i reagenti rimanenti dalla reazione di alchilazione.
ATTENZIONE: Iodoacetamide e ditiotreitolo possono causare gravi danni alla pelle e agli occhi in caso di esposizione diretta. Utilizzare DPI appropriati quando si maneggiano queste sostanze chimiche. - Purificare le proteine attraverso la precipitazione. La precipitazione a base di cloroformio-metanolo ha prestazioni ben31. Questo protocollo è adattabile anche ad altri tipi di approcci alle precipitazioni32.
NOTA: Per l'analisi di singole cellule, dove le perdite di proteine sono preoccupanti, saltare la fase di precipitazione per CE-HRMS. - Essiccare il precipitato proteico in un concentratore sottovuoto (4-37 °C), quindi risospendere il proteoma estratto in 50 μL di bicarbonato di ammonio da 50 mM. Stimare la concentrazione proteica utilizzando un saggio colorimetrico a proteine totali per determinare la quantità di enzima necessaria per la digestione (ad esempio, saggio proteico dell'acido bicinconinico).
- Digerire le proteine in peptidi. Aggiungere tripsina (1 μg/μL di stock) per ottenere un rapporto proteasi/proteina di 1:50 e incubare la miscela a 37 °C per un massimo di 5 ore per campioni monocellulari e fino a 14 ore per campioni di tessuto. Consultare le raccomandazioni specifiche del fornitore per la reazione.
NOTA: La digestione con tripsina per concentrazioni superiori a 14 ore o superiori può introdurre scissioni non specifiche per la sequenza della proteina, sfidando così le identificazioni proteiche33. - Quantificare la concentrazione totale di peptidi utilizzando un saggio colorimetrico.
- FACOLTATIVO: Per la quantificazione del multiplexing, etichettare i peptidi di ciascun campione con un tag di massa isobarica diverso seguendo le istruzioni specifiche del fornitore. Mescolare i peptidi con codice a barre in proporzioni uguali per campione di peptide.
NOTA: Garantire un'etichettatura e una miscelazione accurate per evitare distorsioni quantitative. Per campioni a quantità limitata o campioni a singola cellula, è possibile includere un canale portante basato su TMT composto da tessuti/cellule raggruppati per ridurre al minimo le perdite di campione durante le successive fasi di separazione e per aumentare la sensibilità delle proteine a bassa abbondanza34. - OPZIONALE: Dissalare i peptidi per rimuovere sali e contaminanti (ad esempio, reagenti isobarici di massa non reagiti) su una colonna/punta di spin a fase inversa C18 per proteggere il sistema LC-MS.
- OPZIONALE: Frazionare (ad esempio, frazionamento a fase inversa a medio o alto pH) la miscela peptidica per una rilevazione più approfondita del proteoma tramite piattaforme manuali o automatiche. Utilizzare la fase stazionaria C18 contenente punte per frazionare basse quantità (1-10 μg) di digest peptidici.
- Essiccare la miscela peptidica a 60 °C in un concentratore sotto vuoto.
- Conservare la miscela peptidica a -80 °C fino alla misurazione.
- Per l'analisi a singola cellula mediante CE, riscaldare il campione a 60 °C per ~15 minuti per denaturare le proteine, quindi equilibrare il campione a temperatura ambiente (RT, ~5 min)18,21.
- Separare i peptidi
NOTA: Dopo aver estratto e digerito enzimaticamente le proteine, i peptidi risultanti vengono separati da nanoLC o CE e ionizzati da ESI per il sequenziamento mediante HRMS tandem. La separazione nanoLC in fase inversa è ideale per peptidi che accumulano da ~ 150 ng a ~ 1 μg per analisi. CE fornisce sensibilità complementare per peptidi che vanno dai femtogrammi a <100 ng. Various custom-built and commercial CE-ESI interfaces allow for ready coupling of CE to HRMS with robust performance35 e sono sempre più utilizzati per l'analisi a cella singola18,36,37.- Per separare utilizzando CE, seguire i passaggi 4.2.2-4.2.7.
NOTA: In quanto segue, viene descritto l'uso della piattaforma CE personalizzata per misurare i peptidi. I protocolli per costruire e utilizzare questo strumento CE sono stati forniti in precedenza38, insieme a un esperimento visualizzato sull'uso per piccole molecole20. In alternativa, queste misurazioni possono essere eseguite su un sistema CE commerciale, come AB SCIEX CESI, Agilent 7100 o equivalente. - Ricostituire il digesto proteico in 1-2 μL del solvente del campione, vortice per miscelare il campione e centrifugarlo a 10.000 x g per 2 minuti a pellettare i detriti cellulari.
NOTA: La rimozione dei detriti cellulari riduce al minimo la probabilità di intasamento del capillare CE, prolungando così la durata del sistema di separazione e aumentando la produttività della misura. - Inizializzare lo strumento CE-ESI lavando il capillare CE con il BGE.
- Convalidare le prestazioni strumentali utilizzando uno standard noto (ad esempio, citocromo C o BSA digest, peptidi di angiotensina).
NOTA: Si consiglia di valutare lo strumento in termini di accuratezza di massa, sensibilità di rilevamento, riproducibilità e gamma dinamica lineare di quantificazione prima di misurare campioni preziosi. Ulteriori note sulla convalida e la risoluzione dei problemi delle prestazioni CE-ESI-MS sono elencate altrove18,20,38. - Iniettare ~1-10 nL del campione nel capillare di separazione CE.
NOTA: Questo studio utilizza capillare di silice fusa lungo ~ 1 m (diametro interno / esterno 40/110 μm) con la configurazione del flusso della guaina pompata elettrocineticamente. Gli strumenti CE commerciali di solito richiedono la presentazione di 5-10 μL di campione in una microfiala per iniezione. La piattaforma CE 18,38 costruita su misura è compatibile con ~ 250 nL a 1 μL di campione depositato in una microfiala con caricamento del campione. - Trasferire l'estremità di ingresso del capillare di separazione CE nel BGE.
- Avviare la separazione elettroforetica aumentando gradualmente la tensione di separazione CE dalla terra (ad esempio, gradualmente oltre 1 minuto). Potenziali di 20-28 kV con corrente inferiore a ~10 μA garantiscono prestazioni strumentali stabili e riproducibili per l'analisi.
- Per separare utilizzando nanoLC, seguire i passaggi 4.2.9-4.2.12.
- Risospendere il campione peptidico nella fase A mobile. La concentrazione del campione e il suo volume iniettabile dipendono dal sistema LC e dalla colonna disponibili. In questo studio, ~ 250 ng-1 μg di digest proteico vengono iniettati in 1-20 μL di volume del campione su una colonna a letto impacchettato C18 (diametro interno 75 μm, dimensione delle particelle 2 μm con pori 100 Å, colonna di separazione lunga 25 cm).
- Trasferire il campione in un flaconcino LC.
NOTA: Assicurarsi che non vi siano bolle d'aria nel flaconcino, che potrebbero danneggiare la colonna analitica. Le fiale con inserti potrebbero essere utilizzate per campioni di tessuto a basso volume o di singole cellule. - Caricare ~ 200 ng a 2 μg di campione peptidico sulla colonna analitica C18.
NOTA: Opzionalmente, i peptidi possono essere caricati su una colonna trappola per la desalinizzazione prima della separazione analitica. Ad esempio, una colonna trappola C18 con diametro interno di 0,1 mm, dimensione delle particelle di 5 μm, dimensione dei pori di 100 Å, lunghezza di 20 mm. Dissalare i peptidi con tampone A al 100% ad una portata di 5 μL/min per 5 minuti prima che inizi il gradiente di separazione. - Separare i peptidi usando l'eluizione del gradiente. A una portata di 300 nL/min, il gradiente di 120 minuti utilizzato in questo studio è il seguente: 0-5 min 2% B, 5-85 min 2-35% B, 86-90 min 70% B, 91-120 min 2% B.
- Per separare utilizzando CE, seguire i passaggi 4.2.2-4.2.7.
- Ionizzare i peptidi con ESI
NOTA: Il capillare CE o nanoLC è più tipicamente accoppiato in una sorgente ESI per la ionizzazione. Le interfacce CE-ESI micro-flow (punta smussata) e nano-flusso (punta conica39 e pompa elettrocinetica36 ) sono state sviluppate in precedenza interfacce CE-ESI per il rilevamento ultrasensibile.- Fornire i peptidi di separazione in una sorgente di ioni elettrospray per la ionizzazione utilizzando un'interfaccia ESI commerciale o personalizzata. Per l'analisi CE-ESI-MS a singola cellula negli embrioni di Xenopus , utilizzare un'interfaccia a basso flusso pompata elettrocineticamente in cui l'uscita capillare CE è racchiusa in un emettitore borosilicato tirato.
- Controllare il flusso di liquido attraverso l'emettitore elettrospray utilizzando una telecamera e ispezionare visivamente la configurazione per eventuali perdite.
- Impostare la tensione dell'elettrospray su ~ 2,5 kV per avviare la sorgente ESI (rispetto alla terra terra).
- Garantire un nanospray stabile per l'analisi HRMS monitorando la corrente ionica totale. Regolare la tensione e la distanza dell'elettrospray dall'ingresso dell'HRMS per ottenere uno spruzzo stabile (deviazione standard relativa del <15% nell'intensità totale).
- Rileva i peptidi
NOTA: La rilevazione dei peptidi segue diverse considerazioni strumentali per i peptidi isobarici marcati con e senza tag di massa e dipende dal tipo di spettrometro di massa disponibile. Questo studio utilizza uno spettrometro di massa tribrido orbitrap secondo i seguenti passaggi.- Acquisire eventi MS1 con le impostazioni: Analyzer, orbitrap; Risoluzione spettrale, 120.000 larghezza intera a metà massimo (FWHM); tempo massimo di iniezione (IT), 50 ms; controllo automatico del guadagno (AGC), 4 x 105 conteggi; microscansioni, 1.
- Per sequenziare peptidi, ioni precursori di frammenti per il rilevamento nell'analizzatore trappola ionica utilizzando le impostazioni: modalità di frammentazione, dissociazione di collisione a più alta energia (HCD); gas di collisione, azoto; energia di collisione, 32% di energia di collisione normalizzata (NCE); massimo IT, 70 ms; AGC, 1 x 104 conteggi; microscansioni, 1.
- OPZIONALE: Quantificare i peptidi marcati TMT utilizzando HRMS tandem/multistadio (MS2/MS3). Per MS3 che utilizza la selezione sincrona dei precursori, le impostazioni strumentali tipiche sono le seguenti. Le scansioni monostadio (MS1) che rilevano gli ioni più abbondanti vengono dissociate tramite acquisizione dipendente dai dati utilizzando i parametri: modalità di frammentazione MS2 , dissociazione indotta da collisione (CID); gas di collisione, elio; energia di collisione, 35% NCE; analizzatore per ioni frammento, trappola ionica seguenti impostazioni: massimo IT, 50 ms; AGC, 5 x 104 conteggi; microscansioni, 1. Selezionare 10 ioni frammento MS2 e frammentarli con HCD in azoto (65% NCE). Rileva gli ioni del frammento MS3 utilizzando le seguenti impostazioni: Risoluzione Orbitrap 15.000 FWHM, IT massimo, 120 ms; AGC, 1 × 105 conteggi; microscansioni, 1.
NOTA: Diversi metodi e parametri di acquisizione MS possono essere utilizzati per i campioni etichettati seguendo le raccomandazioni del fornitore come descritto altrove11,40.
- Analizzare i dati
NOTA: Le proteine sono identificate e quantificate utilizzando pacchetti bioinformatici avanzati. La fedeltà delle identificazioni è calcolata utilizzando un database esca, espresso come tasso di falsa scoperta (FDR) a livello di peptidi e proteine.- Elaborare i dati utilizzando pacchetti software commerciali o open source (esaminati nel riferimento41). Confronta i dati grezzi con un database preparato concatenando il proteoma Xenopus 9.2 con il database PHROG derivato dall'mRNA42.
NOTA: I parametri di ricerca sono: enzima digestione, tripsina; scissioni mancate, fino a 2; modificazione variabile, ossidazione della metionina; modificazione statica, carbamidometilazione della cisteina; tolleranza di massa del precursore, 10 ppm; tolleranza di massa del frammento, 0,6 Da; lunghezza minima del peptide, 5; fedeltà di identificazione, FDR <1% per peptidi e proteine. Senza alchilazione ai peptidi, la carbamidometilazione come modifica statica è esclusa durante la ricerca nel database (ad esempio, per l'analisi di singole cellule). - Quantificare l'abbondanza proteica tramite strategie label-free43 o label-based44,45.
- OPZIONALE: Annotare le proteine per l'ontologia genica. È possibile utilizzare PantherDB46, Reactome47 o Xenbase23 .
- OPZIONALE: Quantificare l'abbondanza proteica e le differenze nelle abbondanze proteiche tra i tipi di cellule/tessuti utilizzando pacchetti software / strumenti web, come Trans-Proteomic Pipeline48, Perseus49 e Orange50.
NOTA: Ulteriori considerazioni sulla progettazione sperimentale e sulle opzioni software sono state esaminate altrove41,51. - OPZIONALE: Valutare ulteriormente i risultati utilizzando basi di conoscenza, come STRING52 e BioPlex Display 53 per le interazioni proteina-proteina note e PhosphoSiteplus54 per le fosforilazioni. Per analizzare motivi e domini rappresentati nel proteoma, utilizzare strumenti web come Simple Modular Architecture Research (SMART)55.
- Elaborare i dati utilizzando pacchetti software commerciali o open source (esaminati nel riferimento41). Confronta i dati grezzi con un database preparato concatenando il proteoma Xenopus 9.2 con il database PHROG derivato dall'mRNA42.
Access restricted. Please log in or start a trial to view this content.
Risultati
Questo protocollo ha permesso lo studio delle proteine nelle singole cellule e dei loro lignaggi mentre stabiliscono i tessuti negli embrioni di X. laevis. La Figura 1 illustra una di queste applicazioni dell'approccio per studiare le proteine nelle cellule fatali del tessuto neurale e l'ectoderma neurale appena indotto nell'embrione. Come mostrato nella Figura 1A, il flusso di lavoro bioanalitico ha integrato gli strumenti tradizionali della biologia c...
Access restricted. Please log in or start a trial to view this content.
Discussione
Questo protocollo consente la caratterizzazione dell'espressione proteica in linee cellulari identificate in embrioni della specie Xenopus . Derivando dalla HRMS, la metodologia combina una squisita specificità nell'identificazione molecolare, capacità di rilevamento multi-proteina senza sonde molecolari (di solito centinaia o migliaia di proteine diverse) e una capacità di quantificazione. L'adattabilità agli strumenti e ai flussi di lavoro classici nella (neuro)biologia cellulare e dello sviluppo espande l...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori non dichiarano interessi concorrenti.
Riconoscimenti
Siamo grati a Jie Li (University of Maryland, College Park) per le preziose discussioni sulla dissociazione embrionale e sulla FACS. Ringraziamo Vi M. Quach e Camille Lombard-Banek per l'assistenza nella preparazione dei campioni e nella raccolta dei dati in studi precedenti che esemplificano le applicazioni proteomiche evidenziate in questo protocollo. Parti di questo lavoro sono state sostenute dalla National Science Foundation con il numero di premio IOS-1832968 CAREER (a P.N.), dal National Institutes of Health con il numero di premio R35GM124755 (a P.N.), dall'Università del Maryland-National Cancer Institute Partnership Program (a P.N.) e dai premi di ricerca della COSMOS Club Foundation (ad A.B.B. e L.R.P.).
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| Acetonitrile (LC-MS-grade) | Fisher Scientific | A955 | |
| Agarose | ThermoFisher Scientific | R0492 | |
| Ammonium bicarbonate | Fisher Scientific | A643-500 | |
| Analytical Column | Thermo Scientific | 164941 | |
| Analytical microbalance | Mettler-Toledo | XSE105DU | |
| Automatic peptide fractionation platform | Agilent | 1260 Infinity II | |
| Borosilicate Capillaries | Sutter Instruments Co. | B100-50-10 | |
| Borosilicate Capillaries (for making Emmitters) | Sutter Instruments | B100-75-10 | |
| C18 spin columns (for desalting) | ThermoFisher Scientific | 89870 | |
| Camera ro monitor electrospray | Edmund Optics Inc. | EO-2018C | |
| Combretastatin A4 | Millipore Sigma | C7744 | |
| Commercial CESI system | AB SCIEX | CESI | |
| (Cyclohexylamino)-1-propanesulfonic acid (CAPS) | VWR | 97061-492 | |
| Cytochalasin D | Millipore Sigma | C8273 | |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable | ThermoFisher Scientific | D22910 | |
| Diothiothreitol | Fisher Scientific | FERR0861 | |
| Dumont #5 Forceps | Fine Science Tools | 11252-30 | |
| EDTA | Fisher Scientific | AAJ62786AP | |
| Epifluorescence light source | Lumencore | AURA III | |
| Eppendorf LoBing microcentrifuge tubes: protein | Fisher Scientific | 13-698-793 | |
| Formic acid (LC-MS-grade) | Fisher Scientific | A117-50 | |
| Freezer (-20 °C) | Fisher Scientific | 97-926-1 | |
| Freezer (-80 °C) | Thermo Scientific | TSX40086A | |
| Fused silica capillary | Molex | 1088150596 | |
| Heat Block | Benchmark | BSH300 | |
| High pressure liquid Chromatography System | ThermoFisher Scientific | Dionex Ultimate 3000 RSLC nanosystem | |
| High voltage power supply | Spellman | CZE1000R | |
| High-resolution Mass Spectrometer | ThermoFisher Scientific | Orbitrap Fusion Lumos Tribrid Mass Spectrometer | |
| HPLC caps | Thermo Scientific | C4013-40A | |
| HPLC Vials | Thermo Scientific | C4013-11 | |
| Illuminator e.g. Goosenecks | Nikon | C-FLED2 | |
| Ingenuity Pathway Analysis | Qiagen | ||
| Iodoacetamide | Fisher Scientific | AC122275000 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456-4 | |
| Microcapillary puller | Suttor Instruments | P-2000 | |
| Microinjector | Warner Instrument, Handem, CT | PLI-100A | |
| Micropippette puller | Sutter Instruments Co. | P-1000 | |
| MS data analysis software, commercial | ProteomeDiscoverer | ||
| MS data analysis software, opensource | MaxQuant | ||
| non-idet 40 substitute | Millipore Sigma | 11754599001 | |
| Petri dish 60 mm and 80 mm | Fisher Scientific | S08184 | |
| Pierce 10 µL bed Zip-tips (for desalting) | ThermoFisher Scientific | 87782 | |
| Pierce bicinchoninic acid protein assay kit | ThermoFisher Scientific | 23225 | |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 | |
| Pierce Trypsin Protease (MS Grade) | Fisher Scientific | PI90058 | |
| Protein LoBind vials | Eppendorf | 0030108434 , 0030108442 | |
| Refrigerated Centrifuge | Eppendorf | 5430R | |
| Refrigerated Incubator | Thermo Scientific | PR505755R/3721 | |
| sodium isethionate | Millipore Sigma | 220078 | |
| sodium pyrophosphate | Sigma Aldrich | 221368-100G | |
| Stainless steel BGE vial | Custom-Built | ||
| Stainless steel sample vials | Custom-Built | ||
| Stereomicroscope (objective 10x) | Nikon | SMZ 1270, SZX18 | |
| Sucrose | VWR | 97063-790 | |
| Syringe pumps (2) | Harvard Apparatus | 704506 | |
| Syringes (gas-tight): 500–1000 µL | Hamilton | 1750TTL | |
| Transfer pipettes (Plastic, disposable) | Fisher Scientific | 13-711-7M | |
| Trap Column | Thermo Scientific | 164750 | |
| Tris-HCl (1 M solution) | Fisher Scientific | AAJ22638AP | |
| Vacuum concentrator capable of operation at 4–10 °C | Labconco | 7310022 | |
| Vortex-mixer | Benchmark | BS-VM-1000 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| XYZ translation stage | Thorlabs | PT3 | |
| XYZ translation stage | Custom-Built |
Riferimenti
- Shoemaker, L. D., Kornblum, H. I. Neural Stem Cells (NSCs) and Proteomics. Molecular & Cellular Proteomics. 15 (2), 344-354 (2016).
- Cervenka, J., et al. Proteomic characterization of human neural stem cells and their secretome during in vitro differentiation. Frontiers in Cellular Neuroscience. 14, 612560(2021).
- Christian, J. L. Morphogen gradients in development: From form to function. Wiley Interdisciplinary Reviews. Developmental Biology. 1 (1), 3-15 (2012).
- Gurdon, J. B., Elsdale, T. R., M, F. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 182, 64-65 (1958).
- Harland, R. M., Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 27 (12), 507-515 (2011).
- Moody, S. A. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 119 (2), 560-578 (1987).
- Moody, S. A. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Developmental Biology. 122 (2), 300-319 (1987).
- Dale, L., Slack, J. M. W. Fate map for the 32-cell stage of Xenopus laevis. Development. 99 (4), 527-551 (1987).
- Sun, L. L., et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Analytical Chemistry. 88 (13), 6653-6657 (2016).
- Lombard-Banek, C., Moody, S. A., Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angewandte Chemie-International Edition. 55 (7), 2454-2458 (2016).
- Zhang, Y. Y., Fonslow, B. R., Shan, B., Baek, M. C., Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113 (4), 2343-2394 (2013).
- Sive, H. L., Grainger, R. M., Harland, R. M. Early development of Xenopus laevis: A laboratory manual. , Cold Spring Harbor Laboratory Press. New York. (2000).
- Briggs, J. A., et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 360 (6392), (2018).
- Gupta, M., Sonnett, M., Ryazanova, L., Presler, M., Wuhr, M. Quantitative proteomics of xenopus embryos I, sample preparation. Xenopus. Methods in Molecular Biology. Vleminckx, K. 1865, Humana Press Inc. NY. 175-194 (2018).
- Baxi, A. B., Lombard-Banek, C., Moody, S. A., Nemes, P. Proteomic characterization of the neural ectoderm fated cell clones in the Xenopus laevis embryo by high-resolution mass spectrometry. ACS Chemical Neuroscience. 9 (8), 2064-2073 (2018).
- Moody, S. A. Cell lineage analysis in Xenopus embryos. Methods in Molecular Biology. 135, 331-347 (2000).
- Sater, A. K., Moody, S. A. Using Xenopus to understand human diseases and developmental disorders. Genesis. 55 (1-2), 1-14 (2017).
- Lombard-Banek, C., Choi, S. B., Nemes, P. Enzyme Activity in Single Cells. Methods in Enzymology. Allbritton, N. L., Kovarik, M. L. 628, 263-292 (2019).
- Lombard-Banek, C., Moody, S. A., Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Frontiers in Cell and Developmental Biology. 4, 11(2016).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. Microprobe capillary electrophoresis mass spectrometry for single-cell metabolomics in live frog (Xenopus laevis) embryos. Journal of Visualized Experiments: JoVE. (130), e56956(2017).
- Lombard-Banek, C., Moody, S. A., Manzin, M. C., Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and zebrafish embryos. Analytical Chemistry. 91 (7), 4797-4805 (2019).
- Klein, S. L. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Developmental Biology. 120 (1), 299-304 (1987).
- Karimi, K., et al. Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Research. 46 (1), 861-868 (2018).
- Kakebeen, A. D., Chitsazan, A. D., Wills, A. E. Tissue disaggregation and isolation of specific cell types from transgenic Xenopus appendages for transcriptional analysis by FACS. Developmental Dynamics. 250 (9), 1381-1392 (2021).
- Garcia, B. A. What does the future hold for top down mass spectrometry. Journal of the American Society for Mass Spectrometry. 21 (2), 193-202 (2010).
- Toby, T. K., Fornelli, L., Kelleher, N. L. Progress in top-down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry. (Palo Alto Calif). 9 (1), 499-519 (2016).
- Zhang, Z. B., Dubiak, K. M., Huber, P. W., Dovichi, N. J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis Embryos. Analytical Chemistry. 92 (7), 5554-5560 (2020).
- Wisniewski, J. R. Microbial Proteomics: Methods and Protocols.Methods in Molecular Biology. Becher, D. 1841, Humana Press Inc. NY. 3-10 (2018).
- Hughes, C. S., et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Zhu, Y., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nature Communications. 9, 882(2018).
- Wessel, D., Flugge, U. I. A method for the quantitative recovery of protein in dilute-solution in the presence of detergents and lipids. Analytical Biochemistry. 138 (1), 141-143 (1984).
- Jiang, L., He, L., Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of Chromatography A. 1023 (2), 317-320 (2004).
- Hildonen, S., Halvorsen, T. G., Reubsaet, L. Why less is more when generating tryptic peptides in bottom-up proteomics. Proteomics. 14 (17-18), 2031-2041 (2014).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19, 161(2018).
- Drouin, N., et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: effective electrophoretic mobility for reproducible and robust compound annotation. Analytical Chemistry. 92 (20), 14103-14112 (2020).
- Sun, L. L., Zhu, G. J., Zhang, Z. B., Mou, S., Dovichi, N. J. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. Journal of Proteome Research. 14 (5), 2312-2321 (2015).
- DeLaney, K., Sauer, C. S., Vu, N. Q., Li, L. J. Recent advances and new perspectives in capillary electrophoresis-mass spectrometry for single cell "omics". Molecules. 24 (1), 21(2019).
- Nemes, P., Rubakhin, S. S., Aerts, J. T., Sweedler, J. V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nature Protocols. 8 (4), 783-799 (2013).
- Choi, S. B., Zamarbide, M., Manzini, M. C., Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. Journal of the American Society for Mass Spectrometry. 28 (4), 597-607 (2017).
- Pino, L. K., Rose, J., O'Broin, A., Shah, S., Schilling, B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions. 48 (5), 1953-1966 (2020).
- Chen, C., Hou, J., Tanner, J. J., Cheng, J. L. Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences. 21 (8), 25(2020).
- Peshkin, L., et al. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Developmental Cell. 35 (3), 383-394 (2015).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 13 (9), 2513-2526 (2014).
- Gygi, S. P., et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 17 (10), 994-999 (1999).
- Thompson, A., et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 75 (8), 1895-1904 (2003).
- Mi, H. Y., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive api. Nucleic Acids Research. 49, 394-403 (2021).
- Schmidt, E., et al. On the Move Federated Workshops. , Springer, Verlag. Berlin. 710-719 (2006).
- Deutsch, E. W., et al. Trans-Proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 9 (7-8), 745-754 (2015).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Demsar, J., et al. Orange: Data mining toolbox in Python. Journal of Machine Learning Research. 14, 2349-2353 (2013).
- Oberg, A. L., Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. Journal of Proteome Research. 8 (5), 2144-2156 (2009).
- Jensen, L. J., et al. STRING 8 - a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, 412-416 (2009).
- Schweppe, D. K., Huttlin, E. L., Harper, J. W., Gygi, S. P. BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data. Journal of Proteome Research. 17 (1), 722-726 (2018).
- Hornbeck, P. V., et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research. 43, 512-520 (2015).
- Letunic, I., Khedkar, S., Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Research. 49, 458-460 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie-International Edition. 60 (23), 12852-12858 (2021).
- Lombard-Banek, C., Reddy, S., Moody, S. A., Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Molecular & Cellular Proteomics. 15 (8), 2756-2768 (2016).
- Saha-Shah, A., et al. Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Analytical Chemistry. 91 (14), 8891-8899 (2019).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Analytical Chemistry. 89 (13), 7069-7076 (2017).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47, 442-450 (2019).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati