
Method Article
Uma abordagem simples e eficiente para construir vectores de vírus mutante Vaccinia
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
vírus vaccinia (VV) tem sido amplamente utilizada na investigação biomédica e da melhoria da saúde humana. Este artigo descreve um método simples, altamente eficiente para editar o genoma VV utilizando um sistema CRISPR-Cas9.
Resumo
The CRISPR-associated endonuclease Cas9 can edit particular genomic loci directed by a single guide RNA (gRNA). The CRISPR/Cas9 system has been successfully employed for editing genomes of various organisms. Here we describe a protocol for editing the vaccinia virus (VV) genome in the cytoplasm of VV-infected CV-1 cells using the RNA-guided Cas9. RNA-guided Cas9 induces double-stranded DNA breaks facilitating homologous recombination efficiently and specifically in the targeted site of VV and a transgene can be incorporated into these sites by homologous recombination. By using a site-specific homologous vector with transgene(s), a N1L gene-deleted VV with the red fluorescence protein (RFP) gene incorporated in this region was generated with a successful recombination efficiency 10 times greater than that obtained from the conventional homologous recombination method. This protocol demonstrates successful use of RNA-guided Cas9 system to generate mutant VVs with enhanced efficiency.
Introdução
vírus vaccinia (VV) é um vírus DNA envelopado, pertencente à família poxvírus e tem desempenhado um papel crucial em uma das maiores conquistas da medicina da erradicação da varíola. Na era pós-erradicação da varíola, VV foi desenvolvido como um vetor para a entrega de genes para as vacinas contra o HIV e outras doenças infecciosas 1-7 através da inserção de genes de vários patógenos em vectores VV. VV também tem sido amplamente utilizado como um vector para a imunoterapia do cancro 8-14 especialmente para o desenvolvimento de vírus de vaccinia oncolítico replicar-alvo do tumor. A fim de criar um vector de vacina eficiente com uma selectividade melhorada para as células cancerígenas, são necessárias modificações dentro do genoma viral, incluindo deleções de genes ou introdução de genes terapêuticos.
Com o desenvolvimento da tecnologia de ADN e uma melhor compreensão da biologia molecular e virologia, a inserção de ADN estranho em VV foi originalmente atingird usando recombinação homóloga (HR) em 1980 15. Este método ainda é amplamente utilizado para a construção de vectores VV. Introdução da modificação genética, é conseguido através de um vector de transporte para a AR, que se recombina com o genoma de VV em células pré-infectados. No entanto, este método tem provado ser altamente ineficiente (inferior a 1% de eficiência de recombinação homóloga 16) e muitas vezes resulta na inserção aleatória do marcador de selecção em regiões e / ou perda do marcador sobre a expansão do vírus não-alvo.
A eficiência de ADN de recombinação homóloga para introduzir ADN exógeno em loci genómico pode ser dramaticamente aumentada na presença de quebras de cadeias duplas (DSB) 17. Portanto, a tecnologia que pode induzir LAP em loci alvo tem um grande potencial para a engenharia de genoma de VV.
O sistema CRISPR-Cas9 recentemente desenvolvido mostra a promessa para o desencadeamento de LAP em qualquer região do gene VV. CRISPR-Cas9 é uma RNA-nuclease guiada envolvidos na imunidade adaptativa contra invasores fagos e outros materiais genéticos estranhos 18-20. Existem três sistemas CRISPR-cas em uma variedade de espécies microbianas 21. O sistema CRISPR-cas tipo II é amplamente utilizado para a edição do genoma de células eucarióticas e vírus de grandes dimensões. Consiste na Cas9 endonuclease guiada por ARN (de Streptococcus pyogenes), um ARN de guia (sgRNA) e o crRNA trans-activação (tracrRNA) 22-24. O complexo Cas9 / sgRNA reconhece a sequência genómica de 20 nucleótidos complementares anterior uma sequência 5'-NGG-3 'Protospacer motivo adjacente (PAM) em células de mamífero 22, 23. Ela tem sido usada com sucesso para a geração eficaz de células, vírus geneticamente modificados e modelos animais 22-32.
O sistema CRISPR-Cas9 tem provado ser uma ferramenta eficiente para a segmentação genoma em combinação com recombinação homóloga no citoplasma de VV infectadas células CV-1s para gerar vírus vaccinia mutantes 33, 34. A fim de alargar o potencial de aplicação deste método, apresentamos informações detalhadas sobre metodologia deste sistema. O protocolo descrito pode ser usado para criar um mutante de VV com uma deleção do gene particular e / ou armar o vírus mutante com um transgene terapêutico.
Protocolo
1. Preparação de Alvo RNA e Cas9 Constrói e Reparação Dador de Vector
- Clonagem de gRNAs
- Projetar uma sequência alvo guia-RNA alvo o gene N1L do vírus vaccinia seguindo o princípio afirmado anteriormente 25. Alinhar a sequência alvo de ARN-guia contra o genoma do vírus de vaccinia (neste caso, Lister estirpe de vírus da vacina) para descartar quaisquer sequências alvo gRNA fora do alvo.
- Sintetizar e clonar os oligos gRNA em um vector de clonagem gRNA como previamente descrito 25. Confirmar a sequência de cada indivíduo gRNA no vector resultante por Sanger de sequenciação 33. Nomeie o gRNA construção resultante como gRNA N2 33.
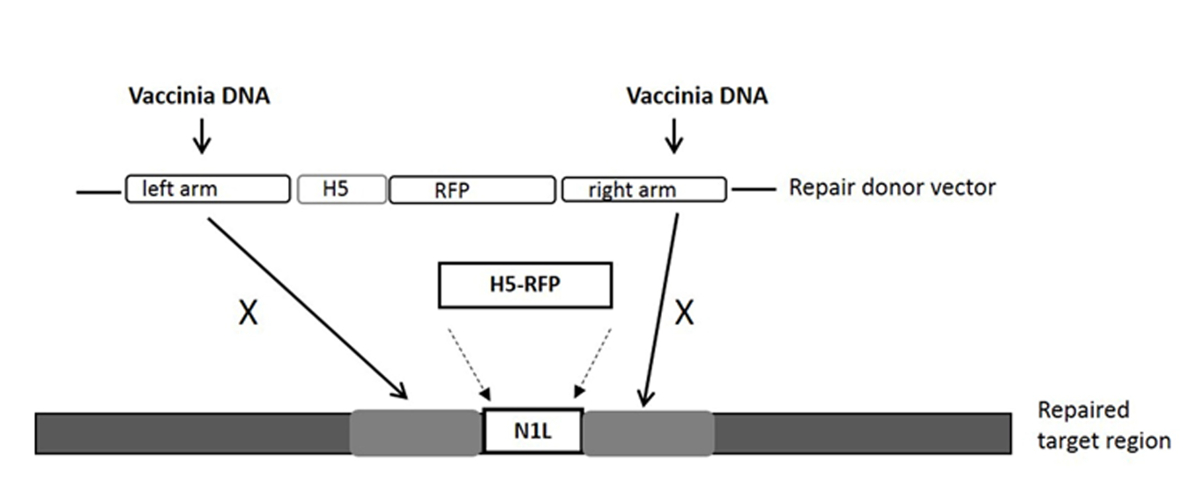
NOTA: O local alvo é gRNA dentro do gene N1L, e é na região entre o braço esquerdo e do braço direito que as tampas vector reparação doador (ver Figura 1).
- Clonar o gene Cas9 falta o sinal de localização nuclear (NLS) navector pST1374 e designar a construção como pST-Cas9 33.
Nota: Quaisquer clonagem de expressão vector de mamífero pode ser utilizado para a construção do vector de expressão Cas9. - Construção de vector de reparação dos doadores para HR
- Use o gene N1L de VV como região-alvo para a modificação 33. Assegure-se que os comprimentos dos braços esquerdo e direito são cerca de 500-600 pb cada, e que flanqueiam a região alvo.
NOTA: O braço / braço direito esquerdo pode ter até 50 sobreposição bp com a região de destino (ver Figura 1). - Clonar o reparo região doadora vector N1L como descrito anteriormente 33, 34 e designar o vetor doador de reparação como pT-N1L.
Nota: Consulte a Figura 1 para o vetor de reparação doador e sua região alvo. Outras regiões da VV pode ser alvejado usando região (gene) espec�ico gRNA (s) e vector de reparação dador específico de região. Qualquer vector de clonagem pode ser utilizado para a construção do vector dador de reparação. - Expansão de plasmídeo
- Transformar o plasmídeo em E. quimicamente competente coli de acordo com as instruções do fabricante. Purifica-se o plasmídeo utilizando um kit de extracção de plasmídeo referindo-se o protocolo fornecido pelo fabricante.
2. Células Sementeira (Dia 1)
- Manter a células CV-1 (fibroblastos de rim de macaco) em Meio de Eagle Modificado por Dulbecco (DMEM) suplementado com 5% de soro fetal de bovino (FBS), 100 U / mL de penicilina, e 100 ug / mL de estreptomicina, a 37 ° C com 5% de CO 2.
- células trypsinize CV-1, que são 80-90% confluentes
- Remover o meio de cultura de células a partir do balão T-175, contendo células CV-1. Lavar a monocamada com 5 mL de PBS estéril para remover o soro residual e aspirar o PBS. Adicionar 2,5 mL de uma solução 1 × Tripsina-EDTA para cobrir as células e colocar o balão em 37 ° C incubadora durante aproximadamente 5 min para auxiliar a separação celular.
- Adicionar10 ml de meio de cultura celular (ver passo 2.1) para suspender as células por acção da pipeta suave. Conte o número de células usando um hemocitômetro.
- Semeando as células em placas de 6 poços
- Seed 2 x 10 5 células CV-1 em 2 mL de meio de cultura de células em cada poço de uma placa de 6 poços um dia antes da transfecção.
NOTA: Um passo adicional de centrifugação para remover a tripsina não é necessária.
- Seed 2 x 10 5 células CV-1 em 2 mL de meio de cultura de células em cada poço de uma placa de 6 poços um dia antes da transfecção.
3. A transfecção do plasmídeo e Cas9 gRNA plasmídeo (Dia 2)
- transfectar células CV-1 (preparado no passo 2.2.3.1) com 0,5 ug de plasmídeo pST-Cas9 e 0,5 ug de plasmídeo gRNA N2 usando um reagente de transfecção de acordo com as instruções do fabricante.
- Incubam-se as células durante 24 h numa incubadora (37 ° C com 5% de CO 2) e, em seguida substituir o meio com 2 mL de meio de cultura celular fresco (descrito no passo 2.1).
4. A infecção de células CV-1 e Shuttle Donor Vector transfecção para HR (dia 3)
- Dilui-se a espinha dorsal vírus VVL15 com meio de cultura celular DMEM a uma concentração de 2 x 10 5 pfu / mL. 24 h pós-transfecção dos plasmídeos Cas9 e gRNA, adiciona-se 100 ul de vírus diluído em cada poço de células CV-1 transfectadas.
- 2 h pós-infecção do vírus, transfectar 1,0 ug de transporte Dador de Vector em células infectadas para VVL15-RH usando um reagente de transfecção de acordo com as instruções do fabricante. Colocar as células transfectadas de uma incubadora a 37 ° C com 5% de CO 2 durante 24 h.
5. recolher o vírus recombinante (Dia 4)
- Após 24 horas de transfecção com o vector de transporte dador para HR na etapa 4.2, separar as células CV-1 transfectadas, utilizando um raspador de células. Recolha a suspensão de células para um frasco de congelação e armazenar a -80 ° C para uso futuro.
NOTA: Limpe qualquer derrame de meio de cultura celular com desinfectante imediatamente e, em seguida, limpe novamente com 70% Ethanol.
6. Purificação da VV Modificado (Primeira Rodada)
- No dia 1, as sementes 15 placas de 6 cavidades com 3 x 10 5 células CV-1 em cada poço.
- No dia 2, descongelar a suspensão de células congeladas a partir do passo 5.1 em um banho de água a 37 ° C durante 3 minutos e agitar com vortex os criotubos vigorosamente durante 30 segundos para obter lisado celular sem remover os detritos celulares.
- Para a infecção de uma placa de 6 poços de células CV-1, diluir 1 mL de lisado de células com 3 mL de DMEM e adicionar 0,5 ml de ligado celular diluído em cada poço da placa de 6 cavidades, na parte superior dos meios de comunicação já presente. Infect todas as placas de 6 poços, preparado no passo 6.1.
NOTA: A remoção de detritos de células não é necessário. - Após dois dias de infecção (isto é, no dia 4), identificar placas RFP-positiva sob um microscópio de fluorescência utilizando 10X lente objectiva e rotular as placas por baixo da placa utilizando uma caneta marcador circundando a localização da placa.
NOTA: Ver Figura 3 para a placa representante RFP-positivo.- Prepare seis cryovials rotulados contendo meio de cultura de células 200 mL sem soro DMEM para pegar seis placas RFP-positivas diferentes. Aspirar o meio de cultura de células a partir da placa bem com rotulada (s) (passo 6.3). Anexar uma ponta de 200 mL de uma pipeta P200, definir o volume em 30 mL e tomar ~ meio de cultura de células 10 mL de uma cryovial.
- Segure o botão pipeta sem liberar a médio e use a ponta a arranhar a área de uma placa rotulada. Solte o botão de pipeta para levantar as células destacadas e transferi-lo para o cryovial contendo 190 mL de DMEM isento de soro.
- Tome um outro 10 � de meio do frasco com as células riscados (da última etapa) e repita o procedimento de pegar a mesma placa RFP-positivas três vezes para recolher o máximo de células riscado quanto possível. Transferir todas as células no mesmo frasco e armazenar o frasco a -80 ° C.
NOTA: Como alternativa, congelar os criotubos em gelo seco durante 10 minutos, se a segunda ronda de purificação é executada imediatamente.
7. A purificação do VV Modificado (Segunda Rodada)
- Seed 3 x 10 5 células CV-1 em cada poço de seis placas de 6 poços e cultura das células durante 24 h.
- Descongelar as criotubos congeladas (a partir do passo 6.3.3) em 37 ° C banho de água durante 3 min, em seguida, os criotubos vortex vigorosamente durante 30 segundos para obter lisado celular.
- Adicionar 0,5 mL do lisado celular mista para um poço de uma placa de 6 poços. Infect uma placa de 6 poços para cada placa. Incubar as placas de 6 poços VV infectadas a 37 ° C com 5% de CO 2 durante 2 dias (Segunda purificação de placa redonda). Em seguida, repita a secção 6.3.
NOTA: Mais de seis placas de RFP-positivos podem ser apanhados; duas ou mais placas de 6 poços pode ser usado para a purificação de cada placa.
- Adicionar 0,5 mL do lisado celular mista para um poço de uma placa de 6 poços. Infect uma placa de 6 poços para cada placa. Incubar as placas de 6 poços VV infectadas a 37 ° C com 5% de CO 2 durante 2 dias (Segunda purificação de placa redonda). Em seguida, repita a secção 6.3.
8. novas rodadas de Plaque Purificação
- Continue mais três a cinco ciclos de purificação de acordo com o mesmo protocolo descrito no passo 7 até que todas as placas formadas a partir de uma placa positiva pareceu ser RFP-positiva sob um microscópio.
NOTA: Esta placa, em seguida, é considerado puro. - Uma vez que a placa é pura (Figura 4, todas as células infectadas que expressam RFP), colher uma placa positiva em um cryovial e armazenar a -80 ° C, tal como descrito na secção 6.3. Verifique a placa seguindo o protocolo descrito no passo 9 e 10.
9. Expanda a placa (s) Purificada
- Seed 3 x 10 5 células CV-1 em cada poço de uma placa de 6 poços um dia antes da infecção pelo vírus.
- Descongelar o criotubo gelado de 8,2 num banho de água a 37 ° C durante 3 minutos para se obter lisado celular. Adicionar todo o lisado (sem necessidade de remover os detritos celulares) para um poço de uma placa de 6 poços de células CV-1 (semeadas no passo 9.1). Incubar as células CV-1 infectadas a 37 ° C com 5% De CO 2 durante 3 dias.
- Expandir todas as placas purificadas (pelo menos três placas).
NOTA: O tempo de infecção varia de 1-3 dias, dependendo da quantidade de vírus no lisado.
- Expandir todas as placas purificadas (pelo menos três placas).
- Colher as células VV infectadas utilizando um raspador de células, quando mais de 50% das células mostram efeito citopático observado sob um microscópio (as células são arredondados, facilmente separado e RFP positivo). Recolher as células destacadas em conjunto com o meio de cultura de células para um tubo de 15 mL.
- Agregar as células por centrifugação a 300 g durante 3 min. Remover o sobrenadante e manter a pelete de células a -80 ° C até à sua utilização ou usar o sedimento celular imediatamente para o passo 10.
10. Verificação da Modificação do Mutant VV Usando PCR
- Extracto de ADN VV a partir do sedimento de células, preparado no passo 9.3.1, utilizando um kit de extracção de ADN seguindo o protocolo do fabricante. Elui-se o ADN com 200 uL de água duplamente destilada.
NOTA: Use da metade do volume do pelete para extrair o ADN e manter o resto para a produção virai se o clone é verificado como contendo a correcta modificação / inserção. - Verificação da deleção do gene de N1L e da inserção do gene para a região de RFP N1L.
- Amplificar um fragmento de DNA abrangendo o gene N1L (L025) e o gene L026 utilizando iniciadores concebidos contra o gene N1L. Amplificar o gene RFP utilizando iniciadores espec�icos RFP. Amplificar um fragmento de ADN de controlo que mede A52R por PCR utilizando iniciadores específicos de A52R.
NOTA: Veja a lista de materiais para primers específicos.- Realizar a PCR usando um kit mestre de mistura de PCR. Defina-se 25 ul de reacção de PCR, utilizando 2 mL de modelo de DNA, desnaturar a reacção de PCR a 94 ° C durante 2 min, e, em seguida, executar 30 ciclos de PCR seguintes etapas: desnaturar o ADN, a 94 ° C durante 15 s, em seguida, recozer os iniciadores para os moldes de ADN a 52 ° C durante 15 s prolongar a reacção de PCR a 72 ° C durante 30 s.
- Amplificar um fragmento de DNA abrangendo o gene N1L (L025) e o gene L026 utilizando iniciadores concebidos contra o gene N1L. Amplificar o gene RFP utilizando iniciadores espec�icos RFP. Amplificar um fragmento de ADN de controlo que mede A52R por PCR utilizando iniciadores específicos de A52R.
- Resolver os produtos de PCR utilizando um gel de agarose a 1% 33. Capturar a imagem do gel usando um doc gel UV. Se o N1L de PCR for negativo e A52R e RFP PCRs são positivos, então a placa está correctamente modificado na região do N1L.
- Use o gene N1L de VV como região-alvo para a modificação 33. Assegure-se que os comprimentos dos braços esquerdo e direito são cerca de 500-600 pb cada, e que flanqueiam a região alvo.
Resultados
Para a construção de um novo vector VV, o ponto de partida chave é projetar e construir um vetor de transporte de doadores que podem alvejar uma região específica do genoma. Neste estudo, o gene de VV N1L foi usado como um exemplo de destino. A cassete do vector de transporte dador para a recombinação e a região alvo no VV são mostrados na Figura 1. A fim de aumentar a eficiência da recombinação homóloga, os plasmídeos que expressam Cas9 e um gRNA específicos de N1L foram co-transfectados na CV -1 células 48 h antes de se realizar a recombinação homóloga 33. Um dia (24 h) após a transfecção, RFP foi expresso em células CV-1 (Figura 2). Após a infecção de células CV-1 com todo o lisado proveniente da recombinação homóloga (passo 5), as placas negativas RFP-positivas e foram observados tanto em células CV-1 (Figura 3). Seguindo o protocolo de purificação descrito, uma placa de co puro uld ser obtido depois de 3-5 ciclos de purificação. Como mostrado na Figura 4, todos os placas após a infecção com lisado de células derivadas a partir de uma placa pura com o vírus vaccinia mutantes apresentaram um sinal RFP. Para verificar se o mutante pura VV tinha a modificação genética correcta, PCR foi utilizado para confirmar a ausência do gene N1L alvo, como se mostra na Figura 5. Por PCR do vírus modificado mostrou um sinal positivo para RFP e um sinal de ausente para N1L ( Figura 5 pista AG), enquanto que os resultados de PCR de N1L para o vírus de controlo (CTR) com uma região intacta N1L é positivo. Os fragmentos de ADN de controlo de A52R foram positivos para todos os vírus recombinantes do vírus e Ctr. A eficiência de RH em placas RFP-positivo é de 100% (6/6) nesta experiência (foi até 94% no trabalho anterior 34). O método aqui descrito melhorou a eficiência de recombinação em mais de 100 vezes em comparação com os protocolos convencionais 33, 34.
conteúdo "fo: manter-together.within-page =" 1 ">
Figura 1:. A cassete do vector de transporte dador para a recombinação homóloga, e a região alvo no genoma VV A purificação RFP gene marcador é dirigido pelo promotor de H5. O vector de reparação dos doadores destinados à região resultados N1L no gene RFP serem incorporadas a região N1L após recombinação homóloga. 'X' indica a sequência homóloga no vector dador de reparação que irá substituir a mesma sequência no genoma do vírus vaccinia. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2:. Imagiologia das células CV-1 um dia após a recombinação homóloga Um dia afteR recombinação homóloga, as células infectadas com VV e transfectadas pelo vector dador de reparação são RFP-positiva A:. imagem de contraste de fase (original 100X). B: A microscopia de fluorescência imagem (original 100X). Barra de escala = 50 mm. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3:. Uma placa RFP-positivo na primeira purificação rodada de vírus mutante Na purificação na primeira rodada do vírus mutante, a placa RFP-positivo (circulado com linha vermelha) com a eliminação região alvo está rodeado por placas formadas pela imagem de contraste de fase (original 100X) B:: Um vírus de tipo selvagem (circulado com linha amarela).. micr fluorescênciaimagem oscopy (original 100X). Barra de escala = 50 mm. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4:. O mutante pura VV Após 3-5 ciclos de purificação, placas puras foram obtidos como todas as placas eram RFP-positiva A:. Imagem de contraste de fase (original 100X). B: A microscopia de fluorescência imagem (original 100X) . Barra de escala = 50 mm. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: VerificatioN pura do mutante VV. O DNA foi extraído a partir de células infectadas com VV CV-1. A deleção da região alvo N1L foi verificada por PCR, utilizando iniciadores N1L, a inserção de SDP na região N1L foi confirmada por PCR utilizando os iniciadores RFP. Lanes AF correspondem a seis placas purificadas, pista G é uma placa de controle com N1L exclusão verificado em trabalhos anteriores 33, pista Ctr (controle) é o vírus vaccinia tipo selvagem (com a região N1L intacta). A52R gene foi amplificado como o gene de controle para todas as amostras. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Há dois objetivos principais relativamente à modificação da VV para fins terapêuticos. Uma consiste em eliminar um gene particular, tal como o gene da timidina quinase (TK) para atenuar o vírus para o anti-tumoral uso terapêutico. A outra é o braço VV com um gene terapêutico desejado (por exemplo, GM-CSF) ou um gene marcador (por exemplo, gene da luciferase). Atingir qualquer um destes envolve a eliminação de uma região alvo / gene. Um método de ligação directa de ADN 35 e um método de empacotamento in vitro 36 Foram utilizados anteriormente para gerar vírus vaccinia mutantes com alterações do gene TK. No entanto, estes métodos têm aplicação limitada em relação a mutação de outras regiões do genoma, devido à falta de locais de enzimas de restrição exclusivos, em todo o genoma. A actual abordagem para a criação de VV mutante envolve a transfecção de um vector de transporte (dador) com um marcador de selecção (RFP ou GFP) em células CV-1 de duas horas após a infecção com VV para induzir uma incorpor recombinação homólogang do marcador de selecção para a região alvo. Colecção de placas de marcadores positivos é seguida por sua h pós-transfecção de purificação 24. O processo de purificação de placa pode levar até 10 rodadas, últimas 3-4 semanas e, muitas vezes leva a vírus recombinantes com marcadores de selecção indesejáveis inseridas em regiões fora do alvo. ADN quebras de cadeia dupla pode efectivamente induzir a recombinação homóloga em células de mamíferos 37, e do aproveitamento deste mecanismo, a eficiência de geração de VV mutante pode ser bastante melhorada. O sistema Cas9 gRNA-guiada tem sido empregada com sucesso na edição do genoma e aumenta a eficiência de recombinação homóloga em organismos eucarióticos 22, 25. Recentemente, em células hospedeiras os genomas de adenovírus e tipo I vírus herpes simplex foram editadas pelo Cas9 gRNA-guiada sistema de 24. Foi demonstrado recentemente que o sistema CRISPR Cas9 é uma ferramenta poderosa para a edição de forma eficiente no genoma VV e construção de um vector para a expressão de um VVgene particular.
Ambos N1L gRNA-guiada ao método tradicional recombinação homóloga (HR) Cas9 e resultou em eventos bem sucedidas de RH no mesmo local alvo de N1L. É interessante notar, no entanto gRNA-guiado Cas9 AR induzida em níveis muito mais elevados de eficiência do que o método tradicional de RH. Assim, a utilização de Cas9 gRNA-guiado elimina a necessidade de purificar o maior número de placas para obter mutantes VV. Neste trabalho, nós melhorou significativamente a eficiência e prazo para geração de VV mutante usando o sistema Cas9 gRNA-guiada 33. De nota, um passo crítico para a obtenção de uma elevada eficiência da recombinação homóloga é para transfectar células CV-1 com os plasmídeos que expressam Cas9 e o gRNA durante 24 h antes de realizar a recombinação homóloga convencional. O intervalo de 24 h permite Cas9 e gRNA a ser expressa em um nível razoável. Além disso, a sequência de direccionamento gRNA de um determinado gene pode ter de ser optimizada, uma vez que descobrimos que os diferentes sequências t gRNAgene argeting N1L variar na sua eficácia 33, 34.
A limitação para a CRISPR / Cas9 na edição VV é a baixa taxa de placas RFP positivo na primeira rodada de recombinação. Para se obter um número suficiente de placas RFP-positivo contendo vírus recombinante, geralmente, pelo menos, dez placas de 6 poços são necessárias, o que torna primeira ronda de rastreio tedioso. Assim, ainda existe uma necessidade para optimizar o protocolo de ultrapassar esta limitação.
O pequeno tamanho de placas RFP-positivo é um outro problema potencial, o que é devido a um elevado volume de lisado utilizado para infectar uma placa de 6 poços. Para superar isto, um menor volume de lisado deve ser utilizado para infectar uma placa de 6 poços para se obter placas de tamanho maior RFP-positiva. Usando um pequeno volume de lisado permitirá também para uma melhor separação das placas RFP-positivo de os RFP-negativo. Consequentemente menos rondas de purificação de placas são necessários para obter placas RFP-positivos puros.
Off-alvo efeitos são sempre um problema indesejado na edição gene. CRISPR-Cas9 tem mostrado efeitos fora do alvo. No entanto, com design cuidadoso de gRNA, longe do alvo efeitos podem ser evitados durante a edição do VV genomas 33, 34. Este é o benefício particular de usar o sistema Cas9 gRNA-guiada para editar o genoma de VV. Dadas as promessas de VV, usando o sistema CRISPR / Cas9 irá acelerar o desenvolvimento de VV mutantes para a investigação biomédica e em medicina translacional. Além disso, um sistema deste tipo também iria acelerar descobertas em biologia celular, tais como a dissecção das vias de sinalização utilizadas por VV para a sua motilidade à base de actina.Divulgações
Authors have no competing financial interests.
Agradecimentos
This work was supported by The MRC DPFS grant (MR/M015696/1) and Ministry of Sciences and Technology of China (2013DFG32080).
Materiais
| Name | Company | Catalog Number | Comments |
| Plasmid #41824 | 41824 | Addgene | gRNA cloning vector |
| pST1374 | 13426 | Addgene | Cas9 cloning vector |
| pGEMT easy | A1360 | promega | repair donor vector cloning |
| One Shot TOP10 Chemically Competent E. coli | C4040-10 | Invitrogen | transformation |
| QIAprep Spin Miniprep Kit | 27106 | Qiagen | plasmid extraction |
| Dulbecco’s Eagle’s Medium (DMEM) | 11965-092 | Life Technologies | cell culture medium |
| fetal bovine serum (FBS) | SH30088.03HI | Hyclone | cell culture serum |
| penicillin, streptomycin | 15070-063 | Thermo Scientific | antibiotics |
| Effectene | 301425 | Qiagen | transfection |
| Thermo Scientific Nalgene Cryogenic vial; 2.0 mL | W-06754-96 | Thermo Scientific | collect virus |
| fluorescence microscope | Olympus BX51 Fluorescence | Olympus | find RFP-positive plque |
| DNeasy Blood & Tissue Kit | 69506 | Qiagen | DNA extraction |
| Extensor Long PCR ReddyMix Master Mix | AB-0794/B | Thermo Scientific | PCR reagent |
| 10 cm culture dish | Z688819-6EA | sigma | cell culture |
| 6-well plate | Z707759 | sigma | cell culture |
| cell scraper | C5981 | sigma | scrap cells |
| 1x Trypsin-EDTA solution | T4299 | sigma | trypsinize cells |
| Virkon | 95015661 | Anachem Ltd | desinfectant |
| Falcon tube | CLS430791-500EA | Sigma | hold cell suspension |
| N1L forward 5’-TATCTAGCAATGGACCGT-3’ | Sigma | PCR primer | |
| N1L reverse 5’-CCGAAGGTAGTAGCATGGA-3' | Sigma | PCR primer | |
| A52R forward 5’-ATAGGATTGTGTGCATGC-3’ | Sigma | PCR primer | |
| A52R reverse 5’-TTGCGGTATATGTATGAGGTG-3’ | Sigma | PCR primer | |
| RFP forward 5’- GCTACCGGACTCAGATCCA-3’ | Sigma | PCR primer | |
| RFP reverse 5’-CGCCTTAAGATACATTGATGAG-3’ | Sigma | PCR primer | |
| Argarose | A7431 | Sigma | resolve PCR product |
| G:BOX F3 | G:BOX F3 | Syngene | UV gel doc |
Referências
- Baroudy, B. M., Venkatesan, S., Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell. 28, 315-324 (1982).
- Madigan, M. T., Martinko, J. M., Parker, J., Brock, T. D. Brock biology of microorganisms, 9th edn. , Prentice Hall. Upper Saddle River, NJ. (2000).
- Cooney, E. L., et al. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet. 337, 567-572 (1991).
- Mackett, M., Yilma, T., Rose, J. K., Moss, B. Vaccinia virus recombinants: expression of VSV genes and protective immunization of mice and cattle. Science. 227, 433-435 (1985).
- Legrand, F. A., et al. Induction of potent humoral and cell-mediated immune responses by attenuated vaccinia virus vectors with deleted serpin genes. J Virol. 78, 2770-2779 (2004).
- Panicali, D., Davis, S. W., Weinberg, R. L., Paoletti, E. Construction of live vaccines by using genetically engineered poxviruses: biological activity of recombinant vaccinia virus expressing influenza virus hemagglutinin. Proc Natl Acad Sci. 80, 5364-5368 (1983).
- Wiktor, T. J., et al. Protection from rabies by a vaccinia virus recombinant containing the rabies virus glycoprotein gene. Proc Natl Acad Sci. 81, 7194-7198 (1984).
- Gallucci, S., Matzinger, P. Danger signals: SOS to the immune system. Curr Opin Immunol. 13, 114-119 (2001).
- Matzinger, P. The danger model: a renewed sense of self. Science. 296, 301-305 (2002).
- Guo, Z. S., Bartlett, D. L. Vaccinia as a vector for gene delivery. Expert Opin Biol Ther. 4, 901-917 (2004).
- Kwak, H., Horig, H., Kaufman, H. L. Poxviruses as vectors for cancer immunotherapy. Curr Opin Drug Discov Devel. 6, 161-168 (2003).
- Tysome, J. R., et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 16, 1223-1233 (2009).
- Kirn, D. H., Wang, Y., Liang, W., Contag, C. H., Thorne, S. H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 68, 2071-2075 (2008).
- Park, B. H., et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 9, 533-542 (2008).
- Panicali, D., Paoletti, E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci. 79, 4927-4931 (1982).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. J Virol. 61, 1788-1795 (1987).
- Rouet, P., Smih, F., Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 14, 8096-8106 (1994).
- Jansen, R., Embden, J. D., Gaastra, W., Schouls, L. M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 43, 1565-1575 (2002).
- Brouns, S. J., et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 321, 960-964 (2008).
- van der Oost, J., Jore, M. M., Westra, E. R., Lundgren, M., Brouns, S. J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 34, 401-407 (2009).
- Jinek, M., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 343, 1247997(2014).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819-823 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-821 (2012).
- Bi, Y., et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 10, 1004090(2014).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339, 823-826 (2013).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 41, 9049-9061 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31, 827-832 (2013).
- Ran, F. A., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 154, 1380-1389 (2013).
- Niu, Y., et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 156, 836-843 (2014).
- Yin, H., et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 32, 551-553 (2014).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153, 910-918 (2013).
- Ma, Y., et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 24, 122-125 (2014).
- Yuan, M., et al. Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J Virol. 89, 5176-5179 (2015).
- Yuan, M., et al. A marker-free system for highly efficient construction of vaccinia virus vectors using CRISPR Cas9. Mol Ther Methods Clin Dev. 2, 15035(2015).
- Merchlinsky, M., Moss, B. Introduction of foreign DNA into the vaccinia virus genome by in vitro ligation: recombination-independent selectable cloning vectors. Virology. 190, 522-526 (1992).
- Timiryasova, T. M., Chen, B., Fodor, N., Fodor, I. Construction of recombinant vaccinia viruses using PUV-inactivated virus as a helper. Biotechniques. 31, 534-540 (2001).
- Johnson, R. D., Jasin, M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 29, 196-201 (2001).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados