
Method Article
U2O5 Film préparation via UO2 dépôt par pulvérisation de courant continu et l’oxydation Successive et réduction avec l’oxygène atomique et de l’hydrogène atomique
Dans cet article
Résumé
Ce protocole présente la préparation de U2O5 couches minces obtenues in situ sous ultravide. Le processus implique oxydation et réduction des films2 UO avec l’oxygène atomique et de l’hydrogène atomique, respectivement.
Résumé
Les auteurs décrivent une méthode pour produire U2O5 films in situ à l’aide de la Labstation, une machine modulaire développée à Karlsruhe JRC. La Labstation, une partie essentielle de la propriétés des Actinides en laboratoire des Conditions extrêmes (PAMEC), permet la préparation de films et d’études des surfaces d’échantillon à l’aide de techniques d’analyse surfaces tels que les rayons x et ultraviolette photoemission spectroscopie (XPS et UPS, respectivement). Toutes les études sont faites sur place, et les films, transférés sous ultravide de leur préparation à une chambre d’analyses, sont jamais en contact avec l’atmosphère. Au départ, un film de UO2 est préparé par dépôt par pulvérisation cathodique courant continu (DC) sur une feuille (Au) or ensuite oxydé par l’oxygène atomique pour produire un film de3 UO. Ce dernier est ensuite réduit à l’hydrogène atomique à U2O5. Des analyses sont effectuées après chaque étape impliquant l’oxydation et réduction, en utilisant la spectroscopie de photoélectrons haute résolution pour examiner l’état d’oxydation de l’uranium. En effet, l’oxydation et réduction de temps et température correspondante du substrat au cours de ce processus ont des effets graves sur l’état d’oxydation résultant de l’uranium. Arrêt de la réduction de l’UO3 U2O5 avec U(V) unique est assez difficile ; Il existe tout d’abord, des systèmes d’uranium et d’oxygène dans nombreuses phases intermédiaires. En second lieu, la différenciation de ces États d’oxydation de l’uranium repose essentiellement sur des pics de satellite, dont les pics intensité sont faibles. Aussi, les expérimentateurs doivent être conscients que la spectroscopie des rayons x (XPS) est une technique avec une sensibilité atomique de 1 à 5 %. Ainsi, il est important d’obtenir une image complète de l’état d’oxydation de l’uranium avec le spectre entier obtenu sur U4f, O1 s et la bande de valence (VB). Programmes utilisés dans la Labstation comprennent un programme de transfert linéaire développé par une société extérieure (voir la Table des matières) ainsi que l’acquisition de données et les logiciels par pulvérisation cathodique, tous deux développés en interne.
Introduction
Oxyde d’uranium est le principal composant des déchets nucléaires, et sa solubilité dans l’eau est liée à l’état d’oxydation de l’uranium, passant de U(IV) à u. Ainsi, l’oxydation de la UO2 + x pendant le stockage géologique est une sécurité importante et cruciale question1,2. Cela motive les études des mécanismes réactionnels qui régissent les interactions de surface entre les oxydes d’uranium et de l’environnement3,4,5,6. Cette connaissance est essentielle pour tous les aspects du traitement des déchets de cycles du combustible nucléaire.
Bien que tétravalent et uranium hexavalent sont bien établies et communs comme des systèmes à l’état solide, ce n’est pas le cas pour l’uranium pentavalent, malgré sa stabilité dans les complexes d’uranyle et survenue en solution aqueuse. En oxydes d’uranium, U(V) est considéré comme un intermédiaire métastable, et il n’est pas signalé comme seul État mais plutôt comme coexistant avec les espèces U(IV) et u. Pour cette raison, rien n’a été signalé sur les propriétés chimiques et physiques de U2O5. C’est aussi en raison d’une caractéristique commune des expériences de la corrosion, dans lequel les échantillons sont exposés à un environnement corrosif. Cela crée une forte pente dans les États d’oxydation entre la surface (exposée aux oxydants) et le volume de l’échantillon. Le changement a lieu au sein de la profondeur d’analyse. Ainsi, les États d’oxydation différents sont observés en même temps, non pas à cause de valence mixte, mais comme un artefact d’une réaction incomplète, engendrant une couche hétérogène. Ces deux problèmes peuvent être résolus en utilisant des couches minces au lieu des échantillons en vrac. Un grand nombre de systèmes différents peut être préparé avec peu de matière première, et le gradient de surface en vrac est évité, car il n’y a pas en vrac.
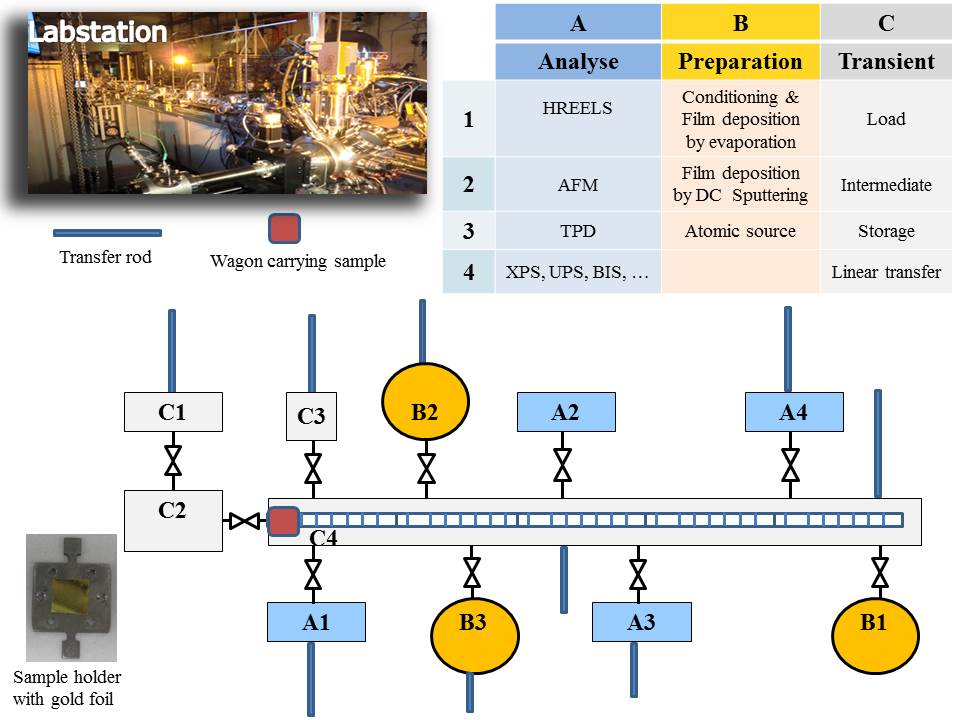
La méthode mentionnée ici permet in situ la préparation d’une couche très mince (quelques dizaines de couches atomiques déposées sur un substrat inerte) et l’analyse de sa surface sans contact avec l’atmosphère. C’est l’un des avantages de la Labstation (Figure 1), qui est un modulaire machine composé de différents compartiments conservés sous vide ultra haut dynamique (UHV), pour atteindre des pressions de 10-9-10-11 mbar. Chambres sont dédiées à la préparation des films minces, traitement de surface (adsorptions gaz) et caractérisation par les techniques de spectroscopie de surface [p. ex. photoélectrons (XPS), la spectroscopie ultraviolet spectrométrie de photoélectrons (UPS), faible Energy electron diffraction spectroscopie (LEED)]. Échantillons sont montés sur des porteurs de spécimen et transférés entre différentes chambres à travers une chambre de transfert linéaire à l’aide d’un chariot de transport. Toutes les chambres sont reliées à cette chambre centrale par une soupape afin qu’ils peuvent être isolés à tout moment (par exemple., pour le gaz de remplissage ou de l’entretien). Récupération de le titulaire/sample de la chambre de transfert linéaire est réalisée par une tige de transfert montée sur chaque chambre. Le système de base de Labstation a été fabriqué par une société externe (voir la Table des matières). Extensions et modifications ont été ajoutées par la suite en fonction des besoins expérimentaux, ayant pour résultat un équipement unique à Karlsruhe JRC. Les extensions sont la source de pulvérisation (un élément de base pour la préparation de la couche mince), qui a été développée en interne ainsi que les programmes d’acquisition par pulvérisation cathodique et données. Le chargement de le titulaire/sample d’une atmosphère ambiante à ultravide se fait via un sas d’écluse de charge spécialement conçu pour effectuer la manipulation des échantillons multiples et minimiser le temps d’atteindre la pression finale d’environ 10-8-10 -9 mbar, limitant ainsi la contamination de l’air du système. La Labstation est le résultat d’années d’expérience et d’expertise dans le domaine de la science des surfaces à Karlsruhe JRC.
Pour passer d’une chambre à l’autre, l’échantillon est monté sur un chariot de transport, conduit par un aimant externe, contrôlé par un programme informatique (Figure 2) et se déplaçant le long de la chambre de transfert linéaire d’environ 7 m à des positions d’arrêt prédéfinis en face de la chambres.
Sans une installation similaire ou proche, l’expérience pourrait être difficile à reproduire. Toutefois, cette installation contribue au laboratoire PAMEC qui contribue au programme libre accès au CCR, dans lequel les utilisateurs externes sont invités à soumettre les propositions examinées par un panel d’experts scientifiques internationaux. Leur évaluation puis permet aux utilisateurs d’accéder à l’infrastructure exploitée par le CCR. Après des demandes et dans le cadre de collaborations, films minces peuvent être préparés pour les utilisateurs externes pour des analyses et des expériences réalisées à l’extérieur de JRC Karlsruhe.
Dans ce rapport, nous fournissons un protocole détaillé de la croissance de single-valence U2O5 films minces, obtenus par des étapes successives impliquant oxydation et réduction de l’UO2 avec l’oxygène atomique et de l’hydrogène atomique, respectivement. Contrairement aux UO2 et UO2 + x, dépôts directs de U2O5 et UO3 films de DC pulvérisation ne peuvent se faire. Par conséquent, nous tout d’abord procéder au dépôt d’un film de2 UO, il s’oxyde en UO3 en utilisant l’oxygène atomique, puis réduisez-la à U2O5 avec l’hydrogène atomique. L’oxydation et réduction de temps et température de l’échantillon au cours du processus ont des effets sur le résultat et sont importants à maîtriser. Composition correcte a été vérifiée par spectroscopie de photoélectrons x à haute résolution, qui constitue une preuve directe et quantitative pour configuration électronique1 5fde l’uranium, comme prévu pour le U(V).
Protocole
1. préparation titulaire
NOTE : Manipulation du porte-échantillon et à l’extérieur de la Labstation sous atmosphère ambiante doit être effectuée avec des gants et pincettes propre.
-
Préparation des échantillons titulaire et ex-situ nettoyage
- Nettoyer une feuille d’or (Au) (0,025 mm d’épaisseur, 99,99 %, voir Table des matières) de taille 8 x 8 mm avec de l’acétone. Place le film d’un acier inoxydable ou un détenteur de molybdène spécialement conçu pour la Labstation et fixer la feuille sur le support en fils de tantale de soudage par points.
- Nettoyer le porte-échantillon et le clinquant à nouveau avec de l’acétone et laissez sécher sous atmosphère ambiante avant l’introduction de la Labstation.
NOTE : Une photo du porte-échantillon et avec une feuille d’or est affichée dans l’encart de la Figure 1.
-
Introduction du porte-échantillon dans la Labstation
- Fermer la vanne d’UHV entre le sas d’écluse de charge et de la chambre de garage (C1/C2). Desserrer la vis de fixation de la porte de la C1 et ouvrir le robinet d’azote pour amener la pression à la pression atmosphérique, permettant l’ouverture de la porte.
- Introduire le porte-échantillon à C1. Fermez la porte du C1 et ouvrir le robinet pour le vide primaire.
- Fermer le robinet de la pompe primaire une fois que le vide primaire atteint environ 1 mbar de pression. Ouvert le robinet relié à la pompe turbomoléculaire UHV immédiatement par la suite. Attendre que la pression atteint 10-7 mbar.
Remarque : Cette dernière étape peut prendre un minimum de 1 à 3 h, selon l’échantillon de dégazage.
-
Transfert à la chambre de préparation (B1)
- Déplacer le chariot de transport de la chambre de transfert vers la chambre intermédiaire et fermer le robinet entre C2 et C3. Ouvrez la valve située entre C1 et C2. Utiliser la tige de transfert de C1 pour transférer le porte-échantillon à C2.
NOTE : C2 est une chambre intermédiaire entre C1 et la chambre de transfert linéaire commune dans toutes les chambres de le Labstation. Il le charge de SAS, qui a un mauvais vide (10-7 mbar) sépare du reste du système (environ 10-9 mbar). Transferts de l’échantillon peuvent être effectuées rapidement à un vide relativement pauvre dans la serrure de charge tout en gardant la Labstation propre. - Après que le porte-échantillon est assis sur le wagon de C2, ramener la tige de transfert à C1 et fermer le robinet entre C1 et C2. Ouvrez le robinet entre C2 et la chambre de transfert linéaire C3.
- Position du wagon dans la chambre de transfert linéaire et connectez-le à l’aimant de conduite avant de fermer le robinet entre C2 et C3.
- Utilisez le programme de contrôle de transfert linéaire (LTC, Figure 2) pour transférer le wagon à la position de la chambre de préparation B1.
- Ouvrez la valve entre la chambre de transfert linéaire et la chambre de préparation B1. La tige de transfert permet de placer l’échantillon dans la chambre de préparation B1. Une fois que la tige de transfert est de retour à sa position initiale, fermer le robinet entre la chambre de transfert linéaire et de la chambre de préparation B1.
- Déplacer le chariot de transport de la chambre de transfert vers la chambre intermédiaire et fermer le robinet entre C2 et C3. Ouvrez la valve située entre C1 et C2. Utiliser la tige de transfert de C1 pour transférer le porte-échantillon à C2.
-
In situ nettoyage du porte-échantillon
- Ouvrir la vanne d’argon pour atteindre une pression de 5 x 10-5 mbar.
- La position de la surface de support d’échantillon à la verticale vers le canon à ions (IG10/35, voir Table des matières).
- Allumez le canon à ions pour commencer la pulvérisation d’ions Ar (2 keV, 10 émissions de mA actuel) et garder le nettoyage pendant 10 min. interrupteur arrêt le canon à ions.
- Portez le thermocouple en contact avec le porte-échantillon, puis allumer l’appareil e-faisceau de recuire l’échantillon à 773 K pendant 5 min. Après une hausse initiale (dans les 10-7 mbar plage), la pression retombe à environ 10-8 mbar, indiquant la fin de dégazage. Éteignez l’appareil e-faisceau et laisser refroidir les échantillons à température ambiante (RT).
- Ouvrez la valve entre la chambre de préparation B1 et de la chambre de transfert linéaire, puis placez le porte-échantillon sur le chariot à l’aide de la tige de transfert. Fermez la valve de la chambre de B1.
-
Caractérisation des échantillons titulaire
Remarque : Des mesures de spectroscopie (XPS) photoélectronique des rayons x à haute résolution ont été effectuées à l’aide d’un analyseur hémisphérique (voir Table des matières). Une source de micro-focus (voir Table des matières) équipé d’un monochromateur et fonctionnant à 120 W a été utilisé pour produire un rayonnement de Al Kα (E = 1 486,6 eV). L’étalonnage du spectromètre a été réalisé à l’aide de la ligne de7/2 de 4fd’un métal d’UA, produisant une valeur d’énergie de liaison 83.9(1) eV (BE) et la ligne de3/2 2pd’un métal Cu à 932.7(1) eV être.- Transfert du wagon par commande à distance à l’aide du programme FLC à la position de la chambre d’analyse A4.
Remarque : La pression de fond dans la chambre d’analyse est de 2 x 10−10 mbar. - Ouvrir la valve de la chambre d’analyses A4 et utilisez la tige de transfert pour prendre l’exemple de la chambre de transfert linéaire à chambre analyses A4. Fermez la valve de la chambre d’analyse A4.
- Transférer l’échantillon dans la position de l’analyse en utilisant le programme d’acquisition (Figure 3).
- Mettez l’eau de refroidissement et de la source de rayons x du spectrophotomètre (tension anode = 15 keV, émission actuelle = 120 mA).
- Commencer la collecte de données en utilisant le programme d’acquisition (Figure 3).
NOTE : Optimisation de la position de l’échantillon peut être effectuée avec le programme d’acquisition pour obtenir l’intensité du signal maximal.
Remarque : Les spectres de photoémission proviennent de la surface de l’échantillon conservée à température ambiante. - Démarrer la mesure d’un spectre d’aperçu en utilisant les paramètres suivants : KEini = 100 eV, KEnageoire = 1 500 eV, scan temps = 300 s, le n° de points = 1 401, pass énergie = 50 eV, fente = 7 x 20 mm diamètre, mode = mode de zone keV/médium 1,5.
Remarque : L’absence d’un pic de1 s C à environ 284,5 eV BE indique que la surface soit propre. - Commencer l’acquisition du spectre niveau Au4f fondamental avec les paramètres suivants : KEini = 1 396,6 eV, KEnageoire = 1 406,6 eV, scan temps = 60 s, nombre de points = 201, pass énergie = 30 eV, fente = 6 mm diamètre, mode = mode de zone keV/médium 1,5 .
Remarque : Cette dernière mesure sera comparée au spectre après la déposition du film UO2 sur la feuille d’or pour évaluer l’épaisseur correspondante du film. - Une fois la surface du porte-échantillon (feuille UA) a été analysée, ouvrir la vanne de la chambre d’analyse A4 pour positionner le porte-échantillon sur le wagon de la chambre de transfert linéaire à l’aide de la tige de transfert.
- Transfert du wagon par commande à distance à l’aide du programme FLC à la position de la chambre d’analyse A4.
2. couches minces préparation
Remarque : Les couches minces d’oxyde d’Uranium sont préparés in situ par courant continu (DC) pulvérisation utilisant un uranium métal cible et gaz mélange d’Ar (6 N) et de la pression partielle de O2 (4,5 N).
- Dépôt de film2 UO
- Transférer le wagon transportant le porte-échantillon dans la position de la chambre de préparation B2 par commande à distance en utilisant le programme FLC.
- Ouvrir la valve de la chambre de préparation B2 et la tige de transfert, placez le porte-échantillon sous la source de la pulvérisation, située au milieu de B2.
Remarque : Avant le processus de pulvérisation, veiller à ce que l’obturateur de la source de pulvérisation est fermé. - Fermer la vanne pour isoler la chambre B2 de la chambre de transfert linéaire. Ensuite, ouvrir la vanne de2 O et ajuster la pression partielle d’oxygène à 2,5 x 10 mbar−5 . Ouvrir le gaz argon de soupape pour atteindre une pression partielle de 5 x 10−3 mbar.
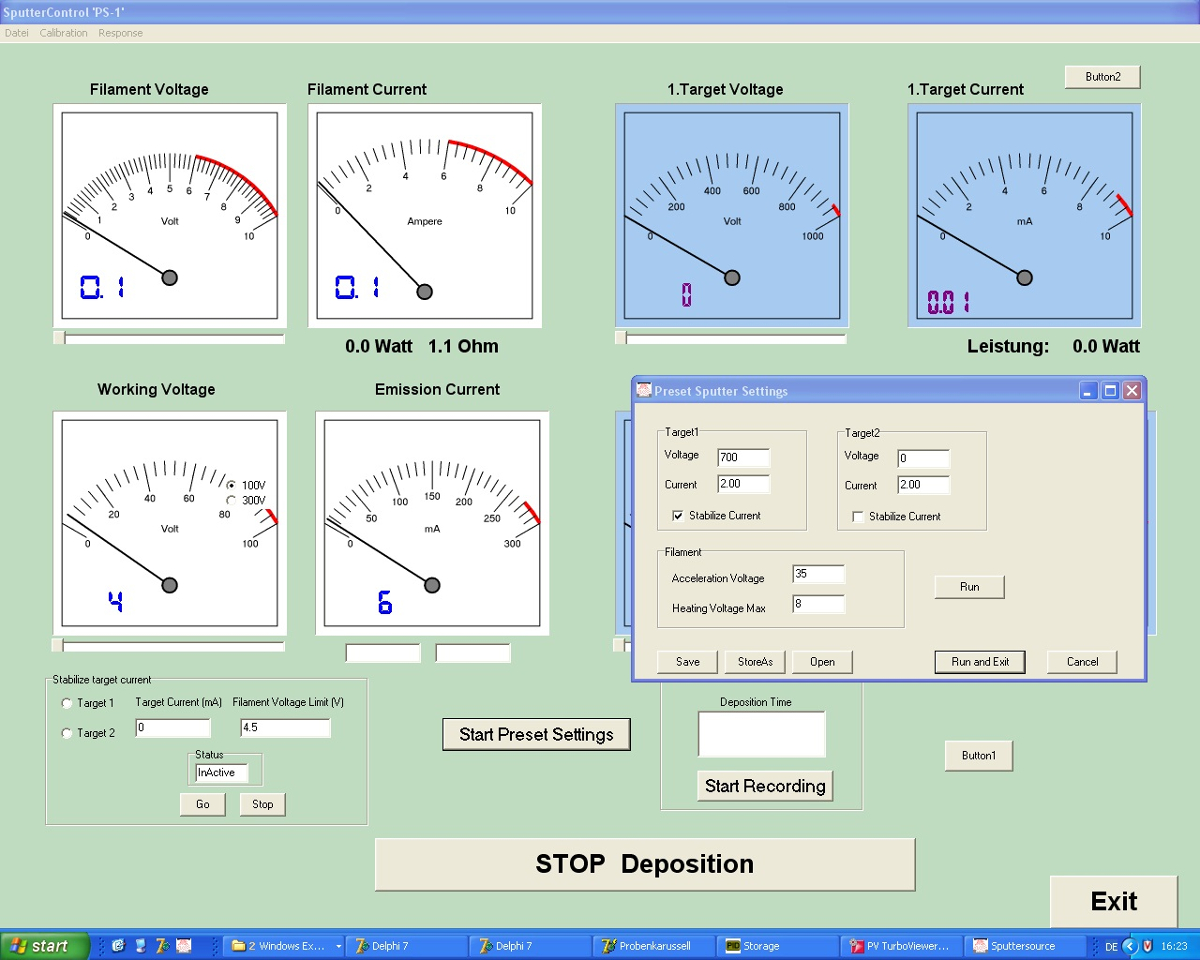
- Le programme de pulvérisation (Figure 4) permet d’entrer les paramètres suivants : temps de dépôt = 300 s, tension cible d’uranium = -700 V, cible d’uranium actuel = 2 mA, filament de chauffage = 3.3 V/3.9 A, tension de filament travail = 40 V.
Remarque : Attendez environ 120 s de pulvérisation avec l’obturateur fermé de la source de la pulvérisation. - Démarrer la minuterie immédiatement après l’ouverture de l’obturateur de la source de pulvérisation afin de permettre la déposition de UO2 sur la grille de l’UA.
Remarque : Pour fonctionner à basse pression Ar sans stabilisation de champs magnétiques, des injections d’énergie de 25 à 50 eV (programme d’installation de la triode) ont pu faire maintenir le plasma dans la diode. - Arrêter la pulvérisation après 300 s, agissez de programme et fermer les vannes de gaz2 Ar et O.
NOTE : La lumière bleue du plasma va disparaître et tous les paramètres de pulvérisation tombera à zéro.
NOTE : Attendez que la pression de la chambre de préparation B2 atteint 10-8 mbar. - Amener l’échantillon dans la chambre de préparation B1 et pour un recuit lisse de l’échantillon, allumer l’appareil e-faisceau et régler la température à 573 K.
Remarque : Attendez 3 à 5 min avant d’arrêter le chauffage.
- Caractérisation de UO2 échantillon
Remarque : Pour la caractérisation de l’échantillon par XPS, la procédure décrite pour la caractérisation de support d’échantillon doit être suivie.- Ouvrez la valve entre la chambre de préparation B1 et de la chambre de transfert linéaire à transférer l’échantillon dans le wagon à l’aide de la tige de transfert. Régler la tige de transfert à la chambre de préparation B1, puis fermez la vanne pour isoler la chambre de transfert linéaire.
- Suivez la procédure décrite dans les étapes 1.5.1 à 1.5.6.
NOTE : Le spectre de la vue d’ensemble permet le contrôle de la qualité du film2 UO en excluant les sommets supplémentaires impureté (C1 s, la contamination croisée du boîtier par pulvérisation cathodique source) et en contrôlant (approximativement) la U:O ratio du film. - Démarrer l’acquisition du niveau de base Au4f (paramètres similaires comme au point 1.5.7).
- Procéder à l’acquisition de U4f, O1 set la bande de valence (VB) en utilisant l’énergie suivant des paramètres : pass = 30 eV, fente = 7 x 20 mm de diamètre, mode = 1,5 keV, zone médium.
U4f: KEini = eV 1 066,6 KEfin = temps de numérisation 1 126,6 eV = 300 s N ° points = 601
O1 s: KEini = eV 946,6 KEfin = temps de numérisation 966,6 eV = 300 s N ° points = 201
VB : KEini = eV 1 473,6 KEfin = temps de numérisation 1 488,6 eV = 1 800 s N ° points = 601
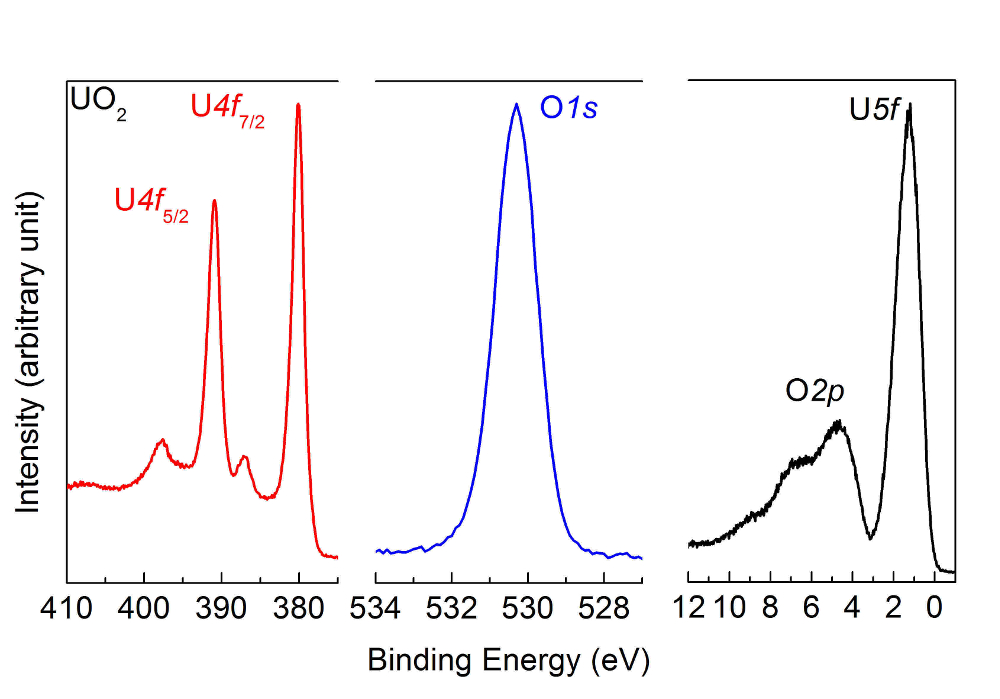
Nota : Les spectres obtenus pour le film de2 UO sont affichées à la Figure 5.
- Oxydation de l’UO2 avec l’oxygène atomique
NOTE : Le flux atom (voir Table des matières) est spécifié pour > 1016 atomes/cm2/s, correspondant à une exposition jusqu'à 20 s d’environ 10 Langmuirs (i.e., 1,33 x 10−3 pa.s).- Transférer le wagon transportant l’échantillon à la chambre de préparation B3. Ouvrir la valve de la chambre de préparation B3 et la tige de transfert, placez l’échantillon à l’intérieur de la B3 devant la source de l’atome. Fermer la vanne pour isoler la chambre de la chambre de transfert linéaire.
- Régler la température de l’échantillon à 573 K. attendre 5 min pour permettre à l’échantillon atteindre la température de l’exploitant.
- Ouvrez le robinet d’oxygène et la valeur de la pression partielle à 1,2 × 10-5 mbar O2. Mettez l’eau de refroidissement pour la source de l’atome.
- Allumez la source atomique, puis affectez-lui le courant de 20 mA. Faites attention à l’heure exacte pour oxyder l’échantillon avec la source atomique. Si le temps d’oxydation est trop court, l’oxydation à l’UO3 peut être incomplète comme indiqué dans la Figure 6.
- Attendre 20 min pour atteindre une oxydation complète de l’UO2 dans UO3 et éteignez la source atomique avant de fermer le robinet d’oxygène. Une fois la pression de la chambre de préparation B3 10-7 mbar, ouvrez le robinet pour transférer l’échantillon dans le wagon présent dans la salle de transfert linéaire. Ensuite, fermez la valve de la chambre de préparation B3.
- Analyse de l’UO3 obtenus après oxydation UO2 avec l’oxygène atomique
- Transférer l’échantillon dans la chambre d’analyse suivant étapes 1.5.1 à 1.5.6 ; puis, pour les analyses, suivez la même procédure comme indiqué au point 2.2.4.
Nota : Les spectres correspondants de UO3 sont affichées dans la Figure 7.
- Transférer l’échantillon dans la chambre d’analyse suivant étapes 1.5.1 à 1.5.6 ; puis, pour les analyses, suivez la même procédure comme indiqué au point 2.2.4.
- Réduction de l’UO3 par l’hydrogène atomique
- Pour transférer l’échantillon dans la chambre de préparation B3, suivez les étapes 2.3.1 et 2.3.2.
- Ouvrez le robinet de l’hydrogène et la valeur de la pression partielle de 3 x 10-5 mbar. Mettez l’eau de refroidissement pour la source de l’atome. Allumez la source atomique et la valeur du courant 30 mA.
- Attendez 60 s de temps de réduction avant d’éteindre la source atomique. Faites attention à l’heure exacte prise pour réduire l’échantillon avec la source atomique. Si réduction temps est trop long, puis UO2 + x est encore réduite à UO2 comme indiqué dans la Figure 8. Dans ce cas, l’échantillon doit être oxydé à nouveau avec l’oxygène atomique (comme nous l’avons fait pour obtenir l’UO3), dont les spectres correspondants sont signalés à la Figure 9.
- Analyse de U2O5 obtenu après réduction de UO3 avec l’hydrogène atomique
- Transférer l’échantillon dans la chambre d’analyse suivant étapes 1.5.1 à 1.5.6 ; puis, pour les analyses, suivez la même procédure comme indiqué au point 2.2.4.
Nota : Les spectres obtenus de U4f, O1 s et le VB sont affichées dans la Figure 10. Comme un exemple de réduction incomplète de U2O5, spectres semblables tel qu’illustré à la Figure 6 sont obtenus.
- Transférer l’échantillon dans la chambre d’analyse suivant étapes 1.5.1 à 1.5.6 ; puis, pour les analyses, suivez la même procédure comme indiqué au point 2.2.4.
Résultats
L’identification des U(V) peut être facilement faite par une énergie caractéristique du satellite d’ébranlement qui accompagnent le doublet de4f caractéristique U. L’énergie de liaison au cours de laquelle apparaît le satellite, associé aux processus de perte d’énergie intrinsèque, dépend de l’état d’oxydation de l’uranium.
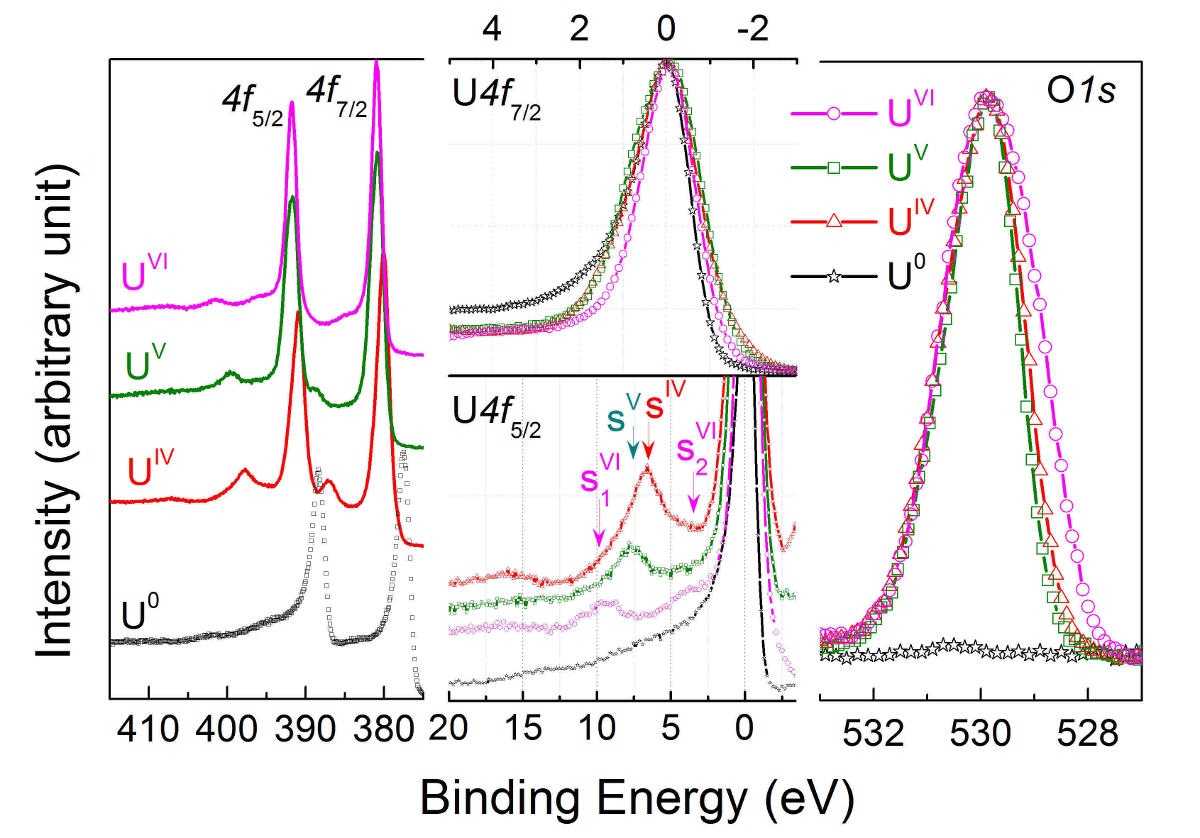
Spectres de la photoémission de la radiographie niveau core 4f des uranium sont enregistrés pour U(IV) UO2 (courbe rouge), U(V) dans U2O5 (courbe verte) et u dans UO3 (courbe rose), puis contre U(0) en uranium métal (courbe noire) à la gauche partie de la Figure 11. Les spectres niveau correspondants O1 s core sont superposées et rapportées dans la partie droite de la Figure 11.
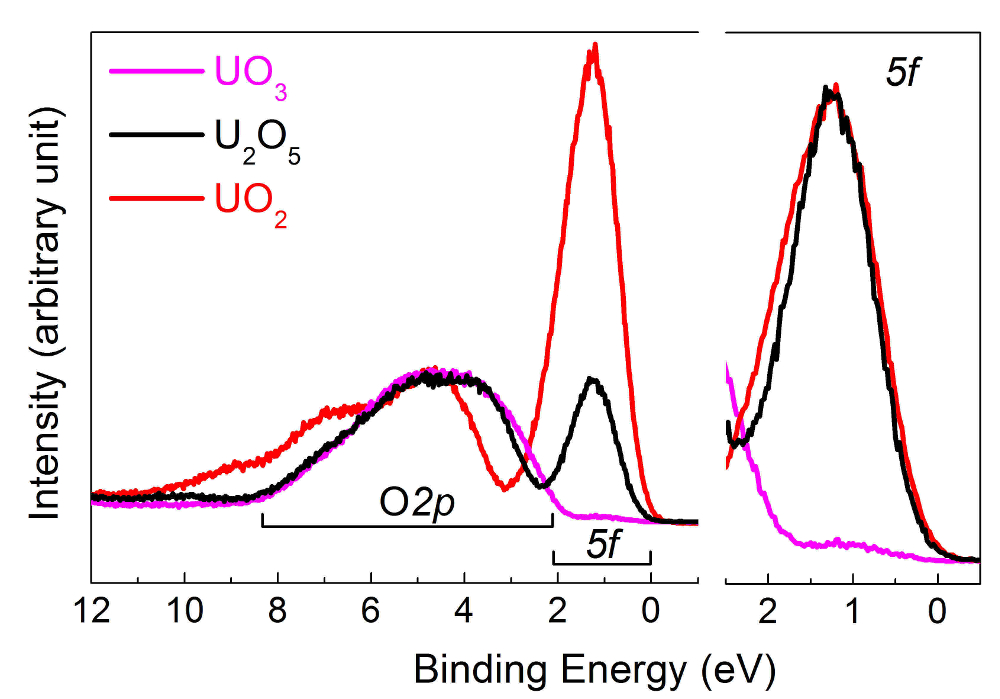
Dans la partie centrale de la Figure 11, les pics U4f7/2 base niveau ont été transférés pour superposer les grandes lignes (moitié supérieure), permettant la visualisation de la séparation de l’énergie (ΔE) entre le satellite et la ligne principale (moitié inférieure). Avec l’état d’oxydation, la séparation de l’énergie augmente, tandis que l’intensité du satellite diminue. Spectres ont été obtenus sur des films minces de monocouches environ 20 d’épaisseur. Le pic d’énergie de satellite et de la raie d’émission de 4f5/2 (4f7/2) ont été utilisés comme une empreinte digitale pour l’état d’oxydation des atomes d’uranium. Les spectres de bande de valence de UO2et U2O5UO3 obtenus sur les mêmes films sont présentés dans la Figure 12.
Les spectres décrites dans le protocole sont correspondant aux films de2 UO (Figure 5) obtenues après dépôt à la chambre de préparation B2. Ce film est ensuite oxydé par l’oxygène atomique. Selon l’heure de l’oxydation, le résultat peut être UO2 + x (comme indiqué dans la Figure 6) ou UO3 (comme indiqué dans la Figure 7). Aussi, si réduction atomique avec de l’hydrogène sur UO3 est trop longue, il retournera à UO2 comme indiqué dans la Figure 8. Dans ce cas, la réoxydation de UO3 comme indiqué à la Figure 9 devait intervenir avant réduisant à nouveau à un moment approprié pour obtenir U2O5, tel qu’illustré à la Figure 10. Les résultats montrent que les processus d’oxydation et de réduction sont complètement réversibles.

Figure 1 : Photographie et schéma de la machine Labstation développé à Karlsruhe JRC pour permettre des études in situ surface science. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 : Capture d’écran du programme de contrôle de transfert linéaire. Le programme permet un transfert du wagon transportant les échantillons (I à V) le long de la chambre de transfert linéaire à des positions différentes de chambre. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : Capture d’écran du programme acquisition. Une fois que les conditions de mesure sont introduites, une série de mesures peut être effectuée automatiquement après la mise en marche du générateur de rayons x. La fenêtre de position d’échantillon permet de positionnement de l’échantillon dans la chambre d’analyse. L’ajustement le long des x, y et z peut être fait pour optimiser l’intensité du signal. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : Capture d’écran du programme de contrôle par pulvérisation cathodique. Les conditions de pulvérisation peuvent être sélectionnées avec ce programme, développé en interne. Parmi les variables à définir sont le chauffage et le travail du filament ainsi que les tensions de jusqu'à deux cibles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : U4f, O1 s et valence spectres de bande après le dépôt d’un film de2 UO, mesuré par spectroscopie à haute résolution de photoémission. Les positions de pointe et par satellite sont caractéristiques d’un échantillon de2 UO. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 6 : U4f, O1 s et valence spectres de bande après oxydation UO2 avec l’oxygène atomique, mesurée par spectroscopie à haute résolution de photoémission. Le temps de l’oxydation est trop court, que donc l’oxydation en UO3 est incomplet. Les positions satellite et pic sont caractéristiques de UOx 2 + et non de l’UO3 indiqué dans la Figure 7. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 7 : U4f, O1 s et valence la bande spectres mesurés après oxydation du film2 UO avec l’oxygène atomique à l’aide de la spectroscopie à haute résolution de photoémission. Les positions de pointe et par satellite sont caractéristiques d’un échantillon de3 UO. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 8 : U4f, O1 s et valence la bande spectres mesurés après réduction de l’UO3 avec l’hydrogène atomique. Le temps de réduction est trop long, donc la U2O5 est encore réduite à UO2. Les positions satellite et pic sont caractéristiques d’une UO2 et pas le U2O5 échantillon indiqué dans la Figure 10. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 9 : U4f, O bande1 s et de valence de l’échantillon obtenu en Figure 8 et ré-oxydé par l’oxygène atomique à UO3. Les positions satellite et pic sont caractéristiques d’un échantillon de3 UO. Les processus de réduction et d’oxydation sont donc réversibles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 10 : U4f, O1 s et valence spectres de bande après réduction du film3 UO avec l’hydrogène atomique, mesurée par spectroscopie à haute résolution de photoémission. Les positions de pointe et par satellite sont caractéristiques d’un échantillon de5 2O U. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 11 : U4f et O1 s core niveau photoémission spectres de rayons x de U(IV) UO2 (courbe rouge), U(V) dans U2O5 (courbe verte) et u dans UO3 (courbe rose), puis contre U(0) en uranium métal (courbe noire). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 12 : Spectres de bandes de Valence de U(IV) UO2 (courbe rouge), U(V) dans U2O5 (courbe noire) et u dans UO3 (courbe de rose). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Discussion
Les premiers résultats obtenus sur les couches minces d’U2O5 d’environ 30 monocouches (ML) d’épaisseur, ainsi que la spectroscopie de niveau noyau correspondant obtenue avec haute résolution spectroscopie de photoémission, ont été signalés dans un précédente publication7. L’évolution de l’état de l’uranium durant le processus d’oxydation de UO2 dans UO3 a été signalée par le biais de spectres de photoélectrons de rayons x obtenues sur des films minces de deux à 50 couches d’épaisseur dans un large éventail de la ratio O:U (Figure 11, Figure 12). Film oxydation et réduction de film ont été obtenus en exposant les films à l’oxygène atomique et de l’hydrogène atomique, respectivement. L’homogénéité des films avec les États d’oxydation de l’uranium de IV à VI pourrait être confirmée en raison de leur faible épaisseur et de la température de réaction. Couches minces d’oxydes d’uranium sont déposés sur un substrat à l’aide de courant continu pulvérisation avec une source de pulvérisation développée à Karlsruhe JRC. La source de pulvérisation est installée dans une enceinte maintenue sous ultravide, comme toutes les chambres de le Labstation. Tandis que UO2 peut être obtenu directement, UO3 et U2O5 films sont uniquement obtenus après traitement supplémentaire avec l’oxygène atomique et de l’hydrogène atomique. L’énergie de liaison des principaux sommets et leurs positions satellites permettent la différenciation entre les États d’oxydation de l’uranium dans un film d’oxyde d’uranium produit in situ. Spectroscopie à haute résolution est nécessaire de différencier les États d’oxydation différents, comme les énergies de liaison par satellite sont proches et ont de faibles intensités.
En 1948, l’uranium pur pentavalent, U2O5, a été identifié pour la première fois8. Sa synthèse a été décrit plus tard, issu des hautes températures (673-1 073 K) et à haute pression (30-60 kbar) d’un mélange de UO2 et U3O89. Toutefois, l’existence et la stabilité de U2O5 aux conditions ambiantes de température et de pression ont été interrogés, ce qui suggère une limite inférieure de x = 0,56 à 0,6 pour la région monophasée dessous U3O810 . Jusqu’ici, la préparation de U2O5 à température et pression élevée ou lors d’un processus de réduction de la thermo n’était pas reproductible ; souvent, il n’était pas possible d’assigner un état d’oxydation simple à échantillons obtenus. Certains d’une préparation de l’échantillon de U2O5 en vrac est apparu sous forme de mélanges de UO2 ou UO3 avec coexistence de U(V) avec U(IV) ou u, en ce qui concerne U4O9 et U3O8. Par exemple, Teterin Al11 déclaré du procédé de lixiviation de U3O8 dans l’acide sulfurique suivie d’un traitement thermique dans une atmosphère d’hélium, affirmant que les résultats étaient associés à U2O5. Cette conclusion pourrait facilement être exclue en raison d’une structure qui en résulte de deux pics dans leurs spectres XPS. Un mélange d’espèces U(V) et u pourrait expliquer le résultat, à l’exclusion de la formation d’un seul état d’oxydation U(V) prévu pour U2O5.
Notre méthode de préparation permet la préparation des couches minces d’oxyde d’uranium avec les États d’oxydation simple de U(IV), u et U(V). L’ensemble du processus de préparation des échantillons a lieu sur place dans un instrument maintenu à ultravide. Il a été constaté que réduction de UO3 par l’hydrogène atomique ne procède pas à l’UO2 mais peut être arrêtée à U(V). Le facteur temps est très important ainsi que la température de l’échantillon pendant la réduction. Avec le spectromètre à haute résolution de photoémission, il a été démontré qu’un échantillon pur de U2O5 peut être préparé in situ. Préparation de films plus épais doit être une étape suivante lorsqu’on examine la structure cristallographique et propriétés en vrac avec ex-situ des techniques.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs n’ont aucun remerciements.
matériels
| Name | Company | Catalog Number | Comments |
| 1ary dry scroll vacuum pump | Agilent | SH-100 | All chambers except B1 |
| 1ary pump | EDWARDS | nXDS10i 100/240V | B1 chamber |
| Acetone | |||
| Acquisition programme | Developed in-house | ||
| Analyser | Specs | Phoibos 150 hemispherical | A4 chamber |
| Argon | BASI | 6N | |
| Atomic source | GenII plasma source | Tectra | B3 chamber |
| Au foil | Goodfellow | ||
| CasaXPS programme | CasaXPS | ||
| Gauge 1ary vacuum | PFEIFFER | TPR 280 (2011/10) | All chambers |
| Gauge 2ary vacuum | VACOM | ATMION ATS40C | All chambers |
| Hydrogen gas | BASI | 6N | |
| Ion gun source | Specs | IG10/35 | B1 chamber |
| Linear transfer programme | Specs | Program delivered with the station | |
| Origin programme | Origin | OriginPro 8.1SRO | |
| Oxygen gas | 6N | ||
| Sampler e-beam heater power supply | Specs | SH100 | B1 chamber |
| Sampler resistance heater | Made in-house | power supply + Eurotherm | B3 chamber |
| Sputtering programme | Developed in-house | ||
| Stainless steal or Molybdenum substrate | in house | ||
| Ta wire | Goodfellow | ||
| turbo pump | PFEIFFER | TC 400 | All chambers |
| Uranium target | in house | in house | Natural uranium target |
| Vacuum gauge controller | VACOM | MVC-3 | All chambers |
| X-ray source | Specs | XRC-1000 MF | Equipped with a monochromator |
Références
- Shoesmith, D. W., Sunder, S., Hocking, W. H. Electrochemistry of UO2 nuclear fuel. Electrochemistry of Novel Materials. Lipkowski, J., Ross, P. N. , New York, N.Y. (1994).
- Shoesmith, D. W. Fuel corrosion processes under waste disposal conditions. Journal of Nuclear Matter. 282, 1-31 (2000).
- Gouder, T., Shick, A. B., Huber, F. Surface interaction of PuO2, UO2+x and UO3 with water ice. Topics in Catalysis. 56, 1112-1120 (2013).
- Cohen, C., et al. Water chemisorption on a sputter deposited uranium dioxide film - Effect of defects. Solid State Ionics. 263, 39-45 (2014).
- Seibert, A., et al. The use of the electrochemical quartz crystal microbalance (EQCM) in corrosion studies of UO2 thin film models. Journal of Nuclear Matter. 419, 112-121 (2011).
- Majumder, I., et al. Syntheses of U3O8 nanoparticles form four different uranyl complexes: Their catalytic performance for various alcohol oxidations. Inorganic Chimica Acta. 462, 112-122 (2017).
- Gouder, T., et al. Direct observation of pure pentavalent uranium in U2O5 thin films by high-resolution photoemission spectroscopy. Scientific Reports. 8, 1-7 (2018).
- Rundle, R. E., Baeziger, N. C., Wilson, A. S., MacDonald, R. A. The structures of the carbides, nitrides and oxides of uranium. Journal of the American Chemical Society. 70, 99(1948).
- Hoekstra, H. R., Siegel, S., Gallagher, F. X. The uranium-oxygen system at high pressure. Journal of Inorganic and Nuclear Chemistry. 32, 3237(1970).
- Kovba, L. M., Komarevtseva, N. I., Kuz'mitcheva, E. U. On the crystal structures of U13O34 and delta-U2O5. Radiokhimiya. 21, 754(1979).
- Teterin, Y. A., et al. A study of synthetic and natural uranium oxides by X-ray photoelectron spectroscopy. Physics and Chemistry of Minerals. 7, 151-158 (1981).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.