
Method Article
Ingegneria genetica di Dictyostelium discoideum cellule basata sulla selezione e crescita sui batteri
In questo articolo
Riepilogo
Dictyostelium discoideum è un organismo modello popolare per studiare i complessi processi cellulari come la migrazione cellulare, endocitosi e sviluppo. L'utilità dell'organismo è dipenda sulla fattibilità di manipolazione genetica. Qui, presentiamo metodi per transfect Dictyostelium discoideum cellule che superare i limiti esistenti delle cellule di coltura in terreno liquido.
Abstract
Dictyostelium discoideum è un intrigante organismo di modello per lo studio dei processi di differenziazione delle cellule durante lo sviluppo, di segnalazione delle cellule e altre domande importanti di biologia cellulare. Le tecnologie disponibili per manipolare geneticamente le cellule Dictyostelium sono ben sviluppate. Transfezioni possono essere eseguite utilizzando differenti marcatori selezionabili e marcatore ri-escursioni in bicicletta, tra cui ricombinazione omologa e mutagenesi inserzionale. Questo è supportato da un genoma ben commentato. Tuttavia, questi approcci sono ottimizzati per linee cellulari axeniche crescendo in colture liquide e sono difficili da applicare per non axeniche cellule wild-type, si nutrono solo di batteri. Le mutazioni che sono presenti nei ceppi axeniche disturbano Ras segnalazione, causando eccessivo Macropinocitosi necessaria per l'alimentazione e alterano la migrazione cellulare, che confonde l'interpretazione della trasduzione del segnale e gli esperimenti di chemiotassi in quegli sforzi. Precedenti tentativi di manipolare geneticamente le cellule non-axeniche hanno mancato efficienza e procedure sperimentali complesse richieste. Abbiamo sviluppato un protocollo di transfezione semplice che, per la prima volta, supera queste limitazioni. Quelli una serie di miglioramenti di grandi dimensioni per la genetica molecolare di Dictyostelium consentire cellule wild-type essere manipolato facilmente come ceppi di laboratorio standard. Oltre ai vantaggi per lo studio dei processi di segnalazione e motilità incorrotto, mutanti che interrompono la crescita basata su Macropinocitosi possono ora essere facilmente isolate. Inoltre, il flusso di lavoro intera transfezione è notevolmente accelerato, con cellule ricombinanti che possono essere generate in giorni anziché in settimane. Un altro vantaggio è che la genetica molecolare ulteriormente può essere eseguita con i campioni di Dictyostelium selvaggio-tipo appena isolati dall'ambiente. Questo può aiutare a estendere l'ambito di approcci utilizzati in queste aree di ricerca.
Introduzione
Il genere di Dictyostelium sono terreno-viventi ameba sociale che principalmente si nutrono di batteri. Essere immessi nel phylum Amoebozoa, un gran numero di specie è stato isolato che possono essere raggruppate in quattro differenti cladi1. Le specie di Dictyostelium discoideum (d. discoideum) sono diventato un organismo modello popolare per studiare i complessi processi cellulari come la migrazione cellulare e la fagocitosi. Di controllare e standardizzare le condizioni sperimentali, axenica cella sono state sviluppate linee che sono in grado di crescere in mezzo liquido complesso o definito in assenza di batteri2. Di particolare importanza sono il Ax2, Ax3, e isolare i ceppi Ax4, che erano tutti generati nel 1970 e infine derivati da un singolo wild NC43. Strumenti per l'ingegneria genetica sono stati sviluppati in questi ceppi axeniche, conseguente il primo Ko pubblicato nel 19874,5. Protocolli sono stati ulteriormente sviluppati e ottimizzati per l'utilizzo sotto condizioni axeniche6,7.
Adattamento di questi protocolli di selvaggio-tipo d. discoideum ceppi che non sono in grado di crescere in brodo liquido sono state tentate da diversi laboratori. Tuttavia, questo non è diventato completamente successo poiché i protocolli di transfezione sono complessi e la mancano di efficienza, in parte a causa della capacità dei batteri di agire come un lavandino per i reagenti selettiva8,9. Di conseguenza, dati essenzialmente tutto molecolari su d. discoideum provengono da discendenti di un singolo isolato di selvaggio-tipo. Abbiamo voluto superare questo limite e sviluppare un metodo per modificare geneticamente le cellule d. discoideum indipendente della loro capacità di crescere in mezzo liquido. La necessità di tale metodo può essere spiegata tramite l'osservazione che è stata assunta in passato che le mutazioni permettendo axenica crescita erano principalmente neutrale e non altera la fisiologia cellulare. Questa supposizione è solo parzialmente corretta. In generale, esistono due differenze notevoli; in primo luogo, tra i diversi isolati ceppi axeniche e secondo, quando questi ceppi axeniche vengono confrontati con non-axeniche selvatici isolati8,9.
Forse il fattore più critico è il gene axenica chiave, asciaB, che è stato identificato recentemente come RasGAP NF1. La funzione principale di NF1 come un RasGAP è di frenare Ras attività3. L'eliminazione dell'enzima in tutti i ceppi axeniche conduce ad un'eccessiva attività di Ras che si è manifestata come la formazione di grandi macchie di Ras attive. Queste patch di Ras allargate portano all'accumulo di PIP3 nella membrana plasmatica. Quelle macchie comparenti coincidente di PIP3 e Ras attivo sono un modello per la formazione di una balza circolare che alla fine si chiude e porta alla formazione di macropinosomes10. La conseguenza è un aumento eccessivo nell'attività macropinocytic. Macropinocitosi sono un processo basato su actina. Un concorso per componenti citoscheletriche per la formazione di macropinosomes o pseudopodi è il risultato. Suo impatto sul comportamento delle cellule si riflette nella prevenzione quasi completa della chemiotassi di cellule vegetative di folato11. Le patch di PIP3 in maniera massiccia ingrandetti sono molto persistenti. Anche nelle celle di morti di fame, le patch di PIP3 rimangono e possono essere erroneamente interpretate come pseudopodi, che possono causare problemi di interpretazione studi sulla chemiotassi al campo.
In alcuni casi, la mutazione NF1 è sperimentalmente utile. Questo ci conduce ad una seconda motivazione per lo sviluppo di un metodo di transfezione per batterico cresciuto d. discoideum cellule, poiché l'aumento nel tasso di Macropinocitosi rende le cellule axeniche prezioso per indagare gli aspetti fondamentali di questo processo12 . Tuttavia, mutazione nei geni necessari per Macropinocitosi, come Ras e PI3-chinasi10, hanno abolito quasi crescita axenica, rendendo necessario per manipolare queste cellule attraverso la crescita sui batteri. Un altro motivo che esegue il rendering basato su batteri transfezioni prezioso è l'uso aumentante di Dictyostelids per esplorare domande nell'evoluzione della multi-cellularità13,14, riconoscimento kin15,16, e il comportamento cellulare altruistico, che dipendono principalmente dall'uso di recente isolate di selvaggio-tipo isola17. Tutti questi ambiti di ricerca possono essere facilitate da metodi efficienti per la manipolazione genetica degli isolati selvatici, che sono non-axeniche e non crescono in brodo liquido.
I nostri protocolli consentono per il superamento delle limitazioni descritte. Presi insieme, la possibilità di effettuare manipolazioni genetiche con batterico cresciuto i benefici d. discoideum cellule prese per tutti i ricercatori di Dictyostelium , anche se è solo la maggiore velocità del processo di selezione grazie alla più veloce crescita dell'ameba (tempo di raddoppiamento di 4 h) sui batteri rispetto alla crescita media axeniche (10 h tempo di raddoppiamento).
Protocollo
1. preparazione delle cellule e materiali
-
Preparazione del tampone di SorMC
- Preparare 100 mL di tampone (buffer di Sorensen incluso MgCl2 e CaCl2) SorMC: 100x sciogliendo 20,36 g di KH2PO4 (15 mM) e 5,47 g di Na2HPO47 H2O (2 mM) in 100 mL di acqua O di ddH2. Agitare la soluzione a temperatura ambiente (TA) e portare il volume a 100 mL con dH2O.
Nota: Il buffer risultante ha un pH di 6 e non ha bisogno di ulteriore regolazione. - Produrre 1000 mL di 1x soluzione di lavoro in ddH2O. aggiungere 50 µ l ciascuna di MgCl2 e CaCl2 per ottenere le concentrazioni finali di 50 µM ciascuna. Filtro sterilizzare la soluzione utilizzando un filtro da 0,22 µm.
Nota: Sempre aggiungere MgCl2 e CaCl2 il buffer 1x per evitare la precipitazione dei sali nel 100 x soluzione di riserva. - In alternativa, preparare KK2 tampone (2,2 g di KH2PO4 e 0,7 g di K2HPO4 per 1 L di tampone) completati con 50 µM MgCl2 e 50 µM CaCl2 (in appresso come KK2MC). Utilizzare questo buffer in tutto invece di SorMC.
- Preparare 100 mL di tampone (buffer di Sorensen incluso MgCl2 e CaCl2) SorMC: 100x sciogliendo 20,36 g di KH2PO4 (15 mM) e 5,47 g di Na2HPO47 H2O (2 mM) in 100 mL di acqua O di ddH2. Agitare la soluzione a temperatura ambiente (TA) e portare il volume a 100 mL con dH2O.
-
Preparazione di batteri come fonte di cibo per d. discoideum
- Utilizzare una singola colonia di K. aerogenes e inoculare 1 L di LB-medium (brodo di Lisogenesi). Utilizzare un pallone da 2 L. Lascia i batteri di crescere durante la notte a 37 ° C con agitazione a 220 giri/min.
Nota: Se grandi quantità di batteri sono necessari, utilizzare mezzi di comunicazione più ricco come 2xTY (Estratto di lievito tryptone Media) o SOB (super ottima brodo) invece di LB. Nel caso in cui l'uso di K. aerogenes batteri non è consentito a causa di restrizioni di sicurezza, BL21 Escherichia coli può essere utilizzato invece. - Raccogliere le cellule il giorno successivo di filatura li giù in due provette da centrifuga 500 mL a ~ 6.600 x g per 20 min. batteri di lavare una volta con 500 mL di buffer di SorMC.
- Risospendere il pellet in 20 mL di SorMC. Verifica il OD600 (densità ottica a 600 nm) utilizzando un fotometro. Diluire con lo stesso buffer per un OD600 di circa 100.
Nota: K. aerogenes batteri sono difficili a pellet, quindi una velocità relativamente elevata per rotazione verso il basso i batteri è necessaria per evitare la perdita di batteri di cibo. La soluzione risultante di stock batterica possono essere memorizzati fino a 4 mesi in frigorifero a 4 ° C e mantenere la propria utilità come fonte di cibo per d. discoideum. 1 L di pernottamento K. aerogenes sospensioni sviluppate in LB medium solitamente produce 20 mL di un OD600 di circa 100.
Attenzione: Per garantire che i batteri preparati sono una monocultura di K. aerogenes, eseguire tutti i passaggi sotto una cappa.
- Utilizzare una singola colonia di K. aerogenes e inoculare 1 L di LB-medium (brodo di Lisogenesi). Utilizzare un pallone da 2 L. Lascia i batteri di crescere durante la notte a 37 ° C con agitazione a 220 giri/min.
-
Preparazione del buffer di elettroporazione H40
- Preparare 100 mL di soluzione tampone, sciogliere g 0,952 di HEPES in ddH2O acqua e aggiungere 100 µ l di MgCl2 da una soluzione madre di 1m. Regolare il pH 7 utilizzando KOH per titolazione. Sterilizzare il buffer utilizzando un filtro da 0,22 µm o in autoclave. Utilizzare HEPES privo di acidi e non il sale di sodio.
- Eseguire la preparazione del plasmide seguendo il protocollo del produttore e utilizzando i kit di riassunti nella Tabella materiali. Utilizzare i plasmidi riassunti nella tabella 1.
Nota: La qualità del DNA usato per la transfezione è cruciale. La selezione di d. discoideum trasfettanti crescendo sui batteri ha requisiti specifici per i promotori della cassetta di selezione ed espressione di guida (vedi discussione).
| nome del plasmide | selezione/resistenza nei batteri | resistenza/selezione in Dictyostelium | Prodotto Tag |
| plasmidi di espressione extracromosomico | |||
| pDM1203 | Ampicilin | G418 | No |
| pDM1207 | Ampicilin | G418 | N-terminale GFP |
| pDM1208 | Ampicilin | G418 | N-terminale mCherry |
| pPI159 | Ampicilin | G418 | N-terminale mNeon |
| pPI437 | Ampicilin | G418 | N-terminale mScarlet |
| pPI54 | Ampicilin | G418 | N-terminale mTurquoise2 |
| pDM1209 | Ampicilin | G418 | C-terminale GFP |
| pDM1210 | Ampicilin | G418 | C-terminale mCherry |
| pPI143 | Ampicilin | G418 | C-terminale mNeon |
| pPI459 | Ampicilin | G418 | C-terminale mScarlet |
| pPI142 | Ampicilin | G418 | C-terminale mTurquoise2 |
| plasmidi di navetta | |||
| pDM344 | Ampicilin | No | No |
| pDM1019 | Ampicilin | No | N-terminale GFP |
| pDM1018 | Ampicilin | No | N-terminale mCherry |
| pPI152 | Ampicilin | No | N-terminale mNeon |
| pPI418 | Ampicilin | No | N-terminale mScarlet |
| pPI150 | Ampicilin | No | N-terminale mTurquoise2 |
| pDM1021 | Ampicilin | No | C-terminale GFP |
| pDM1020 | Ampicilin | No | C-terminale mCherry |
| pPI153 | Ampicilin | No | C-terminale mNeon |
| pPI457 | Ampicilin | No | C-terminale mScarlet |
| pPI151 | Ampicilin | No | C-terminale mTurquoise2 |
| plasmidi di espressione inducibile extracromosomico | |||
| pDM1038 | Ampicilin | Igromicina | No |
| pDM1047 | Ampicilin | Igromicina | N-terminale GFP |
| pDM1046 | Ampicilin | Igromicina | N-terminale mCherry |
| pPI450 | Ampicilin | Igromicina | N-terminale mNeon |
| pPI452 | Ampicilin | Igromicina | N-terminale mScarlet |
| pPI449 | Ampicilin | Igromicina | N-terminale mTurquoise2 |
| pDM1049 | Ampicilin | Igromicina | C-terminale GFP |
| pDM1048 | Ampicilin | Igromicina | C-terminale mCherry |
| pPI470 | Ampicilin | Igromicina | C-terminale mNeon |
| pPI460 | Ampicilin | Igromicina | C-terminale mScarlet |
| pPI469 | Ampicilin | Igromicina | C-terminale mTurquoise2 |
| rifugio sicuro act5 plasmidi di targeting | |||
| pDM1501 | Ampicilin | Igromicina | No |
| pDM1513 | Ampicilin | Igromicina | N-terminale GFP |
| pDM1514 | Ampicilin | Igromicina | N-terminale mCherry |
| pPI231 | Ampicilin | Igromicina | N-terminale mNeon |
| pPI419 | Ampicilin | Igromicina | N-terminale mScarlet |
| pPI228 | Ampicilin | Igromicina | N-terminale mTurquoise2 |
| pDM1515 | Ampicilin | Igromicina | C-terminale GFP |
| pDM1516 | Ampicilin | Igromicina | C-terminale mCherry |
| pPI230 | Ampicilin | Igromicina | C-terminale mNeon |
| pPI458 | Ampicilin | Igromicina | C-terminale mScarlet |
| pPI229 | Ampicilin | Igromicina | C-terminale mTurquoise2 |
| Plasmidi di espressione di REMI | |||
| pDM1220 | Ampicilin | Igromicina | No |
| pDM1351 | Ampicilin | Igromicina | N-terminale GFP |
| pDM1259 | Ampicilin | Igromicina | N-terminale mCherry |
| pPI465 | Ampicilin | Igromicina | N-terminale mNeon |
| pPI468 | Ampicilin | Igromicina | N-terminale mScarlet |
| pPI466 | Ampicilin | Igromicina | N-terminale mTurquoise2 |
| pDM1352 | Ampicilin | Igromicina | C-terminale GFP |
| pDM1305 | Ampicilin | Igromicina | C-terminale mCherry |
| pPI471 | Ampicilin | Igromicina | C-terminale mNeon |
| pPI467 | Ampicilin | Igromicina | C-terminale mScarlet |
| pPI472 | Ampicilin | Igromicina | C-terminale mTurquoise2 |
| mirate in plasmidi telaio | |||
| pDM1355 | Ampicilin | Igromicina | C-terminale GFP |
| pPI461 | Ampicilin | Igromicina | C-terminale mCherry |
| pPI462 | Ampicilin | Igromicina | C-terminale mNeon |
| pPI464 | Ampicilin | Igromicina | C-terminale mScarlet |
| pPI463 | Ampicilin | Igromicina | C-terminale mTurquoise2 |
| plasmidi di knock-out | |||
| pDM1079 | Ampicilin | Dyfed | No |
| pDM1080 | Ampicilin | Nourseothricin | No |
| pDM1081 | Ampicilin | Igromicina | No |
| pDM1082 | Ampicilin | G418 | No |
| Plasmidi di espressione di CRE | |||
| pDM1483 | Ampicilin | Nourseothricin | No |
| pDM1489 | Ampicilin | Igromicina | No |
| pDM1488 | Ampicilin | G418 | No |
Tabella 1: Elenco di plasmide per transfezioni non axeniche.
-
Configurazione di Dictyostelium cellule per la transfezione
- Crescere di K. aerogenes alla confluenza in medio SM (sostanza-ricco medio) pernottamento al RT. culture possa essere memorizzato fino a 2 settimane a 4 ° C.
- Aggiungere circa 400 µ l di questa sospensione batterica su una piastra di agar di SM (peptone 10 g/L; lievito estratto 1 g/L; glucosio 10 g/L; KH2PO4 1,9 g/L; K2HPO4 x 3 H2O, 1,3 g/L; MgSO4 anidro 0,49 g/L; 1,7% agar) e distribuire uniformemente. Prendere un'ansa sterile e inoculare con cellule di Dictyostelium . Diffondere le cellule ad un orlo della piastra.
- Incubare la piastra a 22 ° C per 2 giorni affinché le zone di crescita sufficientemente grande per la transfezione.
Nota: Per i ceppi di Dictyostelium che non rendere grande crescita zone (ad es., Ax3, DH1 o JH10), o per gli sperimentatori inesperti, utilizzare radura piastre invece. Per questo, seguire le istruzioni in procedura 1.5.4 a 1.5.6. - Prendere un'ansa sterile e inoculare con cellule di Dictyostelium (circa 2-4 x 105 celle). Trasferire le cellule a 800 µ l di una densa K. aerogenes sospensione in cellule SM. Mix pipettando su e giù.
- Trasferimento di 400 µ l, 200 µ l, 100 µ l e 50 µ l su piastre di agar SM freschi. Aggiungere ad ogni piatto 400 µ l di sospensioni aggiuntive SM K. aerogenes , distribuiti in modo uniforme e a secco.
- Incubare le piastre a 22 ° C per circa 2 giorni fino a quando le piastre diventano trasparenti.
Nota: A causa del loro tasso di crescita più veloce, i batteri producono inizialmente un prato confluente e le amebe successivamente "cancellare" la piastra dei batteri. Il tempo richiesto per questo processo può variare a seconda della priorità bassa deformazione e la capacità delle cellule mutanti di crescere sui batteri.
2. transfezione delle celle di Dictyostelium basato su selezione batterica

Figura 1 : Flusso di lavoro per la transfezione delle cellule di batteri-grown Dictyostelium . I passaggi per la transfezione sono elencati come segue. Crescere le cellule d. discoideum su una zolla di SM seminata con batteri K. aerogenes (rosso). Raccogliere le cellule solo dalla parte anteriore d'alimentazione (verde), evitando le cellule che si stanno già sviluppando (verde scuro). Lavare le cellule in H40. Risospendere le cellule ad una densità finale di 2-4 x 107 cellule/mL. Mescolare la sospensione cellulare con 1-2 µ g di DNA. Trasferire il composto in un cellule di cuvette e impulsi di elettroporazione. Trasferire le cellule direttamente dopo l'elettroporazione per un piatto con SorMC e batteri. Permettono alle cellule di recuperare per 5 ore prima di aggiungere il marcatore selezionabile. Per plasmidi extracromosomico, aggiungere la selezione direttamente al piatto. Trasfettanti sono di solito visibili dopo ~ 2 giorni. Per i costrutti linearizzati che mirano per singola integrazione nel genoma, impostare tre diluizioni come indicato e aggiungere la selezione. I batteri sono presi dalla OD600 = 100 soluzione di riserva. Mescolare bene i tubi e trasferire le cellule in piastre di coltura del tessuto 96 pozzetti a fondo piatto. Utilizzare due piastre per diluizione. Dispensare 150 µ l di sospensione cellulare in ogni pozzetto. Ci vogliono circa 5 giorni fino a quando le colonie stretti sono visibili. Il rosso wells Visualizza un esempio di solito la quantità di cellule con successo trasformate ottenute (pannello superiore per volta dal precedente pubblicazione22). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Per preparare piatti con sospensione K. aerogenes , aggiungere 10 mL di tampone di SorMC contenente batteri K. aerogenes ad una densità di OD600 = 2 (aggiungere 200 µ l di preparati OD600 = 100 K. aerogenes soluzione di riserva per la desiderata concentrazione di batteri) in un 10 cm coltura del tessuto trattato di Petri.
Nota: Questo piatto è più tardi necessarie per coltivare la transfected le cellule d. discoideum . In alternativa, può essere utilizzata una piastra di coltura del tessuto 6 pozzetti. Nel caso di transfezione dei plasmidi extracromosomico, la piastra di coltura del tessuto 6 pozzetti è più efficiente delle risorse. Utilizzare 2 mL di SorMC K. aerogenes (OD600 = 2) sospensione per pozzetto. -
Preparazione delle cellule di Dictyostelium
- Utilizzando un ciclo di inoculazione 10 µ l, raschiare le cellule delle zone di crescita (circa 3 cm) della piastra di coltura (bordo della zona deselezionata) o piastra di compensazione. Trasferire le cellule in una provetta da 1,5 mL contenente 1 mL di buffer di H40 ghiacciata.
Nota: I tempi di raccolta delle cellule da radura piastre sono cruciali. Raccolta troppo presto rese troppo poco una quantità di cellule, mentre la raccolta alla fine aumenta il rischio di cedere parzialmente sviluppato cellule. - Lavare le cellule riducendo la velocità per 2 minuti a 1.000 x g o flash-spinning per 2 s a 10.000 x g. Eliminare il supernatante e risospendere le cellule in H40 buffer ad una densità finale di 2-4 x 107 cellule/mL. Mantenere le cellule freddo durante la procedura di trasfezione intero. Utilizzare un impasto di acqua e ghiaccio per assicurare il contatto diretto dei tubi e ghiaccio.
- Utilizzando un ciclo di inoculazione 10 µ l, raschiare le cellule delle zone di crescita (circa 3 cm) della piastra di coltura (bordo della zona deselezionata) o piastra di compensazione. Trasferire le cellule in una provetta da 1,5 mL contenente 1 mL di buffer di H40 ghiacciata.
-
Elettroporazione
- Aggiungere 100 µ l di cellule in una provetta con 1-2 µ g di DNA. Miscelare accuratamente pipettando su e giù.
- Trasferire la miscela di DNA e delle cellule in una cuvetta pre-refrigerati elettroporazione (spazio di 2 mm).
- Le celle utilizzando le seguenti impostazioni di onda quadra di impulso: 350 V, 8 ms, 2 impulsi e 1 intervallo di impulso di s.
Nota: Non aggiungere più di 2 µ g di DNA. Importi superiori sono tossici per le cellule e diminuiscono l'efficienza di trasfezione. Aggiunto il totale volume di DNA non deve superare i 5 µ l. - Trasferire immediatamente le cellule alla precedenza preparato 10 cm Petri con K. aerogenes SorMC e permettono alle cellule di recuperare per 5 h.
Nota: Controllare le celle di Petri sotto un microscopio invertito. Le cellule verranno visualizzati rotonde direttamente dopo l'elettroporazione ma tornerà alla loro forma ameboide dopo circa 30 min, quando hanno avuto tempo sufficiente per connettere correttamente alla superficie.
-
Selezione di trasfettanti: a seconda se transfezione è finalizzata a generare un knock-out, knock-in o act5 knock-in, o per esprimere una proteina reporter fluorescente da un plasmide extracromosomico, eseguire il processo di selezione attraverso uno dei di seguito descritti metodi.
Nota: A differenza delle cellule axenically coltivate, batterico coltivate cellule sono altamente resistenti a Consciousness. Selezione, pertanto, viene sempre effettuata utilizzando G418 o igromicina (vedi discussione).- Knock-out, knock-ins e act5 knock-in
- Staccare le cellule attentamente da di Petri forzando ripetutamente il liquido da una pipetta sulla superficie.
- Impostare tre diluizioni la sospensione di SorMC K. aerogenes (OD600 = 2) e aggiungere l'agente selettivo secondo il resistenza (Vedi Figura 1).
- Bassa diluizione: mescolare 9 mL di sospensione cellulare con 20,4 mL di SorMC e 600 µ l di soluzione madre K. aerogenes .
- Diluizione media: mescolare 900 µ l di sospensione cellulare con 28,5 mL di SorMC e 600 µ l di soluzione madre K. aerogenes .
- Alta diluizione: mescolare 90 µ l di sospensione cellulare con 29,3 mL di SorMC e 600 µ l di soluzione madre K. aerogenes .
- Aggiungere 30 µ l di marcatore selezionabile (100 x soluzione di riserva).
- Distribuire le diluizioni preparate in piastre di coltura del tessuto 96 pozzetti a fondo piatto di pipettaggio 150 µ l di sospensione cellulare in ogni pozzetto.
Nota: Questa procedura ha lo scopo di cloni singolo schermo piuttosto che popolazioni. La selezione avviene circa 5-7 giorni, a seconda del costrutto utilizzato.
- Extracromosomico plasmidi
- Aggiungere il marcatore selezionabile direttamente al piatto da 10 mL (Vedi Figura 1).
Nota: Non esiste alcuna necessità di impostare diluizioni, poiché non vi è alcun desiderio per le popolazioni clonali. Il processo di selezione per plasmidi extracromosomico è più veloce dovuto ad alti numeri di copia presenti nelle cellule di d. discoideum . Trasfettanti possono essere preveduto dopo 32 h per 2 giorni.
Attenzione: Gli antibiotici usati come marcatore selezionabile sono tossici. Indossare i guanti.
- Aggiungere il marcatore selezionabile direttamente al piatto da 10 mL (Vedi Figura 1).
- Knock-out, knock-ins e act5 knock-in
-
Schermo i cloni ottenuti per tranfectants positivo. Per i tentativi di knock-out o knock-in, seguire le istruzioni al punto 2.5.1. Verifica il successo di transfezione per plasmidi extracromosomico utilizzando le istruzioni al punto 2.5.2.
- Per knock-out, knock-in e act5 knock-in, eseguire la schermata iniziale tramite PCR per confermare integrazione del costrutto il locus genomico corretto.
Nota: Per massimizzare la probabilità di popolazioni clonali, utilizzare la diluizione più alta possibile che produce trasfettanti dopo selezione. Obiettivo per piatti che sono un massimo un terzo dei pozzi occupati.- Per espandere le popolazioni clonali, trasferire i cloni che sono cresciuti dopo la selezione dalla piastra di coltura del tessuto 96 pozzetti in una piastra di coltura del tessuto 12-pozzetti a crescere abbastanza cellule per l'isolamento di DNA genomic. Fornire ogni pozzetto con 1 mL di SorMC K. aerogenes (OD600 = 2) e fresco marcatore selezionabile.
Nota: 1 giorno è di solito sufficiente per ottenere un confluenti ben adatto per isolamento del DNA. - Per eseguire mini isolamento di DNA genomic, raccogliere le cellule di un pozzo confluente e isolare il DNA di genomic usando un mini kit di estrazione del DNA seguendo le istruzioni del produttore.
- Utilizzare il DNA di genomic isolato insieme con primer adatto e Taq-polimerasi (Vedi Tabella materiali) alla schermata PCR per componenti positivi.
Nota: Dopo la conferma della positiva integrazione il locus genomico corretta, deve essere eseguita un'analisi del sud della macchia. Questo assicura una maggiore fiducia che non si sono verificati eventi di inserimento ulteriori in regioni genomiche aspecifici.
- Per espandere le popolazioni clonali, trasferire i cloni che sono cresciuti dopo la selezione dalla piastra di coltura del tessuto 96 pozzetti in una piastra di coltura del tessuto 12-pozzetti a crescere abbastanza cellule per l'isolamento di DNA genomic. Fornire ogni pozzetto con 1 mL di SorMC K. aerogenes (OD600 = 2) e fresco marcatore selezionabile.
- Per proteine reporter fluorescenti, visivamente identificare cellule fluorescenti positive e controllare con un microscopio a fluorescenza. In alternativa, è possibile eseguire un western blot con anticorpi adatti per identificazione biochimica.
- Per knock-out, knock-in e act5 knock-in, eseguire la schermata iniziale tramite PCR per confermare integrazione del costrutto il locus genomico corretto.
| Programma PCR | ||
| passo | temperatura | tempo |
| Denaturazione iniziale | 94 ° C | 30 s |
| 30 cicli | 94 ° C | 15-30 s |
| 42 ° C | 15-60 s | |
| 68 ° C | 1 min/kb | |
| Estensione finale | 68 ° C | 5 min |
| Tenere premuto | 4-10 ° C | |
| Composizione di reazione | ||
| componente | Reazione di 25 μL | concentrazione finale |
| Primer in avanti di 10 µM | 0,5 Μ l | 0,2 ΜM |
| 10 µM reverse Primer | 0,5 Μ l | 0,2 ΜM |
| Modello del DNA | variabile (ca. 5 µ l) | < 1.000 ng |
| 2 x Master Mix con tampone Standard tra cui polimerasi (Vedi tabella dei materiali) | 12,5 Μ l | 1 x |
| Acqua priva di nucleasi | a 25 µ l | < 1.000 ng |
Tabella 2: composizione di programma e campione PCR per l'amplificazione di D. discoideum DNA genomic.
Risultati
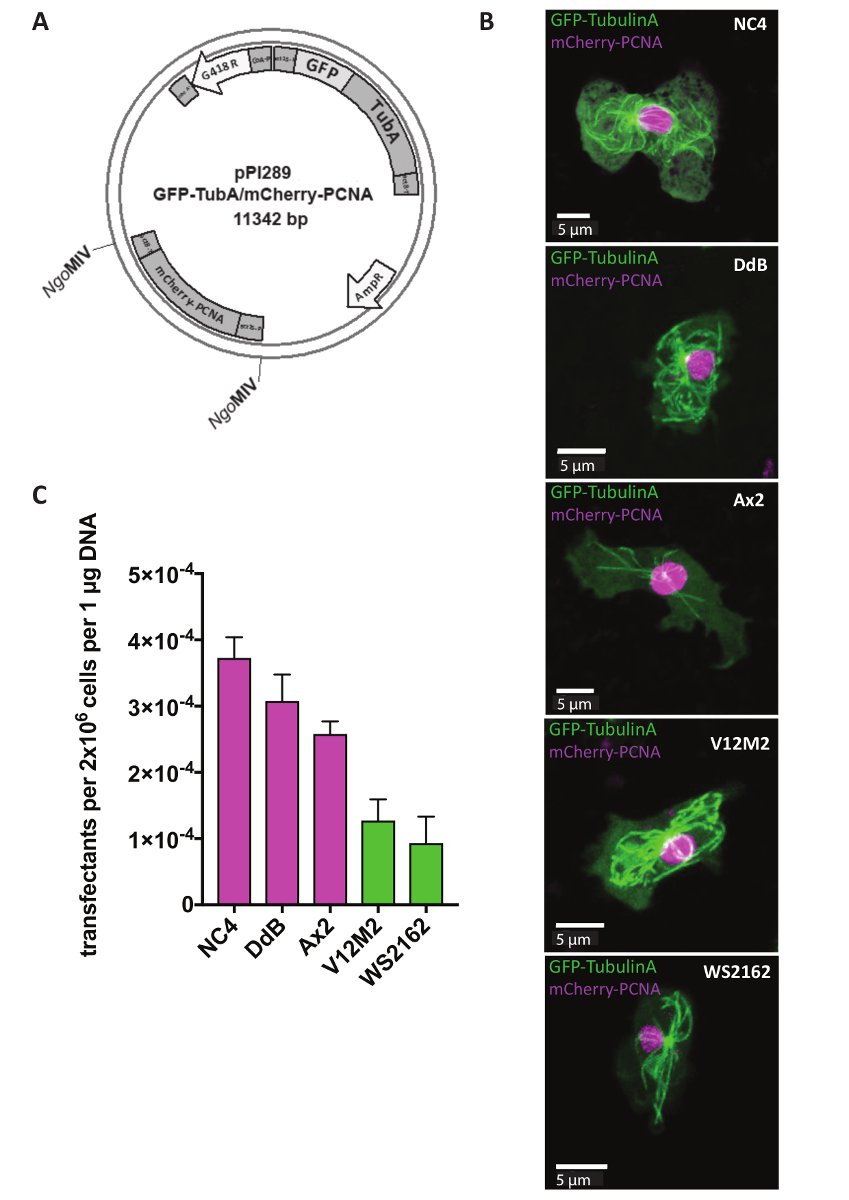
Extracromosomico plasmidi sono utilizzati per studi di reporter, che mirano a identificare la localizzazione di alcune proteine all'interno di una cella o cambiamenti nella struttura cellulare di cellule mutanti. Per molti approcci, quali il monitoraggio del ciclo cellulare, è fondamentale esprimere due reporter allo stesso tempo. Questo è ora possibile utilizzando il nostro sistema di plasmide extracromosomico dual reporter (tabella 1). Il giorno 1, cellule transfected prima di aggiungere il marcatore selezionabile G418 dopo 5 h (Figura 1). Nell'esempio, NC4, DdB, Ax2 e gli isolati selvatici indipendentemente derivati V12M2 e WS2162 (complementare tabella 1) sono state trasfettate con il plasmide pPI289, che codifica per la GFP-TubulinA, un marcatore per i microtubuli e mCherry-PCNA, una proteina che è utilizzato per il monitoraggio del ciclo cellulare (Figura 2A). Dopo 32 h, le cellule sono state osservate al microscopio. La maggioranza delle cellule espresse entrambe proteine di fusione con etichetta fluorescente, coerente con precedenti relazioni che l'espressione di due giornalisti della stesso plasmide spettacoli simili livelli di espressione, che è quasi impossibile quando si utilizzano due diversi plasmidi 18,19. Una cella rappresentanza per ogni linea cellulare (NC4, DdB, Ax2, V12M2 e WS2162) che esprime il reporter dual desiderato è illustrata nella Figura 2B. L'efficienza di trasfezione è riassunti nella Figura2C. Linee cellulari derivate da NC4 mostrano le migliori efficienze di transfezione. Tuttavia, per linee cellulari V12M2 e WS2162, un numero considerevolmente elevato di trasfettanti è stato ottenuto.

Figura 2 : Espressione di un plasmide extracromosomico. (A) extracromosomico plasmidi direttamente transfected in forma circolare. Ad esempio, è mostrato il reporter dual pPI289. I siti MIV ONGindicano inserimento del secondo reporter nel plasmide extracromosomico espressione. (B) Z-proiezione di una rappresentante delle cellule che esprimono GFP-TubulinA (citoplasmatica) e mCherry-PCNA (in gran parte nucleare) per cinque linee cellulari differenti del selvaggio-tipo utilizzato (NC4, DdB, Ax2, V12M2, WS2162). (C), la transfezione efficienze per le linee di cinque cellulari nella foto (b) sono state calcolate. Indicato è la media dei due esperimenti. Barre di errore indicano ± SD. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
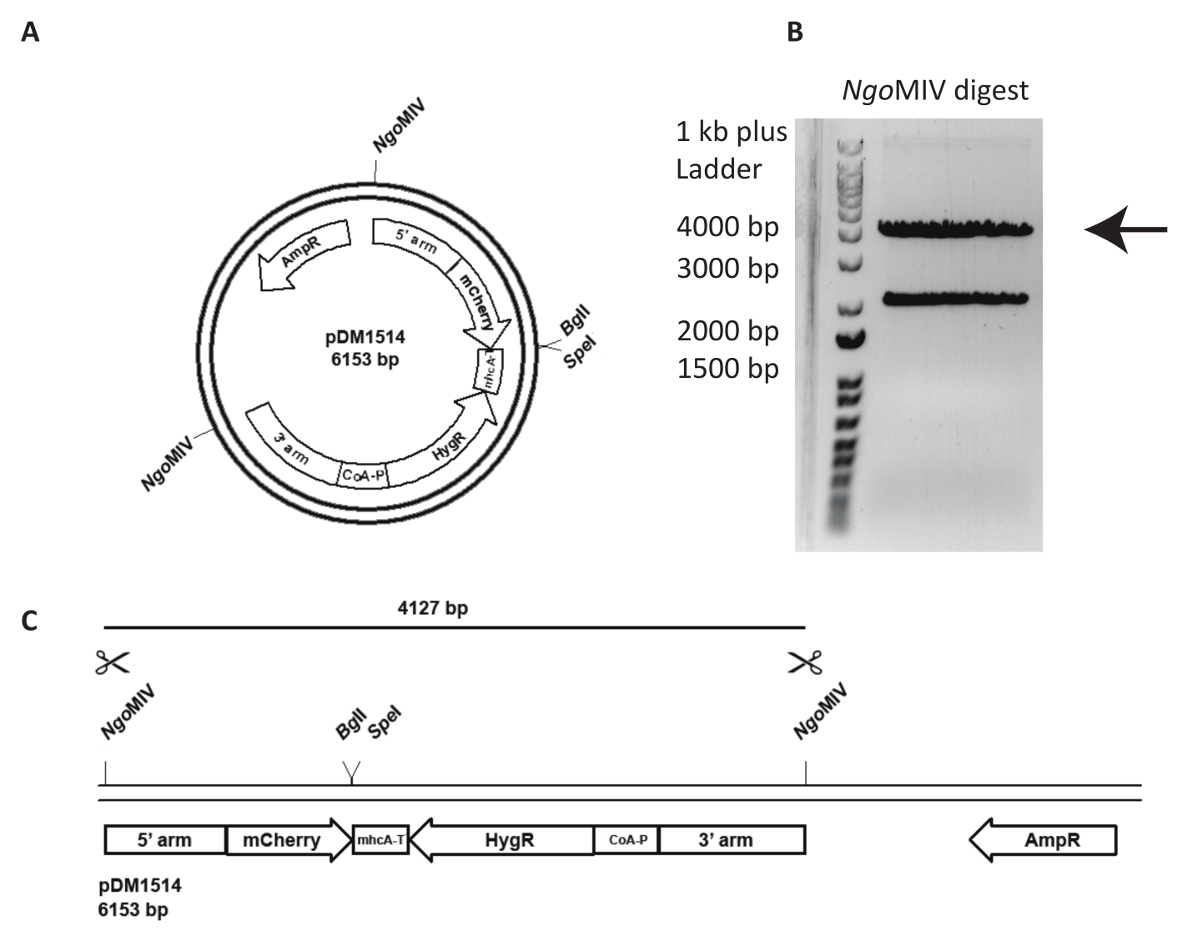
Integrazione di un vettore targeting in un locus genomico specificato è più impegnativo e richiede analisi più attenta della linea cellulare generato. In Figura 3, viene tentata un'act5- mCherry KI in NC4. In primo luogo, il plasmide deve essere linearizzato per aumentare la frequenza di eventi di ricombinazione seguito di transfezione. Per questo, il pDM1514 di plasmide viene tagliato con ONGMIV. Due fasce sono ottenute dopo l'esecuzione il digest su un gel di agarosio. La band di 4127 bp contiene il costrutto desiderato (Figura 3). Per la transfezione, il DNA digerito dovrà essere estratte dal gel e purificato utilizzando un kit di estrazione del gel seguendo le istruzioni del produttore.

Figura 3 : Preparazione di act5 knock-in e del DNA per la transfezione. Il plasmide pDM1514 è riportato un esempio dell'uso di un knock-in atto5. I passaggi per la preparazione sono elencati come segue. (A) prima di elettroporazione, linearizzare il plasmide utilizzando i siti MIV ONGindicati. (B, C) Eseguire il plasmide tagliato su un gel di agarosio, fino a quando le due bande previste correttamente sono separate. Tagliare la banda di bp 4127 contenente le braccia di ricombinazione, mCherry e la cassetta di resistenza, e gel estratto il DNA. Il DNA è ora pronto per la transfezione. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Il DNA purificato è stato utilizzato per la transfezione delle cellule NC4. Dopo 5-6 giorni di selezione, sono stati ottenuti cloni. L'efficienza di trasfezione rappresentativo e la quantità di cloni positivi identificati per diversi act5 knock-in tentativi sono riassunti nella tabella 3. Due cloni di transfezione NC4 erano scelti in modo casuale e analizzati mediante PCR (Figura 4A), ed entrambi hanno mostrato i modelli di banda prevista ci si aspetta da un knock-in e sono stati ulteriormente convalidati con analisi del sud della macchia per garantire una sola integrazione evento del costrutto nel genoma20. Il blot Mostra un chiaro singola integrazione nel luogo desiderato act5 (Figura 4B). La linea di cellulare NC4::act5-mCherry generata può ora utilizzabile negli esperimenti.

Figura 4 : Convalida di act5- mCherry KIs in NC4. (A) schema e controllo PCRs per la validazione delle integrazioni positivi in act5 locus. I primer indicati sono stati utilizzati per analizzare due cloni indipendenti e il genitore. Entrambi i cloni mostrano le bande previste per la resistenza-cassette e a valle (P1) o primer a Monte (P2), che non sono presenti nel ceppo parentale. La combinazione di primer (P1/P2) conferma la corretta integrazione della cassetta mCherry e resistenza il locus di act5 . Il selvaggio-tipo NC4 Mostra la band di bp di 2800 previsto, mentre entrambi i cloni KI mancano di questa band e invece visualizzare un prodotto PCR circa 1400 paia di basi più grandi. Schema (B) per la raccolta di limitazione usato e del sud della macchia. Entrambi i cloni knock-in mostrano la band di bp 3400 più piccola, che è il risultato dell'integrazione del costrutto nel locus agire5, in particolare ulteriori BclI sito nella cassetta di resistenza igromicina. Gli spettacoli del selvaggio-tipo controllo il kb 5,8 attesi derivanti da due situati a valle Bclsiti. La macchia è stata ritagliata e impiombata per chiarezza. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| costrutto mirato nel locus di act5 | nome del plasmide | numero di occupati pozzi | numero di cloni selezionati | Cloni positivi | Cloni corretti (%) | Ceppo di Dictyostelium usato | efficienza di trasfezione (trasfettanti / 2 x 10 ^ 6/1 µ g del DNA) |
| LifeAct-mCherry | pPI226 | 7 | 7 | 1 | 14.2 | AX2 | 3,5 x 10 ^ -6 |
| LifeAct-GFP | pPI227 | 12 | 12 | 5 | 41,6 | AX2 | 6 x 10 ^ -6 |
| GFP | pDM1513 | 3 | 3 | 2 | 66,6 | AX2 | 1,5 x 10 ^ -6 |
| mCherry | pDM1514 | 3 | 3 | 1 | 33,3 | AX2 | 1,5 x 10 ^ -6 |
| GFP | pDM1513 | 66 | 9 | 5 | 55,5 | AX2 | 3.3 x 10 ^ -5 |
| mCherry | pDM1514 | 221 | 12 | 10 | 83,3 | AX2 | 1.1 x 10 ^ -4 |
| H2B-mCherry | pPI420 | 3 | 3 | 1 | 33,3 | AX2 | 1,5 x 10 ^ -6 |
| H2B-mCherry | pPI420 | 7 | 7 | 6 | 85,7 | AX2 | 3,5 x 10 ^ -6 |
| mCherry | pDM1514 | 10 | 10 | 7 | 70 | DdB | 5 x 10 ^ -6 |
| mCherry | pDM1514 | 240 | 12 | 11 | 91,6 | DdB | 1.2 x 10 ^ -4 |
| mCherry | pDM1514 | 320 | 12 | 12 | 100 | NC4 | 1.6 x 10 ^ -4 |
Tabella 3: l'efficienza di trasfezione e la quantità di ottenuti trasfettanti positivo per la generazione di act5 KIs in sfondi di ceppo diverso 22 .
Il locus agire5 offre relativamente omogeneo espressione del giornalista integrato21. Le cellule di NC4::act5-mCherry generate consentono mix esperimenti da eseguire con altri act5 knock-in utilizzando una proteina fluorescente differente come GFP. Per sottolineare il grande vantaggio di questo sistema, sono mostrati miscelazione esperimenti con Ax2::act5-GFP . A causa dell'incapacità a transfect non axeniche cellule wild-type, questo tipo di approccio potrebbe non essere eseguito prima. Mix esperimenti sono uno strumento importante per analizzare il comportamento delle cellule, perché consentono un confronto diretto tra diverse linee cellulari vivendo identiche condizioni sperimentali. NC4 cellule crescono più rapidamente sui prati batterici rispetto alle cellule Ax2 (Figura 5A). Questo può essere dovuto una maggiore capacità di fagocitare batteri o migliorata capacità di spostare e mostrare chemiotassi verso una fonte di cibo. Utilizzando un'analisi di chemiotassi di folato sotto-agar, confronto diretto di una popolazione di NC4::act5-mCherry e Ax2::act5-GFP è stato effettuato, mostrando che NC4::act5-mCherry sono molto più efficiente nel rilevamento di folato. Dopo 4 h, più NC4::act5-mCherry cellule erano in grado di strisciare sotto l'agarosio che le cellule Ax2::act5-GFP (Figura 5B). Analisi delle metriche standard della chemiotassi per le cellule in migrazione sotto l'agarosio ha rivelato che le cellule NC4::act5-mCherry erano più veloci e ha mostrato la più forte risposta chemiotattica delle cellule Ax2::act5-GFP (Figura 5C-E ).

Figura 5 : Usando KIs agire5 per gli esperimenti basati su immagine chemiotassi mix. (A) NC4::act5-mCherry e Ax2::act5-GFP che esprimono la proteina fluorescente indicata dal locus agire5 sono stati analizzati per la capacità di crescere su un prato batterico. Dopo 4 giorni, sono stati misurati il diametro di placca sorto dalle cellule di Dictyostelium placcate solitarie. Non-axeniche act5:: NC4 celle formano placche significativamente più grande rispetto alle cellule Ax2::act5-GFP axeniche (media ± DS, * * * p < 0,0001, n = 3, scala bar 5 mm). (B) per uso della chemiotassi di folato sotto-agarosio dosaggio22 confrontare direttamente le capacità chemiotattiche del NC4::act5-mCherry e Ax2::act5-GFP utilizzato in (A), batteri-cresciuta amebe di entrambi i ceppi sono stati misti in un rapporto di 50: 50. Cellule sono state permesse a strisciare sotto l'agarosio. Dopo 4 h, cellule che stavano migrando il gradiente di folato erano imaged utilizzando un microscopio confocale. Poi è stato determinato il numero di cellule NC4::act5-mCherry e Ax2::act5-GFP . NC4::act5-mCherry cellule erano più efficiente nel rilevamento di folato. Circa 10 volte più NC4::act5-mCherry cellule sono state trovate confrontato alle cellule di Ax2::act5-GFP (media ± DS, * * * p < 0,0001, n = 6, scala bar 100 µm). (C, D) Le cellule sono state filmate per 60 min, e la loro velocità e chemiotattica indice sono stati calcolati. Dopo la pre-selezione delle cellule più chemotactically reattive (solo quelli migrati sotto l'agarosio), Ax2::act5-GFP cellule hanno mostrati valori inferiori per velocità di cella e la chemiotassi. Cinquanta cellule per linea cellulare sono state analizzate. (media ± SD, * p < 0.01, n = 3). (E) le tracce di NC4::act5-mCherry e Ax2::act5-GFPact5 knock-in vengono tracciate oltre 60 min, mostrando il movimento più indirizzato verso la fonte di chemoattractent delle cellule NC4::act5-mCherry (media ± SD, * * * p < 0,0001, n = 6). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Le linee di act5 knock-in cella possono essere utilizzate anche per citometria a flusso. Come con crescita sui batteri, esistono importanti differenze di sviluppo tra ceppi axeniche e tipi selvatici non axeniche. Dopo lo sviluppo, i corpi fruttiferi delle cellule NC4 sono circa due volte grandi come quelli derivati dalle cellule Ax2. Se le cellule sono mescolate, si ottiene un corpo fruttifero di dimensioni intermedio. Per analizzare il contributo delle due linee cellulari più quantitativamente, citometria a flusso è stata utilizzata. Queste analisi hanno mostrato chiaramente che i corpi fruttiferi di intermedio-graduati sono dovuti a diversi livelli di contributo da entrambe le linee cellulari. Mentre le cellule NC4::act5-mCherry è composto da circa il 75% delle spore misurate, Ax2::act5-GFP ha contribuito solo il 25%, rivelando un potenziale vantaggio di fitness per i ceppi non-axeniche (Figura 6). Poiché l'analisi non controlla la popolazione delle cellule di gambo, ci sono due possibilità per spiegare lo squilibrio nello sviluppo tra Ax2 e NC4. Una possibilità è che le cellule Ax2 contribuire principalmente alla popolazione delle cellule del gambo, piuttosto che entrare la popolazione delle cellule di spora. In alternativa, più NC4 cellule possono entrare il ciclo di sviluppo, con Ax2 relativamente in ritardo, e sono quindi incapaci di contribuire per l'assemblaggio di un corpo fruttifero. La possibilità a transfect cellule wild-type non axeniche sviluppa ulteriormente altri approcci e semplifica notevolmente le procedure sperimentali.

Figura 6: atto5 KIs consentono l'analisi di esperimenti di miscela mediante citometria a flusso. (A) NC4::act5-mCherry e Ax2::act5-GFP sono stati sviluppati separatamente o in una miscela 50: 50 sui piatti di agar non-sostanza nutriente. NC4::act5-mCherry cellule formano grandi corpi fruttiferi rispetto Ax2::act5-GFP. Il mix Mostra invece una dimensione intermedia (scala bar 5 mm). Microscopia di fluorescenza confocale suggerisce una maggiore quantità di spore di NC4::act5-mCherry nelle teste spora derivate da miscele. (B) per quantificare questa osservazione, la quantità di spore in raccolta spora teste dall'esperimento miscelazione in (A) sono state analizzate tramite flusso cytometry. Circa il 75% delle spore ha provenuto dalle cellule NC4::act5-mCherry , con solo il 25% da cellule Ax2::act5-GFP . (C) distribuzione rappresentativa delle spore da entrambe le linee cellulari mostrate in una dispersione di citometria a flusso. Circa lo 0.05% Visualizza positivo mCherry e GFP segnali, suggerendo che si sono verificati processi di fusione parasexual o che le spore sono attaccare a vicenda. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Linea cellulare | Geneticamente sfondo | Numero di riferimento | Pubblicato prima | Tipo |
| AX2 (Ka) | DBS0235521 | Bloomfield et al., 2008 | wild-type | |
| NC4 (S) | Bloomfield et al., 2008 | wild-type | ||
| act5::mCherry Clone 5 | NC4 | HM1912 | Paschke et al., 2018 | act5 knock-in |
| act5::GFP Clone 2 | AX2 | HM1930 | Paschke et al., 2018 | act5 knock-in |
| V12M2 | Bloomfield et al., 2008 | wild-type | ||
| WS2162 | Bloomfield et al., 2008 | wild-type |
Complementare tabella 1: diversi ceppi di Dictyostelium studiato.
Discussione
L'uso di non-axeniche, selvaggio-tipo cellule di Dictyostelium è stato molto limitato finora nella ricerca molecolare. Metodi disponibili per l'ingegneria genetica di questi ceppi sono mancata affidabilità ed efficienza23, impedendo la loro adozione generale. I materiali generati e protocolli presentati qui utilizzabile per qualsiasi ceppo di d. discoideum indipendente dalla sua capacità di crescere in mezzo liquido. Va ricordato che questo protocollo è ottimizzato per linee cellulari derivate da NC4. L'efficienza di trasfezione per ceppi di recente isolate dal selvaggio differiscono da NC4, come hanno osservato prima e sono mostrati qui per V12M2 e WS21628,9. Le condizioni di elettroporazione sembrano in particolare hanno una notevole influenza sull'efficienza di trasfezione e possono richiedere un'ulteriore ottimizzazione per alcuni ceppi. In generale, una quantità sufficiente di trasfettanti sono stati osservati in tutti i ceppi testati finora, mostrando che questi metodi sono realizzabili. Il numero di trasfettanti positivi ottenuti utilizzando ceppi NC4-derivato è superiore rispetto ad altri ceppi non-axeniche di selvaggio-tipo, ma in tutti i casi, un numero sufficiente di trasfettanti è ottenuto per consentire ulteriori esperimenti. Questo, oltre alla semplicità del nostro protocollo, è il grande miglioramento rispetto ai precedenti tentativi8,9.
Questi nuovi protocolli non axeniche coprono tutte le procedure standard genetiche e aggiungere ulteriori vantaggi, poiché transfezioni possono essere eseguite rapidamente ed efficientemente in parallelo. Cloni positivi possono essere ottenuti in giorni anziché in settimane, dalla crescita su metà di batteri il tempo di divisione. I plasmidi neoformati anche lavorano in condizioni axeniche e ordinariamente utilizzabile sotto entrambe le condizioni di crescita, che è un altro avanzamento di questa metodologia22. Come introdotto nel protocollo, il plasmide usato per la selezione sui batteri hanno requisiti speciali che sono cruciali per il successo della transfezione. In particolare, i promotori che guida le cassette di espressione e la resistenza sono importanti per la transfezione di successo. Il promotore di spesso usate act6 (actina 6)24 per guidare la resistenza o espressione cassette manca efficienza quando le cellule si sviluppano sui batteri. Nel nostro sistema di plasmide, il promotore altamente attivo act15 (actina 15) unità tutte le cassette di espressione, mentre le cassette di resistenza sono sotto il controllo di un promotore di coA (coactosin A), entrambi dei quali sono attivi in condizioni axeniche e in cellule coltivate sui batteri. Requisiti per l'espressione del reporter costruisce così come knock-in e costrutti di knock-out rendono necessario di utilizzare il nostro repertorio di plasmide, ma purtroppo limita l'uso di vettori già creati che utilizzano il promotore inefficiente act6 .
Promotore efficienze sono particolarmente critiche per transfezioni che dipendono da un evento di integrazione unico nel locus genomico corretto. A causa di nostro miglioramento dei promotori che guida l'espressione di gene di resistenza, abbastanza proteine di resistenza sono prodotto da integrazioni singolo locus. Delle principali preoccupazioni era il favorire più integrazioni nel genoma quando si utilizza igromicina o G418 come descritto. Nessun favorendo di più integrazioni è stato osservato finora, come riportato in precedenza per le selezioni di G418 utilizzando vecchi promotore sistemi25. Ciò significa che entrambi igromicina e G418 sono marcatori selezionabili adatti per la generazione di pulito knock-out e knock-ins. purtroppo, il marcatore selezionabile Consciousness non funziona in condizioni di non-axeniche. Questo è dei principali svantaggi del nostro metodo, poiché la cassetta di resistenza Consciousness è usata ordinariamente per generare costrutti di knock-out d.discoideum. Costrutti che già sono state generate con cassette di Dyfed resistenza dovrà essere riderivato utilizzando uno dei marcatori selezionabili realizzabili. Un'altra possibilità per superare questo limite è quello di combinare la recente costituzione axeniche d. discoideum CRISPR tecnologia con questo protocollo di transfezione26. La generazione di guida adatta, singolo RNAs (sgRNAs) è più semplice e più veloce rispetto alla ricostruzione dei costrutti di knock-out completi. Per orientamenti futuri, la possibilità di generare più knock-out utilizzando CRISPR/Cas9 combinato con questo protocollo di transfezione è attraente e può aprire la strada per molti ricercatori della comunità di Dictyostelium . Tuttavia, la lavorabilità del sistema stabilito espressione transitoria usato per CRISPR/Cas9 in cellule coltivate axeniche dovrebbe essere esaminata con attenzione.
Il act5 knock-in sistema presentato offre un sistema di integrazione affidabile e sicuro per la generazione di linee cellulari stabilizzate in d. discoideum, con il vantaggio di siti simili stabilito in altri organismi22. Inoltre dipende da un evento singolo integrazione nel genoma e offre molte possibilità per le direzioni di ricerca differenti. Il promotore di act5 è fortemente attivo e garantisce quasi omogenea espressione27 indipendente le condizioni di coltivazione. Proteine reporter fluorescenti sono facilmente integrabile in questo luogo di rifugio sicuro usando i plasmidi derivati dal targeting. Questo può essere utile, ad esempio, a fini di rilevamento delle cellule, come illustrato di seguito in un esperimento di mix. Le cellule mostrano variabilità di espressione minima di cellula--cellula, che può aiutare nella cella automatizzata di rilevamento. Soprattutto, l'inserimento di una sequenza desiderata in luogo del agire5 appare fenotipico neutro28,29. Come l'espressione non dipende da un marcatore selezionabile per mantenere l'espressione della proteina, come visto in casuale integranti o linee cellulari di vettore-cuscinetto extracromosomico, il sistema di act5 può essere utile per gli esperimenti di salvataggio, pure. Dal momento che le linee cellulari sono omogenee, tutte le celle possono essere analizzate. Questo non è possibile utilizzando un plasmide extracromosomico per l'espressione, in cui vi è una notevole eterogeneità nell'espressione.
Oltre a questi miglioramenti generali per tutti i ceppi, la possibilità di utilizzare non axeniche cellule wild-type per ricerca molecolare permette una valutazione degli effetti di mutazioni accumulate in ceppi di laboratorio attuali. Riarrangiamenti genici multipli è stato trovato dopo l'adozione del ceppo Ax2 nel 1970, come dimostrato da analisi di microarray precedentemente pubblicati26. A causa della presenza di queste mutazioni si osservano fenotipi sintetici. Ad esempio, un mutante segnalati phg2 Mostra adesione in diminuzione in uno sfondo di ceppo di laboratorio e adesione aumentata in un altro3. Tali dati contrastanti possono ora essere risolti ripetendo l'esperimento nel ceppo antenato comune (in questo caso, DdB). La forza di questo approccio è stato recentemente indicata per il piccolo GTPase RasS30,31.
La possibilità di eseguire esperimenti genetici in qualsiasi ceppo d. discoideum espande il potenziale delle nuove direzioni di ricerca che dipendono dall'uso dei selvatici isolati, in particolare quelle che riguardano l'evoluzione sociale, riconoscimento kin e il ciclo sessuale22.
Divulgazioni
Gli autori non hanno conflitti di interesse a divulgare.
Riconoscimenti
Gli autori ringraziano il laboratorio di Kay per l'input a questo metodo e i servizi di citometria a flusso e microscopia LMB per eccellente sostegno scientifico e tecnico. Questo lavoro è stato finanziato dalla sovvenzione di Medical Research Council grant MC_U105115237 a R. R. Kay, BBSRC (biotecnologie e Biological Sciences Research Council) 1/K009699/BB per R. R. Kay, Cancer Research UK concedere A15672 a R. H. Insall, Wellcome Trust Senior Fellowship 202867/Z/16/Z per Jonathan R Chubb e MRC finanziamenti (MC_U12266B) l'unità di Università MRC LMCB presso UCL.
Materiali
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Eppendorf Microcentrifuges 5424/5424R | ThermoScientific | 05-400-002 | |
| Eppendorf 5702R Centrifuges with A-4-38 Model Rotor | ThermoScientific | 12823252 | |
| Eppendorf Mastercycler Nexus Thermal Cyclers | Sigma Aldrich | EP6331000025 | |
| Gene Pulser X Cell, including CE & PC modules | BioRad | 1652660 | |
| Safety Cabinet Foruna - SCANLAF | Labogene | ||
| BioPhotometer Plus | Eppendorf | ||
| CLSM 710 | Zeiss | ||
| Media & Agaroses | |||
| LoFlo Medium | Formedium | LF0501 | |
| Seakem GTG Agarose | Lonza | 50071 | |
| Low EEO Agarose | BioGene | 300-200 | |
| Purified Agar | Oxoid | LP0028 | |
| SM broth | Formedium | SMB0102 | |
| 2x LB broth | Formedium | LBD0102 | |
| Chemicals | |||
| SYBR Safe DNA Gel Stain | ThermoScientific | S33102 | |
| 1 Kb Plus DNA Ladder | ThermoScientific | 10787018 | |
| 1 Kb DNA Ladder | NEB | N3232L | |
| HEPES free acid | Sigma Aldrich/Merck | 391340 EMD | |
| KH2PO4 | VWR | P/4800/53 | |
| Na2HPO4 * 2 H2O | VWR | 10028-24-7 | |
| MgCl2 * 6 H2O | Sigma Aldrich/Merck | 5982 EMD | |
| CaCl2 * 2 H2O | Sigma Aldrich | 442909 | |
| Folic Acid | Sigma Aldrich | F7876 | |
| KOH | VWR | 26668.263 | |
| Cultur Dishes | |||
| 96 Well Cell Culture Cluster, Flat Bottom with Low Evaporation Lid, Tissue Culture Treated | Corning Incorporated | 3595 | |
| 6 Well Cell Culture Cluster, Flat Bottom with Lid, Tissue Culture Treated | Corning Incorporated | 3516 | |
| 100 x 20 mm style Tissue Culture Dish, Tissue Culture Treated | Corning Incorporated | 353003 | |
| 50 mm Glass Bottom Microwell Dishes No1.5 | MatTek Corporation | P50G-1.5-30-F | |
| Antibiotics | |||
| Geneticin G418-sulphate | Gibco by Life Technologies | 11811-023 | |
| Hygromycin B Gold | InvivoGen | ant-hg-1 | |
| Additional Consumables | |||
| 2 mm gap electroporation cuvettes, long electrode | Geneflow Limited | E6-0062 | |
| 10 µL Inoculating Loop, Blue 10 Micro L | ThermoScientific | 129399 | |
| Spreader, L-shaped, sterile | greiner bio-one | 730190 | |
| Combitips advanced 5 mL | Eppendorf BIOPUR | 30089669 | |
| Kits | |||
| ZR Plasmid Miniprep Classic | Zymoresearch | D4016 | |
| Quick DNA Miniprep Kit | Zymoresearch | D3025 | |
| Zymoclean DNA Recovery Kit | Zymoresearch | D40002 | |
| Enzymes | |||
| Restriction enzymes | NEB | ||
| OneTaq 2X Master Mix with Standard Buffer | NEB | M0482L | |
| T4 DNA Ligase | NEB | M0202S | |
| PrimeSTAR Max DNA Polymerase | TakaraBio | R045A |
Riferimenti
- Schaap, P. Evolutionary crossroads in developmental biology: Dictyostelium discoideum. Development. 138 (3), 387-396 (2011).
- Sussman, R., Sussman, M. Cultivation of Dictyostelium discoideum in axenic culture. Biochemical and Biophysical Research Communications. 29, 53-55 (1967).
- Bloomfield, G., Tanaka, Y., Skelton, J., Ivens, A., Kay, R. R. Widespread duplications in the genomes of laboratory stocks of Dictyostelium discoideum. Genome Biology. 9 (4), 75 (2008).
- Witke, W., Nellen, W., Noegel, A. Homologous recombination in the Dictyostelium alpha-actinin gene leads to an altered mRNA and lack of the protein. The EMBO Journal. 6, 4143-4148 (1987).
- De Lozanne, A., Spudich, J. A. Disruption of the Dictyostelium myosin heavy chain gene by homologous recombination. Science. 236, 1086-1091 (1987).
- Howard, P. K., Ahern, K. G., Firtel, R. A. Establishment of a transient expression system for Dictyostelium discoideum. Nucleic Acids Research. 16, 2613-2623 (1988).
- Knecht, D., Pang, K. M. Electroporation of Dictyostelium discoideum. Methods in Molecular Biology. 47, 321-330 (1995).
- Wetterauer, B., et al. Wild-type strains of Dictyostelium discoideum can be transformed using a novel selection cassette driven by the promoter of the ribosomal V18 gene. Plasmid. 36, 169-181 (1996).
- Lloyd, M. M., Ceccarelli, A., Williams, J. G. Establishment of conditions for the transformation of nonaxenic Dictyostelium strains. Developmental Genetics. 11, 391-395 (1990).
- Bloomfield, G., et al. Neurofibromin controls macropinocytosis and phagocytosis in Dictyostelium. eLife. 4, (2015).
- Veltman, D. M., et al. A plasma membrane template for macropinocytic cups. eLife. 5, (2016).
- Veltman, D. M., Lemieux, M. G., Knecht, D. A., Insall, R. H. PIP(3)-dependent macropinocytosis is incompatible with chemotaxis. The Journal of Cell Biology. 204 (4), 497-505 (2014).
- Hoeller, O., Kay, R. R. Chemotaxis in the absence of PIP3 gradients. Current Biology. 17, 813-817 (2007).
- Chubb, J. R., Wilkins, A., Thomas, G. M., Insall, R. H. The Dictyostelium RasS protein is required for macropinocytosis, phagocytosis and the control of cell movement. Journal of Cell Science. 113, 709-719 (2000).
- Schaap, P., et al. Molecular phylogeny and evolution of morphology in the social amoebas. Science. 314, 661-663 (2006).
- Du, Q., Kawabe, Y., Schilde, C., Chen, Z. H., Schaap, P. The Evolution of Aggregative Multicellularity and Cell-Cell Communication in the Dictyostelia. Journal of Molecular Biology. 427 (23), 3722-3733 (2015).
- Hirose, S., Benabentos, R., Ho, H. I., Kuspa, A., Shaulsky, G. Self-recognition in social amoebae is mediated by allelic pairs of tiger genes. Science. 333 (6041), 467-470 (2011).
- Strassmann, J. E., Queller, D. C. Evolution of cooperation and control of cheating in a social microbe. Proceedings of the National Academy of Sciences of the United States of America. 108 (2), 10855-10862 (2011).
- Wolf, J. B., et al. Fitness Trade-offs Result in the Illusion of Social Success. Current Biology. 25 (8), 1086-1090 (2015).
- Veltman, D. M., Akar, G., Bosgraaf, L., Van Haastert, P. J. A new set of small, extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid. 61 (2), 110-118 (2009).
- Southern, E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. Journal of Molecular Biology. 98 (3), 503-517 (1975).
- Paschke, P., et al. Rapid and efficient genetic engineering of both wild type and axenic strains of Dictyostelium discoideum. PLoS One. 13 (5), 0196809 (2018).
- Woznica, D., Knecht, D. A. Under-agarose chemotaxis of Dictyostelium discoideum. Methods in Molecular Biology. 346, 311-325 (2006).
- Dubin, M., Nellen, W. A versatile set of tagged expression vectors to monitor protein localisation and function in Dictyostelium. Gene. 465 (1-2), 1-8 (2010).
- de Hostos, E. L., et al. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. Journal of Cell Biology. 120, 163-173 (1993).
- Sekine, R., Kawata, T., Muramoto, T. CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium. Scientific Reports. 8 (1), 8471 (2018).
- Sadelain, M., Papapetrou, E. P., Bushman, F. D. Safe harbours for the integration of new DNA in the human genome. Nature Reviews Cancer. 12 (1), 51-58 (2011).
- Rosengarten, R. D., et al. Leaps and lulls in the developmental transcriptome of Dictyostelium discoideum. BMC Genomics. 16, 294 (2015).
- Tunnacliffe, E., Corrigan, A. M., Chubb, J. R. Promoter-mediated diversification of transcriptional bursting dynamics following gene duplication. Proc Natl Acad Sci USA. 115 (33), 8364-8369 (2018).
- Gebbie, L., et al. Phg2, a kinase involved in adhesion and focal site modeling in Dictyostelium. Molecular Biology of the Cell. 15, 3915-3925 (2004).
- Jeon, T. J., Lee, D. J., Merlot, S., Weeks, G., Firtel, R. A. Rap1 controls cell adhesion and cell motility through the regulation of myosin II. Journal of Cell Biology. 176, 1021-1033 (2007).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati