
Method Article
Génie génétique de Dictyostelium discoideum basés sur la sélection et la croissance sur les bactéries
Dans cet article
Résumé
Dictyostelium discoideum est un organisme modèle populaire à étudier les processus cellulaires complexes tels que la migration cellulaire, endocytose et le développement. L’utilité de l’organisme dépend de la possibilité de manipulation génétique. Nous présentons ici les méthodes pour transfecter Dictyostelium discoideum cellules qui surmontent les limites actuelles de culture de cellules dans un milieu liquide.
Résumé
Dictyostelium discoideum est un intrigant organisme modèle pour l’étude des processus de différenciation cellulaire au cours de développement, signalisation cellulaire et d’autres questions importantes de la biologie cellulaire. Les technologies disponibles pour manipuler génétiquement des cellules de Dictyostelium sont bien développées. Transfections peuvent être effectuées à l’aide de différents marqueurs sélectionnables et marqueur de recyclage, y compris la recombinaison homologue et mutagenèse insertionnelle. Ceci est soutenu par un génome bien annoté. Cependant, ces approches sont optimisés pour les lignées de cellules axéniques de plus en plus dans des cultures liquides et sont difficiles à appliquer au non-axéniques cellules de type sauvage, qui se nourrissent seulement de bactéries. Les mutations qui sont présentes dans des souches axéniques troubler Ras signalisation, causant excessive macropinocytose requis pour l’alimentation et nuire à la migration cellulaire, qui confond l’interprétation de la transduction du signal et des expériences de chimiotactisme chez ces souches. Les tentatives antérieures de manipuler génétiquement des cellules non-axéniques ont manqué efficacité et procédures expérimentales complexes requises. Nous avons développé un protocole de transfection simple qui, pour la première fois, permet de surmonter ces limitations. Ces séries de grandes améliorations à la génétique moléculaire Dictyostelium permettre aux cellules de type sauvage à manipuler aussi facilement que des souches de laboratoire standard. Outre les avantages pour étudier les processus de signalisation et de la motilité non corrompues, les mutants qui perturbent la croissance axée sur la macropinocytose peuvent maintenant être facilement isolés. En outre, le flux de travail entière de transfection est grandement accéléré, avec cellules recombinantes pouvant être générés en jours et non en semaines. Un autre avantage est que la génétique moléculaire davantage peut être effectuée qu’avec des échantillons de Dictyostelium sauvage fraîchement isolées de l’environnement. Ceci peut aider à étendre le champ des approches utilisées dans ces domaines de recherche.
Introduction
Le genre de Dictyostelium sont sol vivant amibe sociale qui se nourrissent principalement de bactéries. Étant placé dans le phylum des Amoebozoa, un grand nombre d’espèces ont été isolé qui peuvent être regroupées en quatre différents clades1. L’espèce Dictyostelium discoideum (d. discoideum) est devenu un organisme modèle populaire à étudier les processus cellulaires complexes tels que la migration cellulaire et de la phagocytose. À contrôler et à normaliser les conditions expérimentales, cellule axénique ont été mis au point des lignes qui sont capables de se développer dans un milieu liquide complex ou défini en l’absence de bactéries2. L’Ax2, Ax3, revêtent une importance particulière et isoler les souches Ax4, qui sont toutes produites dans les années 1970 et finalement dérivés dans une nature unique NC43. Outils pour l’ingénierie génétique ont été développés dans ces souches axéniques, résultant en huitièmes publiée en 19874,5. Protocoles ont davantage développés et optimisé pour une utilisation dans des conditions axéniques6,7.
Adaptation de ces protocoles à sauvage d. discoideum souches qui ne sont pas capables de se développer dans un bouillon liquide ont été tentées par plusieurs laboratoires. Cependant, ce n'est pas devenu pleinement réussie puisque les protocoles de transfection sont complexes et manquent d’efficacité, en partie en raison de la capacité des bactéries à agir comme un puits pour les réactifs sélective8,9. Ainsi, essentiellement tout moléculaires données sur d. discoideum proviennent des descendants d’un seul isolat de type sauvage. Nous avons voulu remédier à cette limitation et d’élaborer une méthode pour modifier génétiquement les cellules de d. discoideum indépendamment de leur capacité à se développer dans un milieu liquide. La nécessité d’une telle méthode peut s’expliquer par l’observation qu’il a été assumé par le passé que les mutations permettant la croissance axénique ont été essentiellement neutres et ne portait pas atteinte à la physiologie cellulaire. Cette supposition est seulement partiellement correcte. En général, il y a deux différences notables ; tout d’abord, entre les différents isolement des souches axéniques et, deuxièmement, lorsque ces souches axéniques sont comparées avec des souches sauvages non axéniques8,9.
Peut-être le facteur le plus critique est la clé axénique gène, hacheB, qui a été récemment identifié comme RasGAP NF1. La fonction principale de la NF1 comme un RasGAP est de restreindre l’activité de Ras3. La suppression de l’enzyme chez toutes les souches axéniques entraîne une activité Ras excessive se manifestant par la formation de grandes parcelles de Ras actives. Ces patchs Ras élargies conduisent à l’accumulation de PIP3 dans la membrane plasmique. Ces taches qui apparaissent une coïncidence de PIP3 et Ras active sont un modèle pour la formation d’un volant circulaire qui finalement ferme et conduit à la formation de macropinosomes10. La conséquence est une augmentation excessive de l’activité macropinocytic. Macropinocytose est un processus axé sur l’actine. Un concours pour les composants du cytosquelette de la formation de macropinosomes ou pseudopodes est le résultat. Son impact sur le comportement de la cellule se reflète dans la prévention quasi-complète du chimiotactisme des cellules végétatives de folate11. Les patchs PIP3 massivement élargies sont très persistants. Même dans les cellules affamées, les patchs PIP3 restent et peuvent être mal interprétés comme des pseudopodes, qui peuvent causer des problèmes d’interprétation des études sur le chimiotactisme au cAMP.
Dans certains cas, la mutation de la NF1 est expérimentalement utile. Cela nous amène à une deuxième motivation pour développer une méthode de transfection pour par les bactéries cultivées discoideum d. cellules, puisque l’augmentation du taux de macropinocytose rend les cellules axéniques précieux pour enquêter sur les aspects fondamentaux de ce processus12 . Cependant, mutation dans les gènes nécessaires à la macropinocytose, tels que Ras et PI3-kinase10, ont aboli presque croissance axénique, rend nécessaire de manipuler ces cellules grâce à une croissance sur les bactéries. Une autre raison qui rend transfections axée sur les bactéries utiles est l’utilisation croissante des Dictyostélides pour explorer les questions de l’évolution des multiples de la cellularité13,14, kin reconnaissance15,16, et un comportement cellulaire altruiste qui dépendent principalement de l’utilisation du type sauvage fraîchement isolée isole17. Tous mentionnés aux domaines de recherche peuvent être facilitées par des méthodes efficaces pour la manipulation génétique des isolats de sauvages, qui sont non-axéniques et ne poussent pas en bouillon liquide.
Nos protocoles permettant de surmonter les limitations décrites. Pris ensemble, la possibilité d’effectuer des manipulations génétiques avec bactéries cultivées discoideum d. avantages de cellules cales pour tous les chercheurs de Dictyostelium , même si c’est juste l’augmentation de la vitesse du processus de sélection dû à plus vite croissance de l’amibe (temps de doublement de 4 h) sur les bactéries par rapport à la croissance en milieu axénique (10 h temps de doublement).
Protocole
1. préparation des cellules et matériaux
-
Préparation du tampon SorMC
- Préparer 100 mL 100 x SorMC (Sorensen MgCl2 et CaCl2) tampon en dissolvant 20,36 g de KH2PO4 (15 mM) et 5,47 g Na2HPO4·7 H2O (2 mM) dans 100 mL d’eau de O2ddH. Agiter la solution à température ambiante (RT) et porter le volume à 100 mL avec dH2O.
Remarque : Le tampon résultant a un pH de 6 et n’a pas besoin de plus de réglage. - Produire 1000 mL 1 x solution de travail en FD2O. ajouter 50 µL chaque de MgCl2 et CaCl2 pour atteindre la concentration finale de 50 µM de chaque. Filtre de stériliser la solution en utilisant un filtre de 0,22 µm.
Remarque : Toujours ajouter du MgCl2 et CaCl2 au tampon 1 x pour éviter la précipitation des sels dans 100 x solution mère. - Vous pouvez également préparer KK2 un tampon (2,2 g de KH2PO4 et 0,7 g de K2HPO4 pour 1 L de tampon) additionné de 50 µM MgCl2 et 50 µM CaCl2 (ci-après dénommé "KK2MC"). Utiliser cette mémoire tampon dans l’ensemble au lieu de SorMC.
- Préparer 100 mL 100 x SorMC (Sorensen MgCl2 et CaCl2) tampon en dissolvant 20,36 g de KH2PO4 (15 mM) et 5,47 g Na2HPO4·7 H2O (2 mM) dans 100 mL d’eau de O2ddH. Agiter la solution à température ambiante (RT) et porter le volume à 100 mL avec dH2O.
-
Préparation de bactéries comme source de nourriture pour d. discoideum
- Utiliser une seule colonie de K. aerogenes et ensemencer 1 L de milieu-LB (bouillon de lysogénie). Utiliser un flacon de 2 L. Laisser pousser pendant la nuit à 37 ° C sous agitation à 220 tr/mn les bactéries.
Remarque : Si de grandes quantités de bactéries sont nécessaires, utiliser des supports riches comme 2xTY (extrait de levure tryptone moyen) ou SOB (bouillon super optimal) au lieu de LB. Dans le cas où l’utilisation de K. aerogenes bactéries n’est pas autorisée en raison de restrictions de sécurité, BL21 e. coli peut être utilisé à la place. - Récolter les cellules le lendemain en les tournant vers le bas dans deux tubes à centrifuger 500 mL à ~ 6 600 x g pendant 20 min. des bactéries de lavage une fois avec 500 mL de tampon de SorMC.
- Resuspendre le culot dans 20 mL de SorMC. Vérifier l' OD600 (densité optique à 600 nm) à l’aide d’un photomètre. Diluer avec le même tampon à une OD600 environ 100.
Remarque : K. aerogenes bactéries sont difficiles à pellet, une vitesse relativement élevée pour filer vers le bas de la bactérie est donc nécessaire pour éviter la perte des bactéries alimentaires. La solution bactérienne qui en résultent peuvent être stockés pendant 4 mois au réfrigérateur à 4 ° C et maintenir son utilité comme source de nourriture pour d. discoideum. 1 L de nuit K. aerogenes suspension cultivée dans un milieu LB habituellement donne 20 mL d’une OD600 environ 100.
Mise en garde : Pour s’assurer que les bactéries préparés sont une monoculture de K. aerogenes, exécutez toutes les étapes sous une hotte.
- Utiliser une seule colonie de K. aerogenes et ensemencer 1 L de milieu-LB (bouillon de lysogénie). Utiliser un flacon de 2 L. Laisser pousser pendant la nuit à 37 ° C sous agitation à 220 tr/mn les bactéries.
-
Préparation du tampon d’électroporation H40
- Préparation de 100 mL de solution tampon, dissoudre 0,952 g de HEPES ddH2O eau et ajouter 100 µL de MgCl2 d’une solution mère de 1 M. Ajuster le pH 7 à l’aide de KOH pour le titrage. Stériliser le tampon à l’aide d’un filtre de 0,22 µm ou autoclave. Utilisez HEPES sans acide et pas le sel de sodium.
- Effectuer la préparation de plasmide suivant le protocole du fabricant et en utilisant les kits résumées dans le Tableau des matériaux. Utilisez les plasmides résumées au tableau 1.
Remarque : La qualité de l’ADN utilisée pour la transfection est cruciale. La sélection de d. discoideum transfectants sur les bactéries de plus en plus a des exigences spécifiques pour les promoteurs de la cassette de sélection et d’expression de conduite (voir discussion).
| nom de plasmide | résistance/sélection chez les bactéries | résistance/sélection chez Dictyostelium | balise |
| plasmides d’expression extrachromosomique | |||
| pDM1203 | Ampicilin | G418 | aucune |
| pDM1207 | Ampicilin | G418 | N-terminal GFP |
| pDM1208 | Ampicilin | G418 | MCherry N-terminal |
| pPI159 | Ampicilin | G418 | MNeon N-terminal |
| pPI437 | Ampicilin | G418 | MScarlet N-terminal |
| pPI54 | Ampicilin | G418 | MTurquoise2 N-terminal |
| pDM1209 | Ampicilin | G418 | C-terminal GFP |
| pDM1210 | Ampicilin | G418 | MCherry C-terminal |
| pPI143 | Ampicilin | G418 | MNeon C-terminal |
| pPI459 | Ampicilin | G418 | MScarlet C-terminal |
| pPI142 | Ampicilin | G418 | MTurquoise2 C-terminal |
| plasmides de navette | |||
| pDM344 | Ampicilin | aucune | aucune |
| pDM1019 | Ampicilin | aucune | N-terminal GFP |
| pDM1018 | Ampicilin | aucune | MCherry N-terminal |
| pPI152 | Ampicilin | aucune | MNeon N-terminal |
| pPI418 | Ampicilin | aucune | MScarlet N-terminal |
| pPI150 | Ampicilin | aucune | MTurquoise2 N-terminal |
| pDM1021 | Ampicilin | aucune | C-terminal GFP |
| pDM1020 | Ampicilin | aucune | MCherry C-terminal |
| pPI153 | Ampicilin | aucune | MNeon C-terminal |
| pPI457 | Ampicilin | aucune | MScarlet C-terminal |
| pPI151 | Ampicilin | aucune | MTurquoise2 C-terminal |
| plasmides d’expression extrachromosomiques inductible | |||
| pDM1038 | Ampicilin | Hygromycine | aucune |
| pDM1047 | Ampicilin | Hygromycine | N-terminal GFP |
| pDM1046 | Ampicilin | Hygromycine | MCherry N-terminal |
| pPI450 | Ampicilin | Hygromycine | MNeon N-terminal |
| pPI452 | Ampicilin | Hygromycine | MScarlet N-terminal |
| pPI449 | Ampicilin | Hygromycine | MTurquoise2 N-terminal |
| pDM1049 | Ampicilin | Hygromycine | C-terminal GFP |
| pDM1048 | Ampicilin | Hygromycine | MCherry C-terminal |
| pPI470 | Ampicilin | Hygromycine | MNeon C-terminal |
| pPI460 | Ampicilin | Hygromycine | MScarlet C-terminal |
| pPI469 | Ampicilin | Hygromycine | MTurquoise2 C-terminal |
| refuge de Loi5 ciblant les plasmides | |||
| pDM1501 | Ampicilin | Hygromycine | aucune |
| pDM1513 | Ampicilin | Hygromycine | N-terminal GFP |
| pDM1514 | Ampicilin | Hygromycine | MCherry N-terminal |
| pPI231 | Ampicilin | Hygromycine | MNeon N-terminal |
| pPI419 | Ampicilin | Hygromycine | MScarlet N-terminal |
| pPI228 | Ampicilin | Hygromycine | MTurquoise2 N-terminal |
| pDM1515 | Ampicilin | Hygromycine | C-terminal GFP |
| pDM1516 | Ampicilin | Hygromycine | MCherry C-terminal |
| pPI230 | Ampicilin | Hygromycine | MNeon C-terminal |
| pPI458 | Ampicilin | Hygromycine | MScarlet C-terminal |
| pPI229 | Ampicilin | Hygromycine | MTurquoise2 C-terminal |
| Plasmides d’expression REMI | |||
| pDM1220 | Ampicilin | Hygromycine | aucune |
| pDM1351 | Ampicilin | Hygromycine | N-terminal GFP |
| pDM1259 | Ampicilin | Hygromycine | MCherry N-terminal |
| pPI465 | Ampicilin | Hygromycine | MNeon N-terminal |
| pPI468 | Ampicilin | Hygromycine | MScarlet N-terminal |
| pPI466 | Ampicilin | Hygromycine | MTurquoise2 N-terminal |
| pDM1352 | Ampicilin | Hygromycine | C-terminal GFP |
| pDM1305 | Ampicilin | Hygromycine | MCherry C-terminal |
| pPI471 | Ampicilin | Hygromycine | MNeon C-terminal |
| pPI467 | Ampicilin | Hygromycine | MScarlet C-terminal |
| pPI472 | Ampicilin | Hygromycine | MTurquoise2 C-terminal |
| ciblées dans les plasmides de trame | |||
| pDM1355 | Ampicilin | Hygromycine | C-terminal GFP |
| pPI461 | Ampicilin | Hygromycine | MCherry C-terminal |
| pPI462 | Ampicilin | Hygromycine | MNeon C-terminal |
| pPI464 | Ampicilin | Hygromycine | MScarlet C-terminal |
| pPI463 | Ampicilin | Hygromycine | MTurquoise2 C-terminal |
| knock out plasmides | |||
| pDM1079 | Ampicilin | Blasticidine | aucune |
| pDM1080 | Ampicilin | Nourséothricine | aucune |
| pDM1081 | Ampicilin | Hygromycine | aucune |
| pDM1082 | Ampicilin | G418 | aucune |
| Plasmides d’expression CRE | |||
| pDM1483 | Ampicilin | Nourséothricine | aucune |
| pDM1489 | Ampicilin | Hygromycine | aucune |
| pDM1488 | Ampicilin | G418 | aucune |
Tableau 1 : Liste de plasmide pour non-axéniques transfections.
-
Mise en place pour la transfection des cellules de Dictyostelium
- Atteindre K. aerogenes confluence dans le milieu SM (moyennement riches en éléments nutritifs) nuit à RT. Cultures peut être stockée pendant 2 semaines à 4 ° C.
- Ajouter environ 400 µL de cette suspension bactérienne sur une gélose SM (peptone 10 g/L ; levure extrait 1 g/L ; glucose 10 g/L ; KH2PO4 1,9 g/L ; K2HPO4 x 3 H2O, 1,3 g/L ; MgSO4 anhydre 0,49 g/L ; 1,7 % d’agar) et répartir uniformément. Prenez une boucle stérile et ensemencer avec des cellules de Dictyostelium . Étaler les cellules à un bord de la plaque.
- Incuber les plaques à 22 ° C pendant 2 jours pour s’assurer que les zones de croissance suffisamment grand pour la transfection.
Remarque : Pour les souches de Dictyostelium ne pas rendre les zones de forte croissance (p. ex., Ax3, DH1 ou JH10), ou pour les expérimentateurs inexpérimentés, utilisez compensation plaques au lieu de cela. Pour ce faire, suivez les instructions par étapes 1.5.4 à 1.5.6. - Prenez une boucle stérile et ensemencer avec des cellules de Dictyostelium (environ 2-4 x 105 cellules). Les cellules de transfert à 800 µL d’une dense K. aerogenes suspension dans les cellules SM. Mix par pipetage de haut en bas.
- Transférer 400 µL, 200 µL, 100 µL et 50 µL sur plaques de gélose SM fraîches. Ajouter à chaque µL de plaque 400 de suspension supplémentaire de SM K. aerogenes , répartis uniformément et sécher.
- Incuber les plaques à 22 ° C pendant environ 2 jours jusqu'à ce que les plaques deviennent translucides.
Remarque : En raison de leur taux de croissance plus rapide, les bactéries produisent au départ une pelouse confluente et les amibes par la suite « effacer » la plaque de la bactérie. Le temps nécessaire à ce processus peuvent varier selon le contexte de la souche et la capacité des cellules mutantes de croître sur les bactéries.
2. la transfection de cellules de Dictyostelium basées sur la sélection bactérienne

Figure 1 : "Workflow" pour la transfection de cellules de Dictyostelium bactéries cultivées. Les étapes de la transfection sont répertoriés comme suit. La croissance de cellules de d. discoideum sur une plaque de SM ensemencée par des bactéries de K. aerogenes (rouge). Récolter les cellules uniquement à partir de l’alimentation avant (vert), en évitant les cellules qui sont développent déjà (vert foncé). Laver les cellules en H40. Remettre en suspension les cellules à une densité finale de 2-4 x 107 cellules/mL. Mélanger la suspension cellulaire de 1 à 2 µg d’ADN. Transvaser le mélange dans une cellules électroporation de cuvette et le pouls. Transférer les cellules directement après électroporation dans un plat avec SorMC et les bactéries. Laissez les cellules récupérer pendant 5 h avant d’ajouter le marqueur sélectionnable. Pour les plasmides extrachromosomiques, ajouter la sélection directement au plat. Transfectants sont généralement visibles après environ 2 jours. Pour les constructions linéarisées visant pour une intégration simple dans le génome, mis en place trois dilutions comme indiqué et ajouter la sélection. Les bactéries sont tirées de l' OD600 = 100 solution mère. Bien mélanger les tubes et les cellules de transfert en plaques 96 puits à fond plat culture de tissus. Utiliser deux plaques par dilution. Pipetter 150 µL de suspension cellulaire dans chaque cupule. Il faut environ 5 jours jusqu'à ce que les colonies serrées sont visibles. Les puits rouges montrent un exemple de la quantité habituelle de cellules transformées avec succès obtenus (panneau supérieur modifiée de la précédente publication22). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
- Pour préparer les assiettes avec suspension K. aerogenes , ajouter 10 mL de tampon de SorMC contenant des bactéries à une densité de OD600 K. aerogenes = 2 (ajouter 200 µL de la préparé OD600 = 100 K. aerogenes solution pendant le désiré concentration de bactéries) dans un 10 cm traitée pour la culture de tissus de pétri.
Remarque : Cette plaque est nécessaire plus tard à cultiver la transfected des cellules de d. discoideum . Par ailleurs, une plaque 6 puits culture tissulaire peut être utilisée. Dans le cas de la transfection des plasmides extrachromosomiques, la plaque de 6 puits culture tissulaire est plus économe en ressources. Utiliser 2 mL de SorMC K. aerogenes (OD600 = 2) suspension / puits. -
Préparation des cellules de Dictyostelium
- À l’aide d’une anse à inoculation jetables 10 µL, gratter les cellules provenant des zones de croissance (environ 3 cm) de la plaque de culture (entrée de la surface défrichée) ou plaque de compensation. Cellules de transfert dans un tube de 1,5 mL contenant 1 mL de tampon de H40 glacée.
Remarque : Le moment de la récolte des cellules de compensation des plaques est crucial. Récolte des rendements trop tôt trop peu une quantité de cellules, alors que la récolte tardive augmentations le risque de céder partiellement développé des cellules. - Laver les cellules par rotation vers le bas pendant 2 min à 1 000 x g ou filage éclair pour 2 s à 10 000 x g. Jeter le surnageant et remettre en suspension des cellules dans le tampon de H40 à une densité finale de 2-4 x 107 cellules/mL. Garder au froid les cellules pendant la procédure de transfection ensemble. Un coulis de glace-eau permet d’assurer le contact direct entre les tubes et la glace.
- À l’aide d’une anse à inoculation jetables 10 µL, gratter les cellules provenant des zones de croissance (environ 3 cm) de la plaque de culture (entrée de la surface défrichée) ou plaque de compensation. Cellules de transfert dans un tube de 1,5 mL contenant 1 mL de tampon de H40 glacée.
-
Électroporation
- Ajouter 100 µL de cellules dans un tube avec 1 à 2 µg d’ADN. Mélanger soigneusement par pipetage de haut en bas.
- Transférer le mélange/DNA des cellules dans une cupule d’électroporation préalablement réfrigérées (écart de 2 mm).
- Les cellules en utilisant les paramètres suivants d’ondes carrées d’impulsion : 350 V, 8 ms, 2 impulsions et intervalle d’impulsions 1 s.
Remarque : N’ajoutez pas plus de 2 µg d’ADN. Des quantités plus élevées sont toxiques pour les cellules et diminuent l’efficacité de transfection. Le total a ajouté ADN volume ne doit pas dépasser 5 µL. - Cellules de transfert immédiatement à la précédemment préparé 10 cm boîte de pétri avec K. aerogenes SorMC et laisser les cellules récupérer pendant 5 h.
Remarque : Vérifier les cellules dans la boîte de pétri sous un microscope inversé. Cellules apparaîtront rondes directement après électroporation mais seront de retour à leur forme amiboïde après environ 30 min, quand ils ont eu assez de temps pour fixer correctement à la surface.
-
Sélection de transfectants : selon si la transfection vise à générer un knock out, knock-in ou Loi5 knock-in, ou d’exprimer une protéine fluorescente de reporter d’un plasmide extrachromosomique, mener le processus de sélection par le biais de l’un des ce qui suit décrit les méthodes.
Remarque : Contrairement aux cellules axéniques, cellules par les bactéries cultivées sont très résistantes à la blasticidine. Sélection, donc, est toujours effectuée à l’aide de G418 ou hygromycine (voir discussion).- Les débouchures, knock-ins et Loi5 knock-ins
- Détacher les cellules avec soin à partir de la boîte de pétri en forçant à plusieurs reprises le liquide d’une pipette sur la surface.
- Mettre en place trois dilutions dans la suspension de SorMC K. aerogenes (OD600 = 2) et ajoutez l’agent sélectif selon la résistance appliquée (voir la Figure 1).
- Faible dilution : mélangez 9 mL de suspension cellulaire avec 20,4 mL de SorMC et 600 µL de la solution mère de K. aerogenes .
- Dilution moyenne : mélanger 900 µL de suspension cellulaire avec 28,5 mL de SorMC et 600 µL de la solution mère de K. aerogenes .
- Haute dilution : mélanger 90 µL de suspension cellulaire avec 29,3 mL de SorMC et 600 µL de la solution mère de K. aerogenes .
- Ajouter 30 µL de marqueur sélectionnable (100 x solution mère).
- Distribuer les dilutions préparées en plaques 96 puits à fond plat de vitroplants de pipetage 150 µL de suspension cellulaire dans chaque puits.
Remarque : Cette procédure a pour but de clones unique écran plutôt que des populations. La sélection prend environ 5 à 7 jours selon la construction utilisée.
- Plasmides extrachromosomiques
- Ajouter le marqueur sélectionnable directement au plat 10 mL (voir Figure 1).
Remarque : Il est inutile de mettre en place des dilutions, puisqu’il n’y a aucun désir pour les populations clonales. Le processus de sélection pour les plasmides extrachromosomiques est plus rapide due à un nombre élevé de copies présent dans les cellules de d. discoideum . Transfectants peut s’attendre après 32 h à 2 jours.
Attention : Les antibiotiques utilisés comme marqueur de sélection sont toxiques. Porter des gants.
- Ajouter le marqueur sélectionnable directement au plat 10 mL (voir Figure 1).
- Les débouchures, knock-ins et Loi5 knock-ins
-
Les clones obtenus pour tranfectants positive de l’écran. Des tentatives de knock out ou knock-in, suivez les instructions à l’étape 2.5.1. Vérifier la réussite de transfection pour les plasmides extrachromosomiques en suivant les instructions à l’étape 2.5.2.
- Pour débouchures, knock-ins et Loi5 knock-ins, effectuer le dépistage initial par PCR pour confirmer l’intégration de la construction dans le locus génomique correct.
Remarque : Pour maximiser la probabilité de populations clonales, utiliser la dilution la plus élevée possible qui cède transfectants après sélection. But pour les plaques qui sont un tiers maximum de puits occupés.- Pour développer les populations clonales, transfert de clones qui ont grandi après sélection de la plaque de 96 puits de culture de tissus dans une assiette de 12 puits de culture de tissu de croître suffisamment de cellules pour l’isolement de l’ADN génomique. Fournir à chaque puits 1 ml de SorMC K. aerogenes (OD600 = 2) et marqueur de sélection fraîche.
NOTE : 1 jour est habituellement suffisant pour obtenir un confluent bien adapté pour l’isolation de l’ADN. - Pour effectuer l’isolement d’ADN génomique mini, moissonner les cellules d’un puits confluent et isoler l’ADN génomique à l’aide d’un mini kit d’extraction d’ADN suivant les instructions du fabricant.
- Utiliser l’ADN génomique isolé avec des amorces adaptés et Taq-polymérase (voir Table des matières) pour dépister les PCR positives intégrations.
Remarque : Après la confirmation d’une intégration positive dans le locus génomique correcte, une analyse de Southern blot doit être réalisée. Ceci assure une plus grande confiance que des événements d’insertion supplémentaires dans des régions génomiques non spécifiques n’ont pas eu lieu.
- Pour développer les populations clonales, transfert de clones qui ont grandi après sélection de la plaque de 96 puits de culture de tissus dans une assiette de 12 puits de culture de tissu de croître suffisamment de cellules pour l’isolement de l’ADN génomique. Fournir à chaque puits 1 ml de SorMC K. aerogenes (OD600 = 2) et marqueur de sélection fraîche.
- Pour les protéines fluorescentes journaliste, visuellement identifier les cellules fluorescentes positives et vérifier avec un microscope à fluorescence. Vous pouvez également effectuer une tache occidentale avec des anticorps appropriés pour identification biochimique.
- Pour débouchures, knock-ins et Loi5 knock-ins, effectuer le dépistage initial par PCR pour confirmer l’intégration de la construction dans le locus génomique correct.
| Programme de la PCR | ||
| étape | température | temps |
| Dénaturation initiale | 94 ° C | 30 s |
| 30 cycles | 94 ° C | 15-30 s |
| 42 ° C | 15-60 s | |
| 68 ° C | 1 min/kb | |
| Extension finale | 68 ° C | 5 min |
| Maintenez | 4-10 ° C | |
| Composition de la réaction | ||
| composant | Réaction de 25 μL | concentration finale |
| Primer avant 10 µM | 0,5 ΜL | DE 0,2 ΜM |
| 10 µM amorce inverse | 0,5 ΜL | DE 0,2 ΜM |
| Modèle de l’ADN | variable (ca. 5 µL) | < 1 000 ng |
| 2 x Master Mix avec tampon Standard y compris polymérase (voir table des matières) | 12,5 ΜL | 1 x |
| Eau exempte de nucléase | à 25 µL | < 1 000 ng |
Tableau 2 : composition programme et échantillon PCR pour l’amplification des D. discoideum l’ADN genomic.
Résultats
Extrachromosomiques plasmides sont utilisés pour des études de journaliste, dont le but est d’identifier la localisation de certaines protéines à l’intérieur d’une cellule ou des changements dans la structure cellulaire des cellules mutantes. De nombreuses approches, telles que le suivi du cycle cellulaire, il est essentiel d’exprimer deux reporters en même temps. C’est désormais possible grâce à notre système de plasmide extrachromosomique journaliste double (tableau 1). Jour 1, les cellules ont été transfectées avant d’ajouter le marqueur sélectionnable G418 après 5 h (Figure 1). Dans l’exemple, NC4, DdB, Ax2 et les souches sauvages indépendamment dérivées V12M2 et WS2162 (tableau complémentaire 1) ont été transfectées avec le plasmide pPI289, qui code pour la GFP-TubulinA, un marqueur des microtubules et mCherry-PCNA, une protéine qui est utilisé pour la surveillance du cycle cellulaire (Figure 2A). Après 32 h, les cellules ont été observés au microscope. Exprimée de la majorité des cellules les deux protéines de fusion fluorescent-étiquetées, compatible avec précédents rapports que l’expression de deux reporters des même plasmide spectacles semblables niveaux d’expression, qui est quasiment impossible lorsque vous utilisez deux plasmides différents 18,,19. Une cellule représentative pour chaque lignée cellulaire (NC4, DdB, Ax2, V12M2 et WS2162) exprimant le journaliste double désiré illustrée à la Figure 2B. L’efficacité de transfection est résumées dans la Figure2C. Lignées cellulaires dérivées de NC4 montrent les meilleures efficacités de transfection. Toutefois, pour les lignées de cellules V12M2 et WS2162, un nombre considérablement élevé de transfectants a été obtenu.

Figure 2 : Expression d’un plasmide extrachromosomique. (A) plasmides Extrachromosomique transfected directement en forme circulaire. À titre d’exemple, le journaliste double pPI289 est montré. Les sites MIV ONGindiquent d’insertion du deuxième reporter dans le plasmide expression extrachromosomique. (B) Z-projection d’une cellule représentante exprimant GFP-TubulinA (cytoplasmique) et mCherry-PCNA (en grande partie nucléaire) pour les cinq différentes sauvage lignées cellulaires utilisées (NC4, DdB, Ax2, V12M2, WS2162). (C), la transfection des efficacités pour les cinq lignées illustrées en (B) ont été calculées. Montré est la moyenne des deux expériences. Barres d’erreur indiquent ± SD. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
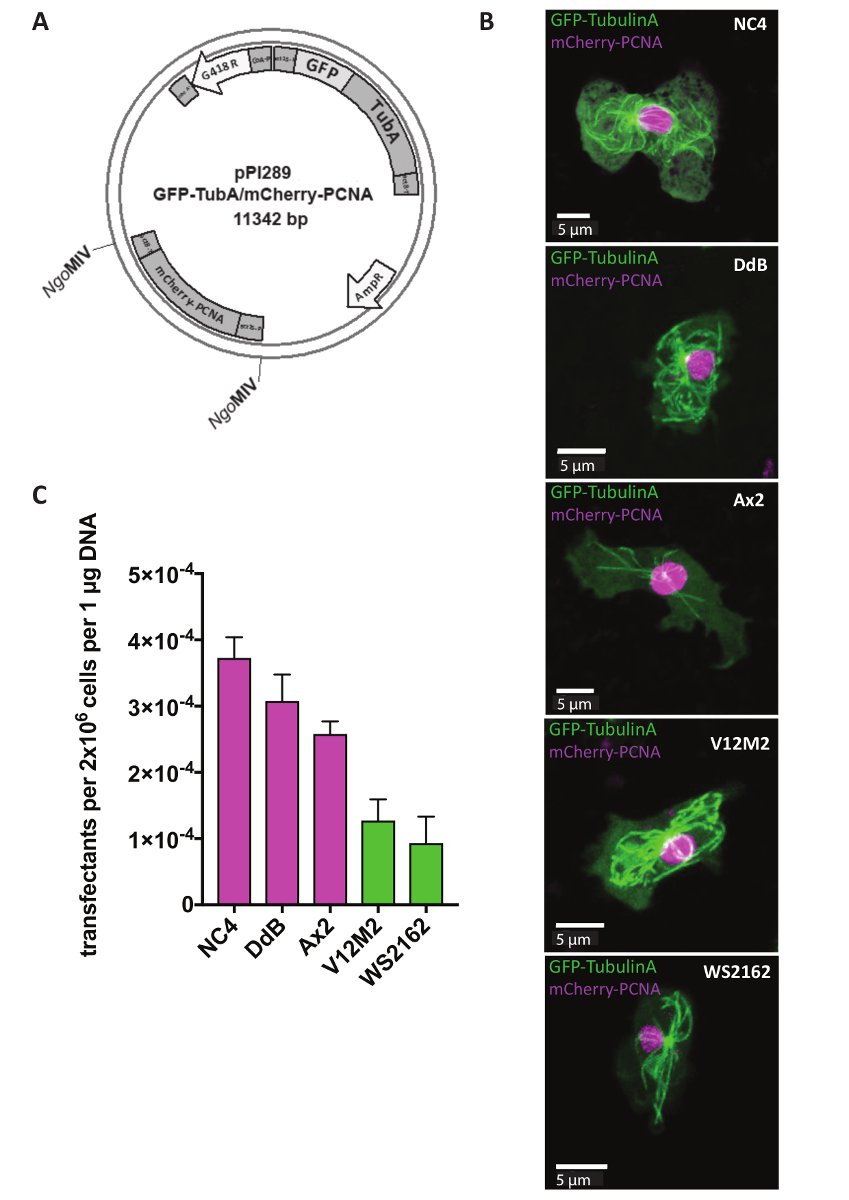{kind=link}
Intégration d’un vecteur de ciblage dans un locus génomique spécifié est plus difficile et nécessite une analyse plus attentive de la lignée cellulaire généré. Dans la Figure 3, un Loi5- mCherry KI en NC4 est tentée. Tout d’abord, le plasmide doit être linéarisé pour augmenter la fréquence des événements de recombinaison après transfection. Pour ce faire, le plasmide pDM1514 est coupé avec des ONGMIV. Deux bandes sont obtenus après l’exécution du digest sur gel d’agarose. Le groupe bp 4127 contient la construction désirée (Figure 3). Pour la transfection, l’ADN digéré doit être extraits du gel et purifié à l’aide d’un kit d’extraction de gel suivant les instructions du fabricant.

Figure 3 : Préparation de Loi5 knock-in et d’ADN pour la transfection. Un exemple de l’utilisation d’un agir5 knock-in, le plasmide pDM1514 est montré. Les étapes de préparation sont répertoriés comme suit. (A) avant d’électroporation, linéariser le plasmide utilisant les sites MIV ONGindiquées. (B, C) Exécutez le plasmide coupé sur gel d’agarose, jusqu'à ce que les deux bandes attendus sont bien séparés. Découper la bande de bp 4127 contenant les armes de recombinaison, mCherry et la résistance-cassette et gel extrait l’ADN. L’ADN est maintenant prêt pour la transfection. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
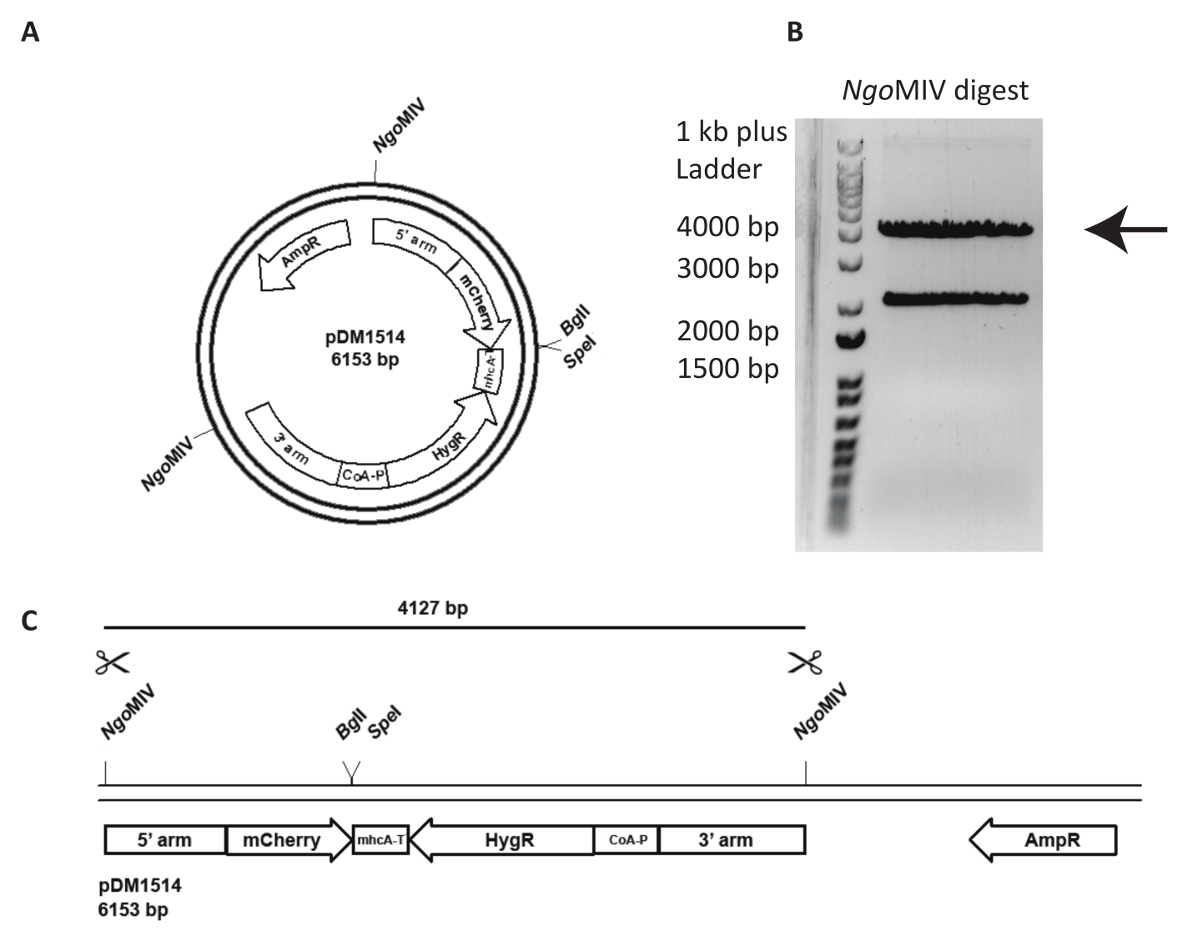{kind=link}
L’ADN purifié a été utilisé pour la transfection des cellules NC4. Après 5-6 jours de sélection, les clones ont été obtenus. Efficacité de transfection représentatif et la quantité des clones positifs identifiés pour plusieurs Loi5 knock-in tentatives sont résumées dans le tableau 3. Deux clones de la transfection NC4 ont été aléatoirement choisis et analysés par PCR (Figure 4A), et les deux montrent les profils de bandes prédit que l'on attend d’un knock-in et ont été validés davantage avec une analyse Southern afin d’assurer une intégration unique en cas de la construction dans le génome de20. La tache montre une intégration unique clairement dans le locus désiré Loi5 (Figure 4B). La lignée de cellules NC4::act5-mCherry générée permet maintenant au cours d’expériences.

Figure 4 : Validation de Loi5- mCherry KIs dans NC4. (A) schéma et contrôle PCRs pour la validation des intégrations positives dans locus Loi5 . Les amorces indiquées ont été utilisées pour analyser deux clones indépendants et le parent. Les deux clones montrent les bandes attendus pour la résistance-cassette et l’aval (P1) ou l’amorce en amont (P2), qui ne sont pas présents chez la souche parentale. La combinaison d’amorces (P1/P2) confirme la bonne intégration de cassette de mCherry et de la résistance dans le locus Loi5 . Le type sauvage NC4 montre la bande attendue de bp 2800, tandis que les deux clones KI n’ont pas cette bande et affichent à la place un produit PCR sur 1400 paires de base plus grandes. (B) régime de sommaire de restriction utilisées et Southern blot. Les deux clones knock-in show la petite bande de bp 3400, qui est le résultat de l’intégration de la construction dans le locus agir5, spécifiquement la supplémentaires Bclje site dans la cassette de résistance à l’hygromycine. Les spectacles de contrôle sauvage le 5,8 Ko attendu résultant des deux situé en aval Bclje sites. La tache a été recadrée et épissée pour plus de clarté. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| construct ciblé dans le locus Loi5 | nom de plasmide | nombre de puits occupés | nombre de clones checked | Clones positifs | Clones correctes (%) | Dictyostelium souche utilisée | efficacité de transfection (transfectants / 2 x 10 ^ 6/1 µg d’ADN) |
| LifeAct-mCherry | pPI226 | 7 | 7 | 1 | 14.2 | AX2 | 3,5 x 10 ^ -6 |
| LifeAct-GFP | pPI227 | 12 | 12 | 5 | 41,6 | AX2 | 6 x 10 ^ -6 |
| GFP | pDM1513 | 3 | 3 | 2 | 66,6 | AX2 | 1.5 x 10 ^ -6 |
| mCherry | pDM1514 | 3 | 3 | 1 | 33,3 | AX2 | 1.5 x 10 ^ -6 |
| GFP | pDM1513 | 66 | 9 | 5 | 55,5 | AX2 | 3.3 x 10 ^ -5 |
| mCherry | pDM1514 | 221 | 12 | 10 | 83,3 | AX2 | 1.1 x 10 ^ -4 |
| H2B-mCherry | pPI420 | 3 | 3 | 1 | 33,3 | AX2 | 1.5 x 10 ^ -6 |
| H2B-mCherry | pPI420 | 7 | 7 | 6 | 85,7 | AX2 | 3,5 x 10 ^ -6 |
| mCherry | pDM1514 | 10 | 10 | 7 | 70 | DdB | 5 x 10 ^ -6 |
| mCherry | pDM1514 | 240 | 12 | 11 | 91,6 | DdB | 1.2 x 10 ^ -4 |
| mCherry | pDM1514 | 320 | 12 | 12 | 100 | NC4 | 1.6 x 10 ^ -4 |
Tableau 3 : efficacité de Transfection et la quantité d’obtient transfectants positive pour la génération de Loi5 KIs en milieux de souche différente 22 .
Le locus agir5 offre une expression relativement homogène du journaliste intégré21. Les cellules NC4::act5-mCherry générés permettent des expériences de mélange à effectuer avec les autres Loi5 knock-ins à l’aide d’une protéine fluorescente différente tels que GFP. Pour souligner le grand avantage de ce système, expérimente le mélange Ax2::act5-GFP est indiqués. En raison de l’incapacité à transfecter non axéniques cellules de type sauvage, ce type d’approche ne peut pas être effectué avant. Expériences de mix sont un outil important pour analyser le comportement de la cellule, car ils permettent une comparaison directe entre les différentes lignées cellulaires connaissent des conditions expérimentales identiques. NC4 cellules croissent plus vite sur les pelouses bactériennes que de cellules Ax2 (Figure 5A). Cela peut être dû à une plus grande capacité à phagocyter les bactéries ou amélioration des capacités de se déplacer et de montrer le chimiotactisme vers une source de nourriture. À l’aide d’une analyse de chimiotactisme agar sous acide folique, une comparaison directe d’une population de NC4::act5-mCherry et Ax2::act5-GFP a été réalisée, montrant que NC4::act5-mCherry sont beaucoup plus efficaces dans la détection de l’acide folique. Après 4 h, plus de cellules NC4::act5-mCherry ont pu ramper sous l’agarose que les cellules Ax2::act5-GFP (Figure 5B). Analyser les mesures standards de chimiotactisme des cellules migrant sous l’agarose a révélé que les cellules NC4::act5-mCherry étaient plus rapides et ont montré une réponse chimiotactique plus forte que les cellules Ax2::act5-GFP (Figure 5C-E ).

Figure 5 : En utilisant agir5 KIs pour expériences de mélange imageur chimiotactisme. (A) NC4::act5-mCherry et Ax2::act5-GFP exprimant la protéine fluorescente indiquée du locus agir5 ont été analysées pour la capacité de croître sur une pelouse bactérienne. Après 4 jours, on a mesuré le diamètre de la plaque de cellules solitaires de Dictyostelium plaqués. Non-axéniques Loi5:: NC4 cellules produisent des plaques significativement plus grandes que les cellules Ax2::act5-GFP axéniques (moyenne ± écart-type, *** p < 0,0001, n = 3, échelle bar 5 mm). (B) pour l’utilisation de la chimiotaxie de folates sous-agarose assay22 de comparer directement les capacités chimiotactiques des NC4::act5-mCherry et Ax2::act5-GFP utilisé dans (A), bactéries-cultivé amibes des deux souches ont été mélangés dans un rapport de 50 : 50. Les cellules étaient autorisés à ramper sous l’agarose. Après 4 h, cellules qui migraient vers le haut de la pente de l’acide folique ont été projetés à l’aide d’un microscope confocal. Le nombre de cellules NC4::act5-mCherry et Ax2::act5-GFP a été déterminé. Les cellules NC4::act5-mCherry ont été plus efficaces dans la détection des folates. Environ 10 fois plus NC4::act5-mCherry cellules ont été trouvés par rapport aux cellules Ax2::act5-GFP (moyenne ± écart-type, *** p < 0,0001, n = 6, échelle bar 100 µm). (C, D) Les cellules ont été filmés pendant 60 min, et leur vitesse et leur indice chimiotactique ont été calculés. Après présélection des cellules plus chimiotaxiquement sensibles (seulement ceux qui ont migré sous l’agarose), Ax2::act5-GFP cellules ont montré des valeurs plus faibles pour la vitesse de la cellule et du chimiotactisme. Cinquante cellules par lignée cellulaire ont été analysés. (moyenne ± écart-type, * p < 0,01, n = 3). (E) les traces de NC4::act5-mCherry et Ax2::act5-GFPact5 knock-ins sont tracés pendant 60 min, retraçant l’évolution plus dirigée vers la source de chemoattractent des cellules NC4::act5-mCherry (moyenne ± écart-type, ** * p < 0,0001, n = 6). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Les lignes de knock-in cell Loi5 permet également pour la cytométrie en flux. Comme pour la croissance sur les bactéries, il y a des différences majeures dans le développement entre souches axéniques et non axéniques sauvage-types. Après développement, les organes de fructification des cellules NC4 sont environ deux fois aussi grands que ceux dérivés de cellules Ax2. Si les cellules sont mélangés, une fructification de taille intermédiaire est obtenue. Pour analyser la contribution des deux lignées cellulaires plus quantitativement, cytométrie en flux a été utilisée. Ces analyses ont montré clairement que les fructifications de taille intermédiaire sont dus à différents niveaux de contribution de deux lignées de cellules. Alors que les cellules NC4::act5-mCherry comptaient d’environ 75 % des spores mesurées, Ax2::act5-GFP a contribué seulement 25 %, révélant un avantage potentiel de remise en forme pour les souches non-axéniques (Figure 6). Étant donné que l’analyse ne surveille pas la population de cellules de tige, il y a deux possibilités pour expliquer le déséquilibre de développement entre Ax2 et NC4. Il est possible que les cellules Ax2 contribuent principalement à la population de cellules de tige, plutôt que d’entrer dans la population de cellules de spore. Sinon, plus de cellules NC4 peuvent conclure le cycle de développement, avec Ax2 relativement retardé, et ils sont donc incapables de contribuer à l’Assemblée d’un organe de fructification. La possibilité de transfecter les cellules de type sauvage non axéniques développe davantage les autres approches et simplifie considérablement les procédures expérimentales.

Figure 6: loi5 KIs permettent une analyse des expériences de mélange à l’aide de cytométrie. (A) NC4::act5-mCherry et Ax2::act5-GFP ont été élaborés séparément ou dans un mélange 50/50 sur milieu gélosé non nutritifs. NC4::act5-mCherry cellules forment des organes de fructification plus grandes que Ax2::act5-GFP. Le mélange montre plutôt de taille intermédiaire (échelle bar 5 mm). Microscopie confocal fluorescence suggère une plus grande quantité de spores de NC4::act5-mCherry dans les têtes de spores provenant de mélanges. (B) afin de quantifier cette observation, les quantités de spores dans spore récolté, chefs de l’expérience de mélange au point A ont été analysés par cytométrie en flux. Environ 75 % des spores provient de cellules NC4::act5-mCherry , avec seulement 25 % des cellules Ax2::act5-GFP . (C) répartition représentative des spores de deux lignées de cellules montrées un scatter de cytométrie de flux. Environ 0,05 % montrent positifs mCherry et GFP de signaux, ce qui suggère que les processus de fusion parasexuel ont eu lieu ou que les spores sont coller les uns aux autres. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Lignée cellulaire | Génétiquement de fond | Numéro de référence | Publié avant | Type de |
| AX2 (Ka) | DBS0235521 | Bloomfield et al., 2008 | type sauvage | |
| NC4 (S) | Bloomfield et al., 2008 | type sauvage | ||
| act5::mCherry Clone 5 | NC4 | HM1912 | Paschke et coll., 2018 | Loi5 knock-in |
| act5::GFP Clone 2 | AX2 | HM1930 | Paschke et coll., 2018 | Loi5 knock-in |
| V12M2 | Bloomfield et al., 2008 | type sauvage | ||
| WS2162 | Bloomfield et al., 2008 | type sauvage |
Supplémentaire tableau 1 : différentes souches de Dictyostelium étudié.
Discussion
L’utilisation de cellules de Dictyostelium non axéniques, sauvage a été très limitée jusqu’ici à la recherche moléculaire. Méthodes disponibles pour le génie génétique de ces souches ont manqué de fiabilité et efficacité23, empêchant leur adoption générale. Les matériaux générés et les protocoles présentés ici peuvent servir pour n’importe quelle contrainte de d. discoideum indépendamment de sa capacité à croître dans un milieu liquide. Il convient de mentionner que ce protocole est optimisé pour les lignées cellulaires dérivées de NC4. Efficacité de transfection des souches fraîchement isolées dans la nature se distinguent NC4, comme nous l’avons observé avant et sont indiqués ici pour V12M2 et WS21628,9. Les conditions d’électroporation en particulier semblent avoir une influence considérable sur l’efficacité de transfection et nécessitent davantage optimisation pour certaines souches. En général, une quantité suffisante de transfectants ont été observées chez toutes les souches testées jusqu'à présent, montrant que ces méthodes sont réalisables. Le nombre de transfectants positifs obtenus à l’aide de souches dérivées NC4 est plus élevé comparé à d’autres souches de type sauvage non axéniques, mais dans tous les cas, un nombre suffisant de transfectants est obtenu pour permettre à d’autres expériences. Cela, plus la simplicité de notre protocole, est l’amélioration majeure par rapport aux précédentes tentatives8,9.
Ces nouveaux protocoles non-axéniques couvrent toutes les méthodes génétiques standards et ajouter d’autres avantages, puisque transfections peuvent être exécutées rapidement et efficacement en parallèle. Les clones positifs peuvent être obtenus en jours et non en semaines, depuis la croissance sur les moitiés de bactéries le temps de la division. Les plasmides nouvellement créés également travaillent dans des conditions axéniques et peuvent être couramment utilisés dans les deux conditions de croissance, qui est un autre développement ultérieur de cette méthodologie22. Tel qu’introduit dans le protocole, le plasmide utilisé pour la sélection des bactéries ont des exigences particulières qui sont cruciales pour le succès de la transfection. En particulier, les promoteurs, les cassettes d’expression et de la résistance au volant sont importantes pour la transfection réussie. Souvent utilisé loi revenu6 (actine 6) promoteur24 pour conduire les cassettes de résistance ou d’expression manque d’efficacité lorsque les cellules sont cultivées sur des bactéries. Dans notre système de plasmide, le promoteur très actif électroniques15 (actine 15) anime toutes les cassettes d’expression, tandis que les cassettes de résistance sont sous le contrôle d’un promoteur de coA (coactosin A), les deux qui sont actives dans des conditions axéniques et dans cellules cultivées sur les bactéries. Exigences pour l’expression du journaliste construit ainsi que knock-in et le knock out constructions rendent nécessaire l’utilisation de notre répertoire de plasmide, mais malheureusement, limite l’utilisation des vecteurs déjà créés qui utilisent le promoteur inefficace loi revenu6 .
Efficacité du promoteur est particulièrement critiques pour transfections qui dépendent d’un événement unique d’intégration dans le locus génomique correct. Grâce à notre amélioration des promoteurs de l’expression de gène de résistance au volant, suffisamment de protéines résistance provient des intégrations locus unique. Une préoccupation majeure était la favorisant des intégrations multiples dans le génome, lorsque vous utilisez l’hygromycine ou G418 comme décrit. Aucun favorisant des intégrations multiples n’a été observé jusqu'à présent, comme indiqué précédemment pour les sélections de G418 à l’aide de systèmes plus anciens promoteur25. Cela signifie que les deux hygromycine et G418 sont marqueurs optionnels appropriés pour la génération des débouchures propres et knock-assur. Malheureusement, la blasticidine marqueur de sélection ne fonctionne pas dans des conditions axéniques-non. Il s’agit d’un inconvénient majeur de notre méthode, étant donné que la cassette de résistance blasticidine est régulièrement utilisée pour générer des constructions knock out chez d. discoideum. Des constructions qui ont été déjà générées avec des cassettes de résistance blasticidine devra être redémontrée à l’aide d’un des marqueurs sélectionnables réalisables. Une autre possibilité pour contourner cette limitation consiste à combiner le axéniques récemment créé d. discoideum CRISPR technologie avec ce protocole de transfection26. La génération d’ARN guide adapté, unique (sgRNAs) est plus simple et plus rapide que la reconstruction des constructions de knock out complets. Pour les orientations futures, la possibilité de générer les débouchures multiples à l’aide de CRISPR/Cas9 avec ce protocole de transfection est attrayant et peut ouvrir la voie à de nombreux chercheurs dans la communauté de Dictyostelium . Cependant, la maniabilité du système établi expression transitoire utilisé pour CRISPR/Cas9 dans des cellules cultivées axéniques devrait être soigneusement examinée.
Le système de knock-in Loi5 présenté propose un système d’intégration fiable et sûre pour la génération des lignées cellulaires stables chez d. discoideum, avec l’avantage de sites similaires établis dans d’autres organismes22. Aussi, il dépend d’un événement unique d’intégration dans le génome et offre de nombreuses possibilités pour les directions de recherche différents. Le promoteur Loi5 est fortement actif et garantit presque homogène expression27 indépendamment des conditions de culture. Les protéines fluorescentes journaliste peuvent s’intègre dans ce locus de refuge sûr en utilisant des plasmides de ciblage machinés. Cela peut être utile, par exemple, à des fins de suivi des cellules, comme illustré ici dans une expérience de mix. Les cellules présentent une variabilité d’expression minimale de cellule à cellule, qui peut aider dans la cellule automatisé de suivi. Ce qui est important, l’insertion d’une séquence souhaitée dans le locus agir5 apparaît phénotypiquement neutre28,29. Comme l’expression ne dépend pas d’un marqueur spécifique de maintenir l’expression de la protéine, comme en intégrants aléatoire ou extrachromosomique vecteur portant les lignées cellulaires, le système de Loi5 peut être utile pour des expériences de sauvetage, ainsi. Étant donné que les lignées cellulaires sont homogènes, toutes les cellules peuvent être analysés. Ce n’est pas possible en utilisant un plasmide extrachromosomique pour l’expression, dans lequel il y a une hétérogénéité considérable dans l’expression.
Outre ces améliorations générales pour toutes les souches, la possibilité d’utiliser des cellules de type sauvage non axéniques de recherche moléculaire permet d’évaluer les effets de mutations accumulées dans les souches de laboratoire actuel. Les réarrangements génétiques multiples a été constaté depuis l’adoption de la souche Ax2 en 1970, tel qu’illustré par microarray publiées antérieurement analyses26. Phénotypes synthétiques sont observées en raison de la présence de ces mutations. Par exemple, un mutant déclarés phg2 montre une diminution d’adhérence dans un contexte de souche de laboratoire et une adhérence accrue dans un autre3. Ces données contradictoires peuvent maintenant être résolues en répétant l’expérience chez la souche d’ancêtre commun (dans ce cas, DdB). La force de cette approche a été récemment démontrée pour la petite GTPase RasS30,31.
La capacité d’effectuer des expériences génétiques dans n’importe quelle contrainte de d. discoideum développe le potentiel des nouvelles orientations de recherche qui dépendent de l’utilisation de souches sauvages, notamment ceux mettant en cause l’évolution sociale, reconnaissance de parenté et le cycle sexué22.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêt à divulguer.
Remerciements
Les auteurs remercient le lab Kay pour l’entrée de cette méthode et les installations LMB microscopie et écoulement cytometry pour excellent soutien scientifique et technique. Ce travail a été financé par Medical Research Council MC_U105115237 à R. R. Kay, BBSRC (Biotechnology and Biological Sciences Research Council) subvention BB/K009699/1 à R. R. Kay, Cancer Research UK accorder A15672 à R. H. Insall, Wellcome Trust Senior Fellowship 202867/Z/16/Z pour Jonathan R Chubb et MRC financement (MC_U12266B) à l’unité de l’Université de LMCB MRC à l’UCL.
matériels
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Eppendorf Microcentrifuges 5424/5424R | ThermoScientific | 05-400-002 | |
| Eppendorf 5702R Centrifuges with A-4-38 Model Rotor | ThermoScientific | 12823252 | |
| Eppendorf Mastercycler Nexus Thermal Cyclers | Sigma Aldrich | EP6331000025 | |
| Gene Pulser X Cell, including CE & PC modules | BioRad | 1652660 | |
| Safety Cabinet Foruna - SCANLAF | Labogene | ||
| BioPhotometer Plus | Eppendorf | ||
| CLSM 710 | Zeiss | ||
| Media & Agaroses | |||
| LoFlo Medium | Formedium | LF0501 | |
| Seakem GTG Agarose | Lonza | 50071 | |
| Low EEO Agarose | BioGene | 300-200 | |
| Purified Agar | Oxoid | LP0028 | |
| SM broth | Formedium | SMB0102 | |
| 2x LB broth | Formedium | LBD0102 | |
| Chemicals | |||
| SYBR Safe DNA Gel Stain | ThermoScientific | S33102 | |
| 1 Kb Plus DNA Ladder | ThermoScientific | 10787018 | |
| 1 Kb DNA Ladder | NEB | N3232L | |
| HEPES free acid | Sigma Aldrich/Merck | 391340 EMD | |
| KH2PO4 | VWR | P/4800/53 | |
| Na2HPO4 * 2 H2O | VWR | 10028-24-7 | |
| MgCl2 * 6 H2O | Sigma Aldrich/Merck | 5982 EMD | |
| CaCl2 * 2 H2O | Sigma Aldrich | 442909 | |
| Folic Acid | Sigma Aldrich | F7876 | |
| KOH | VWR | 26668.263 | |
| Cultur Dishes | |||
| 96 Well Cell Culture Cluster, Flat Bottom with Low Evaporation Lid, Tissue Culture Treated | Corning Incorporated | 3595 | |
| 6 Well Cell Culture Cluster, Flat Bottom with Lid, Tissue Culture Treated | Corning Incorporated | 3516 | |
| 100 x 20 mm style Tissue Culture Dish, Tissue Culture Treated | Corning Incorporated | 353003 | |
| 50 mm Glass Bottom Microwell Dishes No1.5 | MatTek Corporation | P50G-1.5-30-F | |
| Antibiotics | |||
| Geneticin G418-sulphate | Gibco by Life Technologies | 11811-023 | |
| Hygromycin B Gold | InvivoGen | ant-hg-1 | |
| Additional Consumables | |||
| 2 mm gap electroporation cuvettes, long electrode | Geneflow Limited | E6-0062 | |
| 10 µL Inoculating Loop, Blue 10 Micro L | ThermoScientific | 129399 | |
| Spreader, L-shaped, sterile | greiner bio-one | 730190 | |
| Combitips advanced 5 mL | Eppendorf BIOPUR | 30089669 | |
| Kits | |||
| ZR Plasmid Miniprep Classic | Zymoresearch | D4016 | |
| Quick DNA Miniprep Kit | Zymoresearch | D3025 | |
| Zymoclean DNA Recovery Kit | Zymoresearch | D40002 | |
| Enzymes | |||
| Restriction enzymes | NEB | ||
| OneTaq 2X Master Mix with Standard Buffer | NEB | M0482L | |
| T4 DNA Ligase | NEB | M0202S | |
| PrimeSTAR Max DNA Polymerase | TakaraBio | R045A |
Références
- Schaap, P. Evolutionary crossroads in developmental biology: Dictyostelium discoideum. Development. 138 (3), 387-396 (2011).
- Sussman, R., Sussman, M. Cultivation of Dictyostelium discoideum in axenic culture. Biochemical and Biophysical Research Communications. 29, 53-55 (1967).
- Bloomfield, G., Tanaka, Y., Skelton, J., Ivens, A., Kay, R. R. Widespread duplications in the genomes of laboratory stocks of Dictyostelium discoideum. Genome Biology. 9 (4), 75 (2008).
- Witke, W., Nellen, W., Noegel, A. Homologous recombination in the Dictyostelium alpha-actinin gene leads to an altered mRNA and lack of the protein. The EMBO Journal. 6, 4143-4148 (1987).
- De Lozanne, A., Spudich, J. A. Disruption of the Dictyostelium myosin heavy chain gene by homologous recombination. Science. 236, 1086-1091 (1987).
- Howard, P. K., Ahern, K. G., Firtel, R. A. Establishment of a transient expression system for Dictyostelium discoideum. Nucleic Acids Research. 16, 2613-2623 (1988).
- Knecht, D., Pang, K. M. Electroporation of Dictyostelium discoideum. Methods in Molecular Biology. 47, 321-330 (1995).
- Wetterauer, B., et al. Wild-type strains of Dictyostelium discoideum can be transformed using a novel selection cassette driven by the promoter of the ribosomal V18 gene. Plasmid. 36, 169-181 (1996).
- Lloyd, M. M., Ceccarelli, A., Williams, J. G. Establishment of conditions for the transformation of nonaxenic Dictyostelium strains. Developmental Genetics. 11, 391-395 (1990).
- Bloomfield, G., et al. Neurofibromin controls macropinocytosis and phagocytosis in Dictyostelium. eLife. 4, (2015).
- Veltman, D. M., et al. A plasma membrane template for macropinocytic cups. eLife. 5, (2016).
- Veltman, D. M., Lemieux, M. G., Knecht, D. A., Insall, R. H. PIP(3)-dependent macropinocytosis is incompatible with chemotaxis. The Journal of Cell Biology. 204 (4), 497-505 (2014).
- Hoeller, O., Kay, R. R. Chemotaxis in the absence of PIP3 gradients. Current Biology. 17, 813-817 (2007).
- Chubb, J. R., Wilkins, A., Thomas, G. M., Insall, R. H. The Dictyostelium RasS protein is required for macropinocytosis, phagocytosis and the control of cell movement. Journal of Cell Science. 113, 709-719 (2000).
- Schaap, P., et al. Molecular phylogeny and evolution of morphology in the social amoebas. Science. 314, 661-663 (2006).
- Du, Q., Kawabe, Y., Schilde, C., Chen, Z. H., Schaap, P. The Evolution of Aggregative Multicellularity and Cell-Cell Communication in the Dictyostelia. Journal of Molecular Biology. 427 (23), 3722-3733 (2015).
- Hirose, S., Benabentos, R., Ho, H. I., Kuspa, A., Shaulsky, G. Self-recognition in social amoebae is mediated by allelic pairs of tiger genes. Science. 333 (6041), 467-470 (2011).
- Strassmann, J. E., Queller, D. C. Evolution of cooperation and control of cheating in a social microbe. Proceedings of the National Academy of Sciences of the United States of America. 108 (2), 10855-10862 (2011).
- Wolf, J. B., et al. Fitness Trade-offs Result in the Illusion of Social Success. Current Biology. 25 (8), 1086-1090 (2015).
- Veltman, D. M., Akar, G., Bosgraaf, L., Van Haastert, P. J. A new set of small, extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid. 61 (2), 110-118 (2009).
- Southern, E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. Journal of Molecular Biology. 98 (3), 503-517 (1975).
- Paschke, P., et al. Rapid and efficient genetic engineering of both wild type and axenic strains of Dictyostelium discoideum. PLoS One. 13 (5), 0196809 (2018).
- Woznica, D., Knecht, D. A. Under-agarose chemotaxis of Dictyostelium discoideum. Methods in Molecular Biology. 346, 311-325 (2006).
- Dubin, M., Nellen, W. A versatile set of tagged expression vectors to monitor protein localisation and function in Dictyostelium. Gene. 465 (1-2), 1-8 (2010).
- de Hostos, E. L., et al. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. Journal of Cell Biology. 120, 163-173 (1993).
- Sekine, R., Kawata, T., Muramoto, T. CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium. Scientific Reports. 8 (1), 8471 (2018).
- Sadelain, M., Papapetrou, E. P., Bushman, F. D. Safe harbours for the integration of new DNA in the human genome. Nature Reviews Cancer. 12 (1), 51-58 (2011).
- Rosengarten, R. D., et al. Leaps and lulls in the developmental transcriptome of Dictyostelium discoideum. BMC Genomics. 16, 294 (2015).
- Tunnacliffe, E., Corrigan, A. M., Chubb, J. R. Promoter-mediated diversification of transcriptional bursting dynamics following gene duplication. Proc Natl Acad Sci USA. 115 (33), 8364-8369 (2018).
- Gebbie, L., et al. Phg2, a kinase involved in adhesion and focal site modeling in Dictyostelium. Molecular Biology of the Cell. 15, 3915-3925 (2004).
- Jeon, T. J., Lee, D. J., Merlot, S., Weeks, G., Firtel, R. A. Rap1 controls cell adhesion and cell motility through the regulation of myosin II. Journal of Cell Biology. 176, 1021-1033 (2007).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.