
Method Article
Une approche simple et efficace de construire Mutant Vaccinia vecteurs de virus
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
virus de la vaccine (VV) a été largement utilisé dans la recherche biomédicale et l'amélioration de la santé humaine. Cet article décrit une méthode simple, très efficace pour modifier le génome de VV en utilisant un système CRISPR-cas9.
Résumé
The CRISPR-associated endonuclease Cas9 can edit particular genomic loci directed by a single guide RNA (gRNA). The CRISPR/Cas9 system has been successfully employed for editing genomes of various organisms. Here we describe a protocol for editing the vaccinia virus (VV) genome in the cytoplasm of VV-infected CV-1 cells using the RNA-guided Cas9. RNA-guided Cas9 induces double-stranded DNA breaks facilitating homologous recombination efficiently and specifically in the targeted site of VV and a transgene can be incorporated into these sites by homologous recombination. By using a site-specific homologous vector with transgene(s), a N1L gene-deleted VV with the red fluorescence protein (RFP) gene incorporated in this region was generated with a successful recombination efficiency 10 times greater than that obtained from the conventional homologous recombination method. This protocol demonstrates successful use of RNA-guided Cas9 system to generate mutant VVs with enhanced efficiency.
Introduction
virus de la vaccine (VV) est un virus à ADN enveloppé appartenant à la famille des poxvirus et a joué un rôle crucial dans l'une des plus grandes réussites de la médecine de l'éradication de la variole. Dans l'ère post-éradication de la variole, VV a été développé comme un vecteur pour délivrer des gènes pour les vaccins contre le VIH et d' autres maladies infectieuses 1-7 en insérant des gènes provenant de divers agents pathogènes dans des vecteurs VV. VV a également été largement utilisé comme vecteur pour l' immunothérapie du cancer 14/8 en particulier pour le développement du virus de la vaccine oncolytique tumeur ciblée par la réplication. Afin de créer un vecteur de vaccin efficace avec une meilleure sélectivité pour les cellules cancéreuses, des modifications dans le génome viral sont nécessaires, y compris des deletions de gènes ou l'introduction de gènes thérapeutiques.
Avec le développement de la technologie de l'ADN et une meilleure compréhension de la biologie moléculaire et de la virologie, l'insertion d'ADN étranger dans VV a été initialement obtenird en utilisant la recombinaison homologue (RH) dans les années 1980 15. Cette méthode est encore largement utilisé pour la construction de vecteurs de VV. L'introduction de la modification génétique est obtenue en utilisant un vecteur navette pour RH, qui se recombine avec le génome de VV dans les cellules pré-infectées. Cependant, cette méthode est révélée être très inefficace (moins de 1% d' efficacité de la recombinaison homologue 16) et se traduit souvent par insertion aléatoire du marqueur de sélection dans des régions et / ou de perte du marqueur lors de l' expansion du virus non ciblés.
L'efficacité de l' ADN recombinaison homologue pour insérer l' ADN exogène à des loci génomiques peut être considérablement améliorée en présence de cassures double brin (CDB) 17. Par conséquent, la technologie qui peut induire des DSB à la cible loci détient un grand potentiel pour l'ingénierie du génome de VV.
Le système CRISPR-cas9 récemment développé est prometteur pour le déclenchement DSB dans une région du gène VV. CRISPR-cas9 est un ARN-nucléase guidée impliqué dans l' immunité adaptative contre phages envahisseurs et autres matériels génétiques étrangers 18-20. Il existe trois systèmes CRISPR-Cas dans une gamme d'espèces microbiennes 21. Le système de CRISPR-Cas de type II est largement utilisée pour la modification du génome des cellules eucaryotes et de grands virus. Il se compose des endonucléases d'ARN cas9 guidé (Streptococcus pyogenes de), un ARN simple de guidage (ARN sg) et la trans-activatrice crRNA (tracrRNA) 22-24. Le complexe cas9 / ARN sg reconnaît la 20 séquence nucléotidique génomique complémentaire qui précède une séquence 5'-NGG-3 'Protospacer motif adjacent (PAM) dans des cellules de mammifère 22, 23. Il a été utilisé avec succès pour la production efficace des cellules, des virus génétiquement modifiés des modèles et des animaux 22-32.
Le système CRISPR-cas9 a prouvé être un outil efficace pour cibler le génome en combinaison avec la recombinaison homologue dans le cytoplasme de VV infectés cellules CV-1s pour générer des virus de la vaccine mutantes 33, 34. Afin d'étendre l'application potentielle de cette méthode, nous présentons des informations détaillées sur la méthodologie de ce système. Le protocole décrit peut être utilisé pour créer un VV mutant avec une deletion de gène particulier et / ou le bras du virus mutant avec un transgène thérapeutique.
Protocole
1. Préparation de l'ARN cible et cas9 Constructs et réparation Donor Vector
- Clonage des ARNg
- Concevoir une séquence cible guide ARN ciblant le gène N1L du virus de la vaccine suivant le principe énoncé précédemment 25. Aligner la séquence cible guide ARN contre le génome du virus de la vaccine (dans ce cas, la souche Lister du virus de la vaccine) pour écarter toutes les séquences hors cible cibles gARN.
- Synthétiser et cloner oligos gARN dans un vecteur de clonage gARN comme décrit précédemment 25. Confirmer la séquence de chaque individu gARN dans le vecteur résultant par Sanger séquençage 33. Nom du gARN construction résultante comme gARN N2 33.
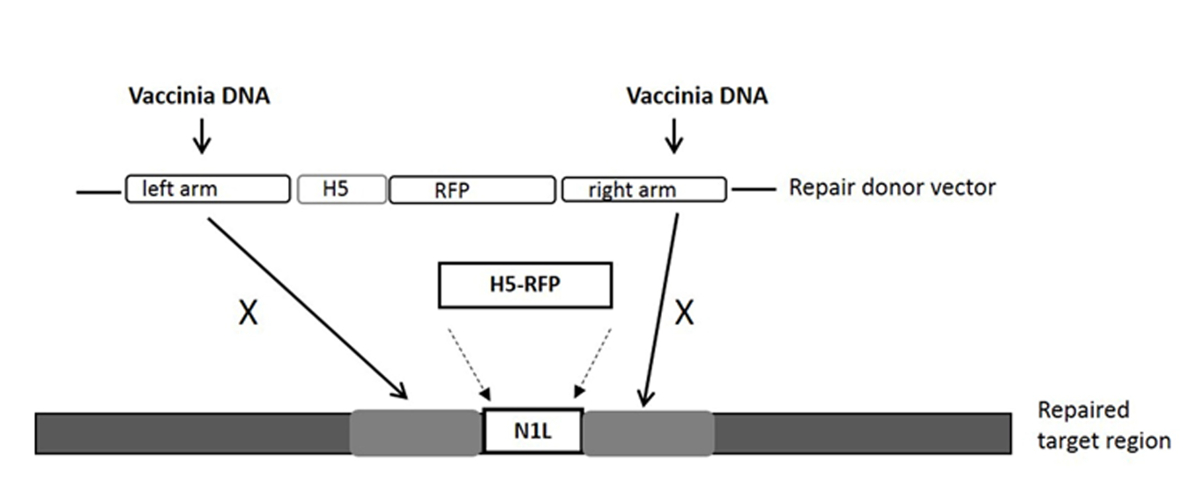
NOTE: Le site cible gARN est dans le gène N1L, et est dans la région entre le bras gauche et le bras droit que les couvertures de vecteur de réparation des donateurs (voir la figure 1).
- Cloner le gène dépourvu cas9 le signal de localisation nucléaire (NLS) dans lapST1374 vecteur et désigner la construction comme pST-cas9 33.
REMARQUE: Toute expression vecteur de clonage de mammifère peut être utilisé pour la construction du vecteur d'expression cas9. - Construction d'un vecteur de réparation des donateurs pour les RH
- Utiliser le gène N1L de VV en tant que région cible pour une modification 33. Faire en sorte que les longueurs des bras gauche et droit sont d'environ 500 à 600 paires de bases chacune, flanquant la région cible.
NOTE: Le bras / bras gauche à droite peut avoir jusqu'à 50 pb chevauchement avec la région cible (voir la figure 1). - Cloner le vecteur donneur région de réparation N1L comme décrit précédemment 33, 34 et désigner le vecteur donneur de réparation comme pT-N1L.
REMARQUE: Voir la Figure 1 pour le vecteur de réparation des donateurs et sa région cible. D'autres régions du VV peuvent être ciblés en utilisant la région (gène) spécifique de gARN (s) et le vecteur de réparation donneur spécifique à la région. Tout vecteur de clonage peut être utilisé pour la construction du vecteur donneur de réparation. - Expansion du plasmide
- Transformer le plasmide dans E. chimiquement compétente coli selon les instructions du fabricant. On purifie le plasmide en utilisant un kit d'extraction de plasmide se référant au protocole fourni par le fabricant.
2. Cellules Seeding (Jour 1)
- Maintenir cellules CV-1 (fibroblastes de rein de singe) en milieu de Eagle modifié par Dulbecco (DMEM) supplémenté avec du sérum de 5% de fœtus bovin (FBS), 100 U / ml de pénicilline et 100 ug / ml de streptomycine à 37 ° C avec 5% de CO 2.
- Trypsiniser cellules CV-1 qui sont confluentes à 80-90%
- Éliminer le milieu de culture de cellules de la fiole T-175 contenant des cellules CV-1. Rincer la monocouche avec 5 ml de PBS stérile pour éliminer le sérum résiduel et aspirez le PBS. Ajouter 2,5 ml de solution 1 × trypsine-EDTA pour couvrir les cellules et placer le ballon dans 37 ° C incubateur pendant environ 5 minutes pour aider le détachement des cellules.
- Ajouter10 ml de milieu de culture cellulaire (voir étape 2.1) pour les cellules en suspension par une action de pipetage doux. Compter le nombre de cellules en utilisant un hémocytomètre.
- Semis cellules dans des plaques 6 puits
- Seed 2 × 10 5 cellules CV-1 à 2 ml de milieu de culture cellulaire dans chaque puits d'une plaque à 6 puits le jour avant la transfection.
NOTE: Une étape de centrifugation supplémentaire pour éliminer la trypsine est pas nécessaire.
- Seed 2 × 10 5 cellules CV-1 à 2 ml de milieu de culture cellulaire dans chaque puits d'une plaque à 6 puits le jour avant la transfection.
3. Transfection de cas9 plasmide et gARN plasmide (Jour 2)
- Transfecter des cellules CV-1 (préparé dans l'étape 2.2.3.1) avec 0,5 pg du plasmide pST-cas9 et 0,5 pg de plasmide gARN N2 en utilisant un réactif de transfection selon les instructions du fabricant.
- Incuber les cellules pendant 24 h dans un incubateur (37 ° C avec 5% de CO 2), puis remplacer le milieu par 2 ml de milieu de culture cellulaire frais (décrit à l' étape 2.1).
4. L'infection de cellules CV-1 et Shuttle Donor Vecteur Transfection pour HR (Jour 3)
- On dilue le virus VVL15 de squelette avec du DMEM milieu de culture cellulaire à une concentration de 2 x 10 5 pfu / ml. 24 h post-transfection de cas9 et gARN plasmides, ajouter 100 ul du virus dilué dans chaque puits des transfectées cellules CV-1.
- infection post-virus 2 h, transfecter 1,0 pg vecteur donneur de navette dans les cellules infectées par VVL15 pour HR en utilisant un réactif de transfection selon les instructions du fabricant. Placer les cellules transfectées dans un incubateur à 37 ° avec 5% de CO2 pendant 24 heures.
5. récolte le virus recombinant (Jour 4)
- Après 24 h de transfection avec le vecteur navette donateurs pour les RH à l'étape 4.2, détacher les transfectées cellules CV-1 en utilisant un grattoir à cellules. Recueillir la suspension cellulaire dans un cryovial et stocker à -80 ° C pour une utilisation future.
REMARQUE: Essuyer tout milieu de culture cellulaire déversement avec un désinfectant immédiatement et essuyez ensuite avec 70% ethanol.
6. Purification de la VV modifiée (Premier tour)
- Au jour 1, des graines de 15 plaques à 6 puits avec 3 x 10 5 cellules CV-1 dans chaque puits.
- Le jour 2, décongeler la suspension cellulaire congelée de l'étape 5.1 dans un bain d'eau à 37 ° C pendant 3 min et vortexer les cryotubes vigoureusement pendant 30 s pour obtenir un lysat cellulaire sans enlever les débris cellulaires.
- Pour l'infection d'une plaque à 6 puits de cellules CV-1, diluer 1 pl de lysat cellulaire avec 3 ml de DMEM et ajouter 0,5 ml de lysat cellulaire dilué dans chaque puits de la plaque de 6 puits, au-dessus des médias déjà présent. Infect toutes les plaques de 6 puits préparés à l'étape 6.1.
REMARQUE: L'élimination des débris de cellules n'a pas été nécessaire. - Après deux jours de l' infection (ie, le jour 4), identifier les plaques de la DP-positif sous un microscope à fluorescence en utilisant 10X objectif et étiqueter les plaques sous la plaque à l' aide d' un stylo marqueur en encerclant l'emplacement de la plaque.
NOTE: Voir Figure 3 pour la plaque de DP positif représentatif.- Préparer six cryotubes étiquetés contenant 200 ul DMEM milieu de culture cellulaire dépourvu de sérum pour ramasser six plaques de DP-positives différentes. Aspirer le milieu de culture cellulaire provenant du puits de plaque marquée (s) (étape 6.3). Fixer une pointe de 200 pi à une pipette P200, régler le volume à 30 pi et de prendre ~ 10 pi de milieu de culture cellulaire d'un cryovial.
- Maintenez le bouton poussoir pipette sans relâcher le moyen et utilisez la pointe à gratter la surface d'une plaque marquée. Relâchez le bouton pipette de poussée pour soulever les cellules détachées et le transférer dans le cryovial contenant 190 pi de DMEM sans sérum.
- Prenez un autre 10 moyen ul du flacon avec les cellules grattées (de la dernière étape) et répétez la procédure de prise en charge de la même plaque de DP positif à trois reprises pour collecter autant de cellules grattées que possible. Transférer toutes les cellules dans le même flacon et conserver le flacon à -80 ° C.
NOTE: Vous pouvez également geler les cryotubes sur glace sèche pendant 10 minutes si le second tour de purification est effectuée immédiatement.
7. Purification de la VV modifiée (deuxième ronde)
- Seed 3 × 10 5 cellules CV-1 dans chaque puits de six plaques de 6 puits et la culture des cellules pendant 24 h.
- Décongeler les cryotubes congelés (de l'étape 6.3.3) à 37 ° C bain d'eau pendant 3 min, puis vortexer les cryotubes vigoureusement pendant 30 secondes pour obtenir un lysat cellulaire.
- Ajouter 0,5 pi de lysat cellulaire mixte à un puits d'une plaque à 6 puits. Infect une plaque de 6 puits pour chaque plaque. Incuber les plaques à 6 puits VV-infectées à 37 ° C avec 5% de CO 2 pendant 2 jours (Deuxième purification de plaque ronde). Ensuite, répétez la section 6.3.
NOTE: Plus de six plaques de DP-positifs peuvent être ramassés; deux ou plusieurs plaques de 6 puits peuvent être utilisées pour la purification de chaque plaque.
- Ajouter 0,5 pi de lysat cellulaire mixte à un puits d'une plaque à 6 puits. Infect une plaque de 6 puits pour chaque plaque. Incuber les plaques à 6 puits VV-infectées à 37 ° C avec 5% de CO 2 pendant 2 jours (Deuxième purification de plaque ronde). Ensuite, répétez la section 6.3.
8. Autres rounds suivants de Plaque Purification
- Continuez encore trois à cinq cycles de purification en suivant le même protocole décrit à l'étape 7 jusqu'à ce que toutes les plaques formées à partir d'une plaque positives semblent être RFP positifs au microscope.
NOTE: Cette plaque est alors considérée comme pure. - Une fois que la plaque est pure (Figure 4, toutes les cellules infectées exprimant RFP), récolte une plaque positive dans un cryovial et conserver à -80 ° C comme décrit dans la section 6.3. Vérifiez la plaque en suivant le protocole décrit à l'étape 9 et 10.
9. Développez le Purifié Plaque (s)
- Seed 3 × 10 5 cellules CV-1 dans chaque puits d'une plaque à 6 puits un jour avant l' infection par le virus.
- Décongeler le cryovial congelé de 8,2 dans un bain d'eau à 37 ° C pendant 3 min pour obtenir un lysat cellulaire. Ajouter tous les lysat (sans enlever les débris cellulaires) dans un puits d'une plaque à 6 puits de cellules CV-1 (tête de série à l'étape 9.1). Incuber les cellules CV-1 infectées à 37 ° C avec 5% De CO2 pendant 3 jours.
- Développer toutes les plaques purifiées (au moins trois plaques).
REMARQUE: La durée de l'infection varie de 1-3 jours en fonction de la quantité de virus dans le lysat.
- Développer toutes les plaques purifiées (au moins trois plaques).
- Récolter les cellules infectées par VV à l'aide d'un grattoir cellulaire lorsque plus de 50% des cellules montrent un effet cytopathique observé sous un microscope (cellules sont arrondies, facilement détaché et RFP positif). Recueillir les cellules individuelles avec un milieu de culture cellulaire dans un tube de 15 ml.
- Sédimenter les cellules par centrifugation à 300 g pendant 3 min. Retirer le surnageant et conserver le culot cellulaire à -80 ° C jusqu'à utilisation ultérieure ou d'utiliser immédiatement le culot cellulaire pendant l'étape 10.
Vérification de la modification du Mutant VV Utilisation PCR 10.
- Extraire l'ADN de VV le culot de cellules préparée à l'étape 9.3.1 en utilisant un kit d'extraction d'ADN en suivant le protocole du fabricant. Eluer ADN avec 200 pi d'eau distillée deux fois.
REMARQUE: Utiliser la moitié du volume de la pastille pour extraire l'ADN et le reste pour maintenir la production virale si le clone est vérifiée comme contenant la modification / insertion correcte. - Vérification de la deletion du gène N1L et l'insertion du gène de DP dans la région N1L.
- Amplifier un fragment d'ADN recouvrant le gène N1L (L025) et le gène L026 en utilisant des amorces conçues contre le gène N1L. Amplifier le gène de la DP en utilisant des amorces spécifique de la DP. Amplifier un fragment d'ADN de contrôle couvrant A52R par PCR en utilisant des amorces spécifiques de A52R.
REMARQUE: Voir la liste des matériaux pour des amorces spécifiques.- Effectuer la PCR en utilisant un kit maître mélange PCR. Mettre en place 25 pi de réaction de PCR en utilisant 2 pi de matrice d'ADN, dénaturer la réaction PCR à 94 ° C pendant 2 min, puis exécutez 30 cycles de PCR suivants comme suit: dénaturer l'ADN à 94 ° C pendant 15 s, puis recuit les amorces aux matrices d'ADN à 52 ° C pendant 15 s étendent la réaction de PCR à 72 ° C pendant 30 s.
- Amplifier un fragment d'ADN recouvrant le gène N1L (L025) et le gène L026 en utilisant des amorces conçues contre le gène N1L. Amplifier le gène de la DP en utilisant des amorces spécifique de la DP. Amplifier un fragment d'ADN de contrôle couvrant A52R par PCR en utilisant des amorces spécifiques de A52R.
- Résoudre les produits de PCR en utilisant un gel d'agarose à 1% 33. Capturez l'image de gel en utilisant un doc de gel UV. Si le PCR N1L est négatif et A52R et RFP RFP sont positifs, puis la plaque est correctement modifiée dans la région N1L.
- Utiliser le gène N1L de VV en tant que région cible pour une modification 33. Faire en sorte que les longueurs des bras gauche et droit sont d'environ 500 à 600 paires de bases chacune, flanquant la région cible.
Résultats
Pour la construction d'un nouveau vecteur de VV, le point de départ essentiel est de concevoir et de construire un vecteur navette donneur qui peut cibler une région particulière du génome. Dans cette étude, le gène VV N1L a été utilisé comme exemple cible. La cassette du vecteur navette de donneur pour la recombinaison et la région ciblée dans le VV sont présentés dans la figure 1. Afin d'améliorer l'efficacité de la recombinaison homologue, des plasmides exprimant cas9 et un gARN N1L spécifique ont été co-transfectées dans le CV -1 cellules 48 h avant d'effectuer la recombinaison homologue 33. Un jour (24 h) post-transfection, DP a été exprimée dans les cellules CV-1 (figure 2). Après l' infection des cellules CV-1 avec le lysat entier à partir de la recombinaison homologue (étape 5), DP-positives et les plaques négatives ont été observées à la fois dans les cellules CV-1 (figure 3). En suivant le protocole de purification décrit, un co pur de la plaque ULD être obtenue après 3-5 tours de purification. Comme on le voit sur la figure 4, toutes les plaques après l' infection par lysat cellulaire dérivée d'une plaque pur avec le virus mutant de la vaccine ont présenté un signal DP. Pour vérifier si le mutant pur VV avait la modification génétique correcte, la PCR a été utilisée pour confirmer l'absence du gène N1L spécifique, telle que représentée sur la figure 5. PCR du virus modifié a montré un signal positif pour DP et un signal d' absence pour N1L ( Figure 5 voies AG), tandis que les résultats de la PCR de N1L pour le virus de contrôle (Ctr) avec une région intacte N1L est positive. Les fragments d'ADN de contrôle de A52R ont été positifs pour tous les virus recombinants et le virus Ctr. L'efficacité des ressources humaines dans les plaques de la DP-positif est de 100% (6/6) dans cette expérience (il a été jusqu'à 94% dans les travaux antérieurs 34). Le procédé décrit ici a amélioré l'efficacité de la recombinaison par plus de 100 fois par rapport aux protocoles classiques , 33, 34.
contenu "fo: keep-together.within-page =" 1 ">
Figure 1. La cassette du vecteur navette donneur pour la recombinaison homologue, et la région cible dans le génome de VV le gène marqueur DP de purification est dirigée par le promoteur H5. Le vecteur de réparation des donateurs ciblant les résultats de la région N1L dans le gène DP étant incorporé dans la région N1L après recombinaison homologue. 'X' indique la séquence homologue dans le vecteur donneur de réparation qui remplacera la même séquence sur le génome du virus de la vaccine. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 2:. L' imagerie du CV-1 cellules un jour après recombinaison homologue Un jour AFTEimage de contraste de phase (100X de grossissement) B:. image de microscopie de fluorescence (100X de grossissement) r recombinaison homologue, les cellules infectées par VV et transfectées par le vecteur donneur de réparation A sont RFP-positive.. Barre d'échelle = 50 pm. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 3:. Une plaque de RFP-positif dans la première purification ronde de virus mutant Dans la purification du premier tour du virus mutant, la plaque de la DP-positive (encerclée avec la ligne rouge) avec la région cible de suppression sont entourés par des plaques formées par virus de type sauvage (entouré avec la ligne jaune) A:. image de contraste de phase (100X de grossissement) B:. fluorescence micrl'image oscopy (100x de grossissement original). Barre d'échelle = 50 pm. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Image de contraste de phase (100X de grossissement) B:. Image de microscopie de fluorescence (100X de grossissement) Figure 4:. A Le mutant pur VV Après 3-5 tours de purification, plaques pures ont été obtenues comme toutes les plaques ont été RFP-positive. . Barre d'échelle = 50 pm. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 5: Verification du mutant VV. ADN pur a été extrait de VV infecté cellules CV-1. La suppression de la région cible N1L a été vérifiée par PCR en utilisant des amorces N1L, l'insertion de la DP dans la région N1L a été confirmée par PCR en utilisant des amorces de la DP. Lanes AF correspondent à six plaques purifiées, voie G est une plaque de commande avec N1L suppression vérifié dans les travaux antérieurs 33, voie Ctr (contrôle) est le virus de la vaccine de type sauvage (avec la région N1L intacte). A52R gène a été amplifié en tant que gène de contrôle pour tous les échantillons. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Discussion
Il existe deux principaux objectifs en ce qui concerne la modification de VV à des fins thérapeutiques. La première consiste à supprimer un gène particulier, tel que le gène de la thymidine kinase (TK) pour atténuer le virus pour une utilisation thérapeutique anti-tumorale. L'autre consiste à armer VV avec un gène thérapeutique souhaité (par exemple, GM-CSF) ou un gène marqueur (tel que le gène de la luciférase). La réalisation ou l'autre de ceux-ci implique la suppression d'une région cible / gène. Une méthode de ligature directe de l' ADN 35 et un procédé d'encapsidation in vitro 36 ont été utilisées précédemment pour générer des virus de la vaccine mutants avec des modifications du gène TK. Cependant, ces méthodes ont une application en ce qui concerne la mutation d'autres régions dans le génome limité en raison d'un manque de sites d'enzymes de restriction uniques dans tout le génome. L'approche actuelle de la création VV mutant implique la transfection d'un vecteur navette (donneur) avec un marqueur de sélection (RFP ou GFP) dans les cellules CV-1 deux heures après l'infection par VV pour induire une recombinaison homologue incorporating le marqueur de sélection dans la zone cible. Sélection de plaques positives au marqueur est suivie de leur h après la transfection 24 purification. Le procédé de purification de plaque peut prendre jusqu'à 10 tours, 3-4 dernières semaines et conduit souvent à des virus recombinants indésirables avec des marqueurs de sélection insérés dans les régions hors-cible. ADN cassures double brin peuvent effectivement induire la recombinaison homologue dans des cellules de mammifères 37, et en mettant à profit ce mécanisme, l'efficacité de la génération VV mutant peut être grandement améliorée. Le système cas9 gARN-guidée a été utilisée avec succès dans l' édition du génome et améliore l'efficacité de la recombinaison homologue dans des organismes eucaryotes 22, 25. Récemment, dans les cellules hôtes les génomes d'adénovirus et de type I virus de l' herpès simplex ont été révisés par le cas9 gARN-guidée système 24. Il a récemment été démontré que le système CRISPR cas9 est un outil puissant pour l'édition de façon efficace le génome de VV et la construction d'un vecteur d'expression d'un VVnotamment gène.
Les deux cas9 N1L gARN-guidée et la méthode traditionnelle recombinaison homologue (RH) ont donné lieu à des événements RH réussies sur le même site cible de N1L. Fait intéressant, toutefois gARN guidée cas9 HR induite à des niveaux beaucoup plus élevés d'efficacité que la méthode RH traditionnelle. Ainsi, l'utilisation de cas9 gARN guidée élimine la nécessité de purifier le plus grand nombre de plaques pour obtenir VVS mutant. Dans ce travail, nous avons considérablement amélioré l'efficacité et le délai pour la génération de VV mutant en utilisant le système cas9 gARN guidée 33. Il convient de noter, une étape cruciale pour l'obtention d'une grande efficacité de la recombinaison homologue est de transfecter des cellules CV-1 avec les plasmides exprimant cas9 et gARN pendant 24 h avant d'effectuer la recombinaison homologue classique. L'intervalle de 24 h permet cas9 et gARN à être exprimées à un niveau raisonnable. En outre, la séquence gARN ciblage d'un gène particulier peut avoir besoin d'être optimisé car nous avons trouvé que les différentes séquences de t gARNgène argeting N1L varier leur efficacité 33, 34.
La limitation de la CRISPR / cas9 dans l'édition VV est le taux de plaques DP-positives dans le premier cycle de recombinaison faible. Pour obtenir un nombre suffisant de plaques de DP-positif contenant le virus recombinant, habituellement au moins dix plaques de 6 puits sont nécessaires, ce qui rend le dépistage du premier tour fastidieux. Par conséquent, il existe encore un besoin d'optimiser le protocole de surmonter cette limitation.
La petite taille des plaques de la DP-positif est un autre problème potentiel, ce qui est dû à un volume élevé de lysat utilisé pour infecter une plaque de 6 puits. Pour y remédier, un plus petit volume de lysat doit être utilisé pour infecter une plaque de 6 puits pour obtenir des plaques grand format de la DP-positive. L'utilisation d'un plus petit volume de lysat permettra également pour une meilleure séparation des plaques de la DP-positives de celles de la DP-négative. Par conséquent moins de cycles de purification de plaque sont nécessaires pour obtenir des plaques de DP-positives pures.
Off-cible effets sont toujours un problème indésirable dans l'édition du gène. CRISPR-cas9 a montré des effets hors-cible. Cependant, avec une conception soignée de gARN, hors cible effets peuvent être évités lors de l' édition du VV génomes 33, 34. Ceci est l'avantage particulier de l' utilisation du système cas9 gARN guidée pour l' édition du génome de VV. Compte tenu des promesses de VV, en utilisant le système CRISPR / cas9 permettra d'accélérer le développement de VVS mutantes pour la recherche biomédicale et la médecine translationnelle. En outre, un tel système permettrait également d'accélérer les découvertes de la biologie cellulaire, telles que la dissection des voies de signalisation utilisées par VV pour sa motilité à base d'actine.Déclarations de divulgation
Authors have no competing financial interests.
Remerciements
This work was supported by The MRC DPFS grant (MR/M015696/1) and Ministry of Sciences and Technology of China (2013DFG32080).
matériels
| Name | Company | Catalog Number | Comments |
| Plasmid #41824 | 41824 | Addgene | gRNA cloning vector |
| pST1374 | 13426 | Addgene | Cas9 cloning vector |
| pGEMT easy | A1360 | promega | repair donor vector cloning |
| One Shot TOP10 Chemically Competent E. coli | C4040-10 | Invitrogen | transformation |
| QIAprep Spin Miniprep Kit | 27106 | Qiagen | plasmid extraction |
| Dulbecco’s Eagle’s Medium (DMEM) | 11965-092 | Life Technologies | cell culture medium |
| fetal bovine serum (FBS) | SH30088.03HI | Hyclone | cell culture serum |
| penicillin, streptomycin | 15070-063 | Thermo Scientific | antibiotics |
| Effectene | 301425 | Qiagen | transfection |
| Thermo Scientific Nalgene Cryogenic vial; 2.0 mL | W-06754-96 | Thermo Scientific | collect virus |
| fluorescence microscope | Olympus BX51 Fluorescence | Olympus | find RFP-positive plque |
| DNeasy Blood & Tissue Kit | 69506 | Qiagen | DNA extraction |
| Extensor Long PCR ReddyMix Master Mix | AB-0794/B | Thermo Scientific | PCR reagent |
| 10 cm culture dish | Z688819-6EA | sigma | cell culture |
| 6-well plate | Z707759 | sigma | cell culture |
| cell scraper | C5981 | sigma | scrap cells |
| 1x Trypsin-EDTA solution | T4299 | sigma | trypsinize cells |
| Virkon | 95015661 | Anachem Ltd | desinfectant |
| Falcon tube | CLS430791-500EA | Sigma | hold cell suspension |
| N1L forward 5’-TATCTAGCAATGGACCGT-3’ | Sigma | PCR primer | |
| N1L reverse 5’-CCGAAGGTAGTAGCATGGA-3' | Sigma | PCR primer | |
| A52R forward 5’-ATAGGATTGTGTGCATGC-3’ | Sigma | PCR primer | |
| A52R reverse 5’-TTGCGGTATATGTATGAGGTG-3’ | Sigma | PCR primer | |
| RFP forward 5’- GCTACCGGACTCAGATCCA-3’ | Sigma | PCR primer | |
| RFP reverse 5’-CGCCTTAAGATACATTGATGAG-3’ | Sigma | PCR primer | |
| Argarose | A7431 | Sigma | resolve PCR product |
| G:BOX F3 | G:BOX F3 | Syngene | UV gel doc |
Références
- Baroudy, B. M., Venkatesan, S., Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell. 28, 315-324 (1982).
- Madigan, M. T., Martinko, J. M., Parker, J., Brock, T. D. Brock biology of microorganisms, 9th edn. , Prentice Hall. Upper Saddle River, NJ. (2000).
- Cooney, E. L., et al. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet. 337, 567-572 (1991).
- Mackett, M., Yilma, T., Rose, J. K., Moss, B. Vaccinia virus recombinants: expression of VSV genes and protective immunization of mice and cattle. Science. 227, 433-435 (1985).
- Legrand, F. A., et al. Induction of potent humoral and cell-mediated immune responses by attenuated vaccinia virus vectors with deleted serpin genes. J Virol. 78, 2770-2779 (2004).
- Panicali, D., Davis, S. W., Weinberg, R. L., Paoletti, E. Construction of live vaccines by using genetically engineered poxviruses: biological activity of recombinant vaccinia virus expressing influenza virus hemagglutinin. Proc Natl Acad Sci. 80, 5364-5368 (1983).
- Wiktor, T. J., et al. Protection from rabies by a vaccinia virus recombinant containing the rabies virus glycoprotein gene. Proc Natl Acad Sci. 81, 7194-7198 (1984).
- Gallucci, S., Matzinger, P. Danger signals: SOS to the immune system. Curr Opin Immunol. 13, 114-119 (2001).
- Matzinger, P. The danger model: a renewed sense of self. Science. 296, 301-305 (2002).
- Guo, Z. S., Bartlett, D. L. Vaccinia as a vector for gene delivery. Expert Opin Biol Ther. 4, 901-917 (2004).
- Kwak, H., Horig, H., Kaufman, H. L. Poxviruses as vectors for cancer immunotherapy. Curr Opin Drug Discov Devel. 6, 161-168 (2003).
- Tysome, J. R., et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 16, 1223-1233 (2009).
- Kirn, D. H., Wang, Y., Liang, W., Contag, C. H., Thorne, S. H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 68, 2071-2075 (2008).
- Park, B. H., et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 9, 533-542 (2008).
- Panicali, D., Paoletti, E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci. 79, 4927-4931 (1982).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. J Virol. 61, 1788-1795 (1987).
- Rouet, P., Smih, F., Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 14, 8096-8106 (1994).
- Jansen, R., Embden, J. D., Gaastra, W., Schouls, L. M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 43, 1565-1575 (2002).
- Brouns, S. J., et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 321, 960-964 (2008).
- van der Oost, J., Jore, M. M., Westra, E. R., Lundgren, M., Brouns, S. J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 34, 401-407 (2009).
- Jinek, M., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 343, 1247997(2014).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819-823 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-821 (2012).
- Bi, Y., et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 10, 1004090(2014).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339, 823-826 (2013).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 41, 9049-9061 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31, 827-832 (2013).
- Ran, F. A., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 154, 1380-1389 (2013).
- Niu, Y., et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 156, 836-843 (2014).
- Yin, H., et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 32, 551-553 (2014).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153, 910-918 (2013).
- Ma, Y., et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 24, 122-125 (2014).
- Yuan, M., et al. Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J Virol. 89, 5176-5179 (2015).
- Yuan, M., et al. A marker-free system for highly efficient construction of vaccinia virus vectors using CRISPR Cas9. Mol Ther Methods Clin Dev. 2, 15035(2015).
- Merchlinsky, M., Moss, B. Introduction of foreign DNA into the vaccinia virus genome by in vitro ligation: recombination-independent selectable cloning vectors. Virology. 190, 522-526 (1992).
- Timiryasova, T. M., Chen, B., Fodor, N., Fodor, I. Construction of recombinant vaccinia viruses using PUV-inactivated virus as a helper. Biotechniques. 31, 534-540 (2001).
- Johnson, R. D., Jasin, M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 29, 196-201 (2001).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.