
Method Article
大規模なSNPジェノタイピングアプリケーション用インフィニウムアッセイ
要約
プロトコルは、大規模な遺伝子型決定を行うためにイルミナインフィニウムアッセイを使用することが記載されている。これらのアッセイは、確実に3日間で、個々のDNAサンプルの数百人を越えSNPの何百万人の遺伝子型を決定することができます。一度生成された、これらの遺伝子型は異なる疾患または表現型の様々な関連性をチェックするために使用することができる。
要約
ヒトゲノムにおける遺伝子型決定の変異体は、表現型と遺伝的関連を同定する効率的な方法であることが証明された。家族又は集団内の変異の分布は、疾患の遺伝因子の同定を容易にすることができる。遺伝子型決定BeadChip製品のイルミナのパネルは、研究者は、一塩基多型(SNP)の数千または数百万の遺伝子型を決定するか、またはそのようなコピー数のような他のゲノムの変異体を分析するために、DNAの多数のサンプルにわたって可能にする。これらのSNPは、ゲノム全体に広がるか、潜在的な発見を最大化するために、特定の領域に標的とすることができる。のInfiniumアッセイは、迅速に、高品質で正確な結果を得るために最適化されている。適切なセットアップを使用すると、単一の技術者は、アレイの種類に応じて、週に1000人以上のDNAサンプルには数百から処理することができます。このアッセイは、ゲノムDNAから開始して、アレイのスキャンで終わる、すべてのステップを使用してユーザーをガイドします。適否レアージュを使用してNTSは、サンプルが増幅され、断片化し、沈殿した再懸濁し、染色された単一の塩基により拡張チップにハイブリダイズされ、のiScanまたはハイスキャン高分解能光学イメージングシステムのいずれかでスキャン。一晩のステップは、DNAを増幅するために必要とされている。 DNAを変性し、等温的に全ゲノム増幅によって増幅され、従って、全くPCRは必要とされない。試料は、第一晩ステップ中にアレイにハイブリダイズされる。 3日目までに、サンプルがスキャンされ、分析される準備ができている。増幅されたDNAは、ビーズアレイ、それにより、スループットを最大化する、毎日を処理することができ、大量に貯蔵されてもよい。
概要
一塩基多型(SNP)を入力すると、疾患に関連するリスク変異体を同定する重要な方法である。歴史的に、遺伝子型決定実験の範囲は、利用可能な技術によって制限されてきた。ゲル電気泳動に基づく遺伝子型決定方法は、サンプルSNPスループットが制限されている[1]。これらのアッセイの開発は、多くの場合、最適化のためのバリアントを囲む領域のメイクや構造に依存する、労働集約的であることができます[1]。ライフテクノロジーズが開発したTaqManジェノタイピングアッセイは、すぐに大量のサンプルを実行し、最小限の技術者の関与と[2]が、SNP多重化の制限は一日だけでなく下に百万に遺伝子型の総数を制限し続けることができます[3,4 ]。未満百SNPが一緒に多重化することができるようにシーケノムのIPLEXプラットフォームはまた、一度に多くのサンプルを実行することができますが、スループットが全体的に比較的低い[5]。ベックマン·コールターのSNPストリーム技術理論的には一日あたり300万人以上の遺伝子型を生産するが、反応あたりわずか四〇から八SNPの最大化するためにこの技術を制限し、プロジェクトの範囲[4,6]ことができます。 GoldenGateのアッセイは、サンプルあたりのSNP数百または数千に毎日ほぼ200 DNAサンプルを処理することができますが、遺伝子型あたりの価格は、高度な、超高スループット技術と競合しないで、一度に3000以上のSNPを入力するときに[4,7 ]。一日あたり数百万の遺伝子型を処理するために、大規模なゲノムワイド関連研究のために必要なスケールは、アレイハイブリダイゼーションアッセイは、市場で最もコスト効率の良い選択肢となっている。
ハイブリダイゼーションアレイのAffymetrix社のラインとのInfiniumベースのアレイのイルミナのラインは、サンプルの潜在的に何百ものパラレル[4,8]内のSNPの数千または数百万、数百で入力することができます。これらのSNPは、元のように利益の地域でローカライズさ、ゲノム全体に散在することができますomes、またはユーザの好みに合わせてカスタマイズ。これらのアレイは、正確に一度試料100万のSNPの遺伝子型を決定することが、また、潜在的に発表染色体異常をコピー数の変化を測定することができない唯一の利点を有する。インフィニウムアラインOMNIビーズチップアレイは現在、毎日ほぼ百サンプルまでに50万カスタム座を含むサンプルあたりほぼ500万マーカーまでの遺伝子型と能力を持っている。
ほとんどの疾患は遺伝的要素を持っているように、これらの大規模な実験は、疾患に関連する遺伝子を発見する上で重要であることができる。ハイスループット遺伝子型決定は、試料中の効率的な遺伝子型生成を可能にする説得力の低いマイナーアレル頻度で遺伝的関連を検出するのに十分な大きさに設定する。全ゲノムジェノタイピングプロジェクトは、統計的に有意な症例対照対立遺伝子頻度やコピー数の違いで領域を特定するために使用することができる[9,10,11]。国家人権のGeによると、ノーム研究所は、ゲノムワイド関連研究は、P-1未満の値×10 -5で8283個のSNPの発見に端を発する、2008年11月25日およ び2013年1月25日の間で1490の別々の出版につながった(http://を参照してくださいwww.genome.gov/gwastudies/)。高さから精巣癌に至るまでの条件を調査し、これらの研究は、ゲノムワイドな解析によってもたらされる幅広いアプローチから恩恵を受けた。タイピングの範囲が厳しすぎるされていたこのような場合には、対象となる全領域は、予告を免れた可能性があります。したがって、大規模なアソシエーション分析のため、ゲノムワイドな遺伝子型決定技術は、選択された技術である。
のInfiniumアッセイの異なるバージョンは、それぞれが特定のタイプのアレイで使用するためのものが存在する。または24サンプルアレイチップ - 以下に詳しく説明InfiniumUltraアッセイは、多くの12に適している。これらは多くの場合、DNAサンプルあたりの10万以上のSNPを遺伝子型とTに焦点このようなexomeやカスタムパネルのようargeted地域。他のアッセイのバージョンでは、全ゲノムジェノタイピングアレイのような他のチップタイプのために必要になることがあります。すべてのインフィニウムアッセイは共通の基盤を共有し、主に試薬名、試薬容量、または正確な染色試薬手順のみが異なるしかし、1アッセイのバージョンに完成の技術は、多くの場合、普遍的に適用することができます。このようなメチル化アレイのような他の配列は、だけでなく、ほぼ同一のプロトコルを使用する場合があります。お手入れは、使用中のチップタイプに必要なアッセイのバージョンを使用するように注意しなければならない。このような遺伝子発現レベルを測定するものなど、いくつかのタイプは、nonInfiniumプロトコルの使用を必要とする場合があります。
サンプルは、バッチで処理しなければならない。例えば、InfiniumUltraアッセイで、プレハイブリダイゼーション試薬チューブは、96のサンプルを実行するのに十分な量が含まれており、チューブが再凍結することはできません。そのため、サンプルは一度に96サンプルのバッチで実行する必要があります。サンプルはモミ上で増幅されますTの日。ベンチワークの約1時間後、サンプルを20〜24時間、対流式オーブン内で加熱しなければならない。次の日、ほぼ4時間で沈殿させること、断片化過ごし、サンプルはいずれの将来の使用のために凍結またはチップにハイブリダイズさせることができる、その時点でサンプルを再懸濁します。チップをロードすると、サンプルは16〜24時間、一晩ハイブリダイズされるまでの、ほぼ2時間かかります。三日目に、染色および拡張手順は、〜4時間かかります。さらに1時間の洗浄、コーティング過ごし、チップを乾燥されます。最後に、配列は使用されるタイプに応じて、15〜60分/チップからかかることがあり、スキャンされます。
標準的な実験室の安全性と清潔予防措置が適用されます。増幅はPCRベースないが、前およびpostamplification手順のための別個のワークステーションは、汚染の可能性を最小限にするために必要である。使用されているすべてのキットが提供する試薬の識別番号は、追跡シートにログインする必要があります。レアージュ国税庁は、使用直前に解凍し、分配する前に数回反転する必要があります。タイプされるDNAは、高品質のゲノムDNA(280分の260 1.6〜2.0の吸光度比、230分の260以下の3.0の吸光度比)、標準的な方法で単離し、蛍光光度計で定量化しなければならない。 DNAの分解は、多くの場合、低品質のアッセイ結果における寄与因子である。この量は、いくつかのチップの種類によって異なる場合がありますが、通常、DNAの200 ngの、必要とされている。テカンの液体処理ロボットはプロトコルの多くのステップを自動化し、要素として、ヒューマンエラーを最小限に抑えることができます。
プロトコル
初日
1。準備
- ディープウェル、96サンプルプレートにDNA 200ngの分注する。少なくとも96のサンプル(完全版)がない試薬が無駄にされません確実にするためにめっきされている必要があります。このキットにより供給されたバーコードステッカーとプレートにラベルを付けて、それを遠心する。
- ボリュームを正規化するために、液体を蒸発させるために、引き出しやヒュームフード晩でサンプルを残して。埃が入らないようにふたや紙タオルで緩くプレートをカバーしています。
2。増幅
警告:4時間は、断片化、沈殿、および再懸濁の手順については、次の日に利用できるようになりますしない限り、サンプルは増幅を受けるべきではありません。
- -20℃の冷凍庫(MA1、MA2、とMSMの1管は96サンプルの各セットのために十分である)から「プレ」ラベルしたチューブの企業が提供するボックスやパックを取り外します。解凍ベンチにMA2とMSMのチューブを設定します。正しくキャリブレーションさオンにオーブン、37℃に設定
- 試薬流域および10μL、8チャンネルピペットを使用して、サンプルを再水和するために、各ウェルにDNA再懸濁バッファーを4μlを分注する。それは注意が液体に触れないように注意されている場合、各列の間にピペットチップを廃棄する必要はありません。
- 8チャンネルピペットを用いて、プレートの各ウェルにMA1を20μlを分注する。新しい試薬は、新しい試薬流域を使用しています。再利用可能なシール、パルス遠心機、およびマイクロプレートシェーカーで1,600 rpmで1分間ボルテックスでプレートをカバーしています。
- 2.4)で少なくとも30分間室温でインキュベートする。
- プレートの各ウェルに0.1 N NaOHを4μLを分注する。再利用可能なシール、パルス遠心し、1,600 rpmで1分間ボルテックスでプレートをカバーしています。
- 10分間室温でインキュベートする。
- プレートの各ウェルにMA2の34μLを分注する。
- プレートの各ウェルに、MSMの38μLを分注する。でプレートをカバー再利用可能なシール、パルス遠心し、1,600 rpmで1分間ボルテックス。
- 20〜24時間、オーブンに入れます。
二日目
3。フラグメンテーション
- 「ポスト·1」からFMSチューブを外し-20℃の箱(1管は96サンプルの各セットのために十分である)。ベンチにまたは室温の水浴中で解凍する。卓上マイクロサンプルのインキュベーションシステムにMIDIプレートインサートを挿入した後、ヒートブロックをオンにし、37℃に設定
- チューブを解凍したら、オーブン、パルス遠心分離機からサンプルプレートを取り外します。 DNAのプレートの各ウェルに、FMSの25μLを分注する。 1分間1,600 rpmで蓋、パルス遠心分離機、渦を交換してください。
- 1時間ヒートブロックに静置します。
4。降水量
- 4℃の箱「ポスト3」からPM1チューブを削除するか、パックし、室温に戻し(1管は96サンプルの各セットのために十分である)。熱圏からプレートを取り外しKとパルス遠心。
- プレートの各ウェルにPM1の50μLを分注する。 1分間1,600 rpmで蓋、パルス遠心分離機、渦を交換してください。
- 5分間ヒートブロック中でプレートをインキュベート。
- ヒートブロックからプレートを取り外し、それをオフにし、プレートの蓋を捨てる。プレートの各ウェルに100%イソプロパノール155μlずつ分注する。新しい蓋をしっかりとカバーして、手動で混合するプレートを複数回転倒。
- 最低30分間、4℃でプレートを置きます。冷却遠心機の電源を入れ、冷却するために4℃に設定してください。
- 3000 X gで20分間4℃でプレート遠心分離のバランスをとる。
- それを反転させずにプレートの底を点検し、サンプルが青色のペレット中に沈殿していることを確認します。何ペレットが見られない場合は、再度遠心する。ペーパータオルを取得します。
- ふたを捨て、すぐにプレートを反転し、ペーパータオルでカバーベンチトップの表面を強くそれをタップすることで、液体を除去。プレートは私がしたらnverted、任意の液体が残っている間、それを元に戻すしないように注意してください。すべての液体が除去されるまで繰り返し卓上に対してプレートをタップします。
- 乾燥させるために、反転し、試験管ラックにプレートをセット。 1時間室温でインキュベートする。
5。再懸濁
- 「ポスト-2」からRA1を除去-20 Cボックスと解凍°室温の水浴中。オーブンの電源を入れ、48℃に設定してください
- 一度解凍したサンプルプレートの各ウェルにRA1の23μLを分注する。盆地にRA1の全体のボトルを入れないでください。後で30ミリリットルを保存します。試料はその日の配列にハイブリダイズされる場合は、4℃のクーラーにRA1を置く。サンプルは後日実行される場合には、ボトルにラベルを付け、RA1を再凍結。
- プレートの上に新しいふたをヒートシール。プレートを、パルス遠心し、1時間、オーブンに入れてください。
- 1分間1800回転でオーブン渦からプレートを取り外します。
サンプルは、安全にHすることができます最長1週間、この段階でELD。プレートを備蓄することができ、サンプルは、必要に応じて、ビードチップアプリケーションの準備のために再編成されてもよい。手続きを進める場合は、室温でプレートを出て、ヒートブロックをオンにします。それ以外の場合は、-20℃でプレートを保存する
6。混成
警告:5.5時間染色し、洗浄工程のために、次の日に利用できない限り、サンプルのハイブリダイゼーションを受けるべきではありません。
- ヒートブロックの電源を入れ、95℃に設定してくださいオーブンの電源を入れ、48℃に設定してください
- 温度が安定したら、20分間、ヒートブロック上でプレートをインキュベートする。
- プレートが変性している間、4℃からビーズチップのボックスを削除し、ベンチの上に置きます。 (室温キットから)XC4のボトルを取り、100%エタノール330ミリリットルを追加。それをよく振って、室温で一晩インキュベートする。ホルムアミド/ EDTA混合物(95%ホルムアミド、0.2%調製EDTA(0.5 M)、体積で4.8%H 2 O)と別個の15mlの増分で凍結する。過剰のホルムアミドを備蓄してもよい。
- ヒートブロックからサンプルプレートを取り外します。 30分間室温でインキュベートする。
- プレートが冷却されている間、HYB室を準備します。一方の室は処理ごとに4ビーズのチップのために必要とされます。
ベースの前面に厚い穴を合わせて、HYB室ベースの上にゴム製のマットを置きます。ベース内にエッチングバーコードシンボルは、まだ( 図2参照) を表示する必要がある。 - 千μlのピペットを用いて、ベースに刻まれた8加湿貯水池のそれぞれにPB2の400μLを分注する。 PB2は「ポスト3」ボックスまたはパックに記載されています。ベースにHYB室蓋を置き、最初の対角線上反対側に2留め金を閉じて両端を握る。
- チップ「万博を最小限に抑えるために、それぞれのボックスからビーズチップの個々の銀のパックを削除するが、空気や光にしてください、それらをまだ開いていない。慎重にチップのバーコードをスキャンして、彼らが来た箱のIDと一緒に、トラッキング·シート上にロードされる順序を記録します。それぞれの個々のDNAサンプルは、ビーズチップ上に分配される場所の十分な理解が必要である。
- それぞれのボックスからビーズチップの個々の銀のパックを削除するが、空気や光にチップの暴露を最小限に抑えるために、それらをまだ開いていない。慎重にチップのバーコードをスキャンして、彼らが来た箱のIDと一緒に、トラッキング·シート上にロードされる順序を記録します。それぞれの個々のDNAサンプルは、ビーズチップ上に分配される場所の十分な理解が必要である。
- 30分には、クールダウン直前に銀ビーズチップパックを開き、完了しています。その透明なプラスチックスリーブからチップを取り外します。ビーズに触れることなく、チップを配向、HYB室インサート上のすべてのビーズチップを配置インサートの上部表面にエッチングされたバーコードシンボルをバーコード。
- はがれやサンプルプレートの蓋を捨てる。サンプルプレートからサ ンプルを15μlを取り、ゆっくりと、アレイへの入口に分配する( 図3を参照)。臨時ケアは以前に記録したビーズチップの順序と一致する、正しい位置に正しいビーズチップ上の正しいサンプルを置くように注意しなければならない。マルチチャンネルピペットチップ上の液体を分配するために用いることができるが、先見の明は、プレート上の行の数は、常に数と一致しないように、安全に、ビードチップ上に配置することができるサンプルの数のみを吸引するように注意しなければならないチップ上の行。
- 一度サンプルは、対応するアレイ上に置かれた、視覚的に液体に塗布されていない気泡や地域のチップを検査します。これらの問題が存在する場合は、ゆっくり挿入を揺する。必要であれば、より多くの液体がアレイに添加してもよい。正しいDNAサンプルを追加するように注意してください。
- HYB室ANを開く加湿貯水池の上にインサートを配置dは。チップのバーコードはHYB室基板にエッチングされたバーコードの上に配置する必要があります。 HYB室カバーを元に戻し、最初の対角線上反対側に留め金を閉じて、すべての4つの留め金を閉じます。
- 16〜24時間にわたりオーブンでHYB室をインキュベートする。それらを移動するときにチャンバーを傾けないように注意してください。
- 複数のプレートが処理される場合、6.2のステップに戻る。一日に24以上のビーズチップを処理する医療マストなどの消耗品またはチップを大量に取り扱う場合、ヒューマンエラーの可能性を最小限にし、将来の工程でチップを処理するのに必要な時間の量を最小にするために両方のために推奨されないできるだけ多くの空気への曝露を制限するために取られる。 XC4一瓶には、最大24のビーズチップで十分である。
一日三
7。染色の準備
- オーブンからHYB室を取り外し、室内TEでインキュベート25分間mperature。二つ以上のHYBチャンバを処理する場合は、それらの二重の除去10分毎にずらす。乾燥からチップを維持するために、まだHYB室を開けないでください。
- HYB室が冷却されていますが、Cの箱-20「ポスト1」からXC1、XC2、TEM、STM、およびATMのチューブを取り外して、解凍するベンチに設定してください。 (前日から試薬を再利用する場合は4℃)-20℃で「ポスト2」ボックスからRA1のボトルを取り出し、室温の水浴中で解凍する。だけでなく、95%のホルムアミドのチューブを解凍する。 8ビーズチップのため、各試薬の2チューブ、10ミリリットルのRA1、15ミリリットルのホルムアミドを処理し、(室温、同社が提供するボックスにある)150ミリリットルのXC3が必要になります。
- 処理されたチップ毎に、一枚のガラススライド、一つのプラスチックフロースルーブレース、二つの金属の留め金、および一つのプラスチックスペーサが必要となる。プラスチック·スペーサの場合は、唯一の明確な1を使用し、不透明なスペーサーを分離し、廃棄します。加えて、2ビーズチップ洗浄料理と一方のビードチップトレイは、一液アセンブリ局と1つのプラスチック組立バーと共に、必要とされるであろう。
- エタノールを噴霧し、乾燥して拭いてガラススライドを清掃してください。
- フロースルーチャンバーラックに取り付けるのは湯浴の電源を入れ、44℃に設定してください優しく気泡を取り除くために、チャンバーのラックを横に振る。
- HYB室は25分間冷却したときに、PB1(約200ミリリットル)で洗浄ディッシュを埋める。 PB1は、「ポスト4」は、室温箱に記載されています。
- 1 HYB室を開きます。 1コーナーでシールをつかみ、ゆっくりと( 図4を参照)斜めに剥離することにより、ビーズチップからカバーシールを取り外します。シールが除去されると、すぐにビードチップトレイ内のチップを配置し、ビーズに触れることなく、PB1に沈める。トレイは任意のチップを乾燥させないように注意しながら、ビーズチップで満たされるまで繰り返します。
- 優しくあらゆる気泡を除去し、1分間水没のチップを残して、トレイを攪拌。PB1と第二の洗浄の皿を埋める。もう一度トレイを攪拌。
- 第二の洗浄皿にビーズチップのトレイを移動します。静かに攪拌し、1分間染み込ませ、再度攪拌する。
- 液体フロースルー組立ステーションでは、溝の中に4黒いプラスチックのフロースルー中括弧を置く。液体はほぼ8組立金具(約150ミリリットル)の高さに達するまで、PB1とステーションを埋める。
- トレイからビーズチップを取り外し、プラスチックのフロースルーブレースに組立ステーションに配置します。チップバーコードはアセンブリステーションにエッチングされたバーコードシンボルの上に揃っている必要があります。組立ステーション内に収まることができ、他の3つのチップについて、この手順を繰り返します。
- ビーズチップの上に透明なプラスチックのスペーサを配置します。スペーサの外周縁部は、組立ステーション内にブラケットを取り囲むべきである。 PB1に沈め他のチップについて、この手順を繰り返します。
- 溝の上に嵌合することにより、組立ステーションでのプラスチック組み立てバーを配置。優しく組立バーに対してスライドの後端を押し、ゆっくりと液体にフロントを下げることによって、プラスチックスペーサーの上にガラススライドを配置。スライドの上部の溝は、ビーズチップとバーコードの末尾のスライドの間に隙間を残して、直接チップのバーコードの上に、下方向に向くようにします。 PB1に沈め他のチップについて、この手順を繰り返します。完全なフロースルー組立図については、 図5を参照してください。
- チップとスライドの間に気泡がないか確認してください。気泡が表示された場合は、静かにバーコード終了時にスライドを高め、再試行してください。永続的な泡は、紙の研究室タオル(キムワイプ)で、ガラスから拭き取ることができます。
- ガラススライドの周囲に二つの金属の留め金、バーコード終わりに向かって1と後方に1スナップ。留め金の端べきグリップチップの下にプラスチック製のフロースルーブレース。 PB1に沈めチップ毎に繰り返します。
- 完成したフロースルー·アセンブリを取り外し、B上に水平に置きますench。ガラススライドの下に液体が逃がす、縦にアセンブリを傾けないように注意してください。より多くのチップを洗浄トレイに待機している場合、それらは同じPB1下で組み立てることができる。他のチップが元HYB室で待機している場合は、組立ステーション内のすべての液体を廃棄し、皿を洗って、7.6段階に戻ります。
- 一旦全てのビーズチップはできるだけスライドガラスに近い、外科用はさみを取り、オフスペーサの両端を切断し、それらのフロースルーアセンブリである。
8。染色および拡張
- フロースルーチャンバーラックは温度プローブを複数の位置で44℃に達したことを確認します。温度が半分以上程度ではオフになっている場合は、水浴の温度を調整します。
- ダウンアセンブリをスライドさせ、上部にブレースをフックして、チャンバラックにビーズチップフロースルーアセンブリを配置します。ビーズチップの裏面は、チャンバラックに触れなければならない。 G少女のスライドは、試薬リザーバーを形成するために、上部の溝に、外を向いている必要があります。すべてのビーズチップアセンブリのために繰り返します。
- 200μlのピペットを用いて、ガラスリザーバーに液体を分配することによって、フロースルーアセンブリRA1to 150μLを加える。 30秒間インキュベートする。 5倍を繰り返します。
- 千μLピペットを用いて、ガラスリザーバーに液体を分配することによって、フロースルーアセンブリXC1to 450μlを加える。 10分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルー·アセンブリに450μlのXC2を追加します。 10分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルー·アセンブリに200μlのTEMを追加します。 15分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルーアセンブリに450μlの95%ホルムアミド/ EDTAミックスを追加します。 1分間インキュベートする。 1Xを繰り返します。
- 5分間のフロースルー·アセンブリをインキュベートする。
- 温度lに熱水浴を設定istedは、STMのチューブに(または37℃いずれも図示されていない場合)。各STM管が記載されている同じ温度を持っていることを確認します。
- フロースルーアセンブリに450μlのXC3を追加します。 1分間インキュベートする。 1Xを繰り返します。
- 湯浴の温度が所望の温度に達したときに、ガラスリザーバ内に液体を分配することによって、フロースルーアセンブリ250μLSTMを追加する。 10分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルー·アセンブリに450μlのXC3を追加します。 1分間インキュベートする。 1Xを繰り返します。
- 5分間のフロースルー·アセンブリをインキュベートする。
- ガラスリザーバーに液体を分配することによって、フロースルー·アセンブリに250μLのATMを追加します。 10分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルー·アセンブリに450μlのXC3を追加します。 1分間インキュベートする。 1Xを繰り返します。
- 5分間のフロースルー·アセンブリをインキュベートする。
- フロースルーに250μlのSTMを追加ガラスリザーバーに液体を分配することによって、アセンブリ。 10分間インキュベートします。
- ガラスリザーバーに液体を分配することによって、フロースルーアセンブリとADD450μlのXC3。 1分間インキュベートする。 1Xを繰り返します。
- 5分間のフロースルー·アセンブリをインキュベートする。
- 繰り返して8.14から8.19 1Xを繰り返します。
- チャンバーラックからフロースルー·アセンブリを取り外し、水浴をオフにしてください。
9。洗濯およびシーリング
- PB1(約315ミリリットル)で染色ビーズチップ洗浄の皿を埋める。二つの垂直洗浄皿と一つの垂直ビードチップトレイは、少なくとも1つの真空マニホールドと共に、必要とされるであろう。
- 留め金と括弧の間に薄い金属棒を挿入して旋回させることによって、フロースルーアセンブリを分解します。ガラススライドを脇に置き、ビードチップを取り外します。すぐに垂直ビーズチップトレイにビーズチップを配置し、PB1に沈める。 EACに直面するように注意しながら、すべてのフロースルーアセンブリに対して繰り返しHビーズ同じ方向にチップと空気への曝露を最小限に抑えることができます。
- 穏やかに気泡を除去するためにビーズチップトレイを攪拌する。 5分間のPB1に沈めビーズチップをインキュベートする。 XC4と第二の垂直洗浄の皿を埋める前日用意しました。再びビーズチップトレイを攪拌。
- XC4で満たされた洗浄皿にビーズチップトレイを移動します。優しく泡を除去するために、トレイを攪拌。 5分間インキュベートし、再び攪拌する。
- そのため、すべてのビーズチップ面にビーズが上向き、1滑らかな動きでは、洗浄皿からビーズチップトレイを引き出し、試験管ラックに設定してください。セルフロックピンセットで、トレイからビーズチップをスライドさせて、試験管ラックに配置します。すべてのビーズチップについて、この手順を繰り返します。
- 慎重に真空デシケーターにチップの試験管立てを移す。閉じて、適切なシールを確実にする、真空をオンにします。 50〜55分間インキュベートします。必要に応じて、インキュベーション中にスキャナをウォームアップ。
10。スキャニング
- TU真空からRnとゆっくり大気圧まで圧力を戻す。ビーズチップが乾燥していることを確認してください。必要に応じて、任意の液体や破片を除去するためにペーパータオルでエッジとチップの下部を拭く。
- スキャンソフトウェアをアクティブにして、スキャントレイにビーズチップを移動させる。デコードファイルのクライアントソフトウェアを起動すると、それに対応する箱のIDと一緒に、必要なビーズチップバーコードを入力することにより、チップのデコードファイル(dmaps)をダウンロードします。スキャンされていないビーズチップが安全に最大72時間、乾燥した、暗い領域に格納することができる。
- チップが正しくトレイに装着されているとデコードファイルはソフトウェアによって認識されると、スキャンを開始します。
結果
スキャンしながら、適切に処理されたビーズチップは明るく明瞭な赤と緑のレーザー光強度を表示する必要があります。 図6では、のiScanスキャンソフトウェアが正常カスタムパネルのSNPジェノタイピングアレイにハイブリダイズ標準ゲノムDNAサンプルが表示されます。延長および染色工程の間、付着させた標識ヌクレオチドは、2つのレーザの光の下に蛍光を発する。これらのヌクレオチドは、選択的に断片化したDNA鎖にハイブリダイズしたビーズのオリゴヌクレオチド鎖を延長し、オリゴヌクレオチド鎖が変異の部位で終端するように設計されているように、得られた信号の色及び強度は、存在する対立遺伝子を決定するために使用することができるようにSNP部位。
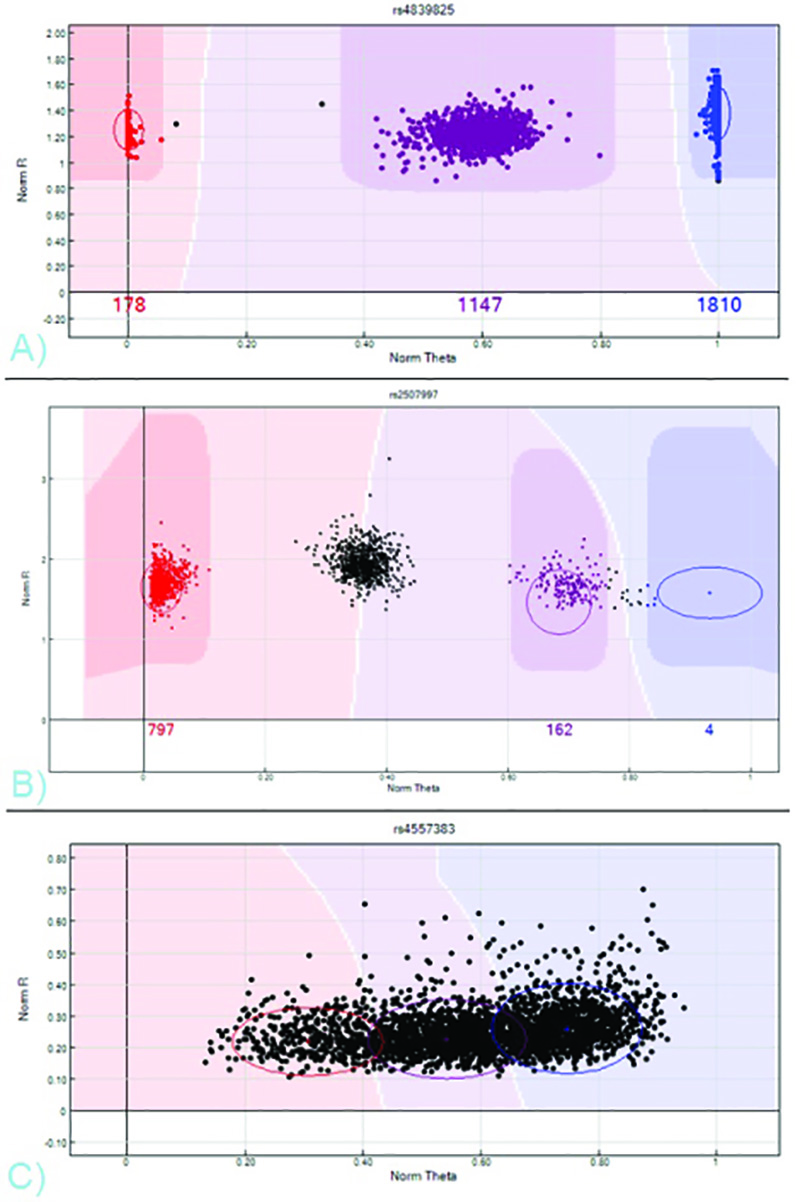
会社から直接得CHIPIDと位置に、サンプルIDと臨床情報と一致するユーザが生成したサンプルシート、およびアレイ固有のSNPマニフェストは、SCAをインポートするために、両方の必要なGenomeStudio解析ソフトウェアへのnner出力ファイル。例のサンプルシートは、同社のウェブサイトに掲載されています。 GenomeStudioプロジェクトが構築されると、遺伝子型はソフトウェアによって自動的に生成された結果の強度クラスタから取得することができます。複数の表示オプションが存在しますが、デフォルトのビュー、NORM-シータ(対立遺伝子の強度比)対ノーム-R(正規化された強度値)は、多くの場合、三つの異なるクラスタを区別するための最も簡単なプロットである。大部分のSNPは、マイナーアレル頻度に応じて、1つ、2つ、または3つのクラスターを示すべきである。
3クラスタは( 図7を参照)、AA、AB、またはBBの対立遺伝子を実証するサンプルを表す。これらのクラスタは、ソフトウェアの分析ツールバーから「すべてのSNPをクラスタ化」を選択することにより、会社から直接、標準的なクラスタリング位置テンプレートをインポートすることによって、またはクリックして手動でEDI強度プロット上で色付きの円をドラッグしてどちらかの遺伝子型を割り当てることができます T·コール。 (赤、紫、青)強度プロット上のサンプルの色はコール(AA、ABまたはBBのそれぞれ)を示し、黒はコールがないことを示します。ソフトウェアのフルデータペインにリストされているのSNPをスクロールすることにより、各SNPのためのクラスタリングは、表示または呼び出すことができます。クラスタは、満足の呼び出しを割り当てられると、ソフトウェアのフルデータペインには、各々の特定のSNPにおける各々のサンプルのための遺伝子型を示しています。テーブルの上の列の選択オプションは、データ形式を切り替えることができます。
データは、詳細な分析のための分析ツールバーを介して、または直接フルデータテーブルのいずれかからエクスポートできます。

図1。概要-インフィニウムアッセイプロトコル。683/50683fig1highres.jpg "ターゲット=" _blank ">拡大画像を表示するにはここをクリックしてください。

図2。完全Hybをチャンバーベースとマットに加え、蓋[12] 拡大画像を表示するには、ここをクリックしてください 。
{kind=link}

図3ビーズチップのロード-入口ポート上のサンプルを分注[12] で拡大表示するには、ここをクリックしてください画像。
{kind=link}

図4ビーズチップCoversealの削除 - 。コーナーでシールをつかみ、ゆっくりと斜めに剥離[12] 拡大画像を表示するには、ここをクリックしてください 。
{kind=link}

図5。完全なビーズチップフロースルーアセンブリ -ビーズチップは、スペーサを用いてガラススライドから分離し、留め金でバインドされている。>拡大画像を表示するにはここをクリックしてください。

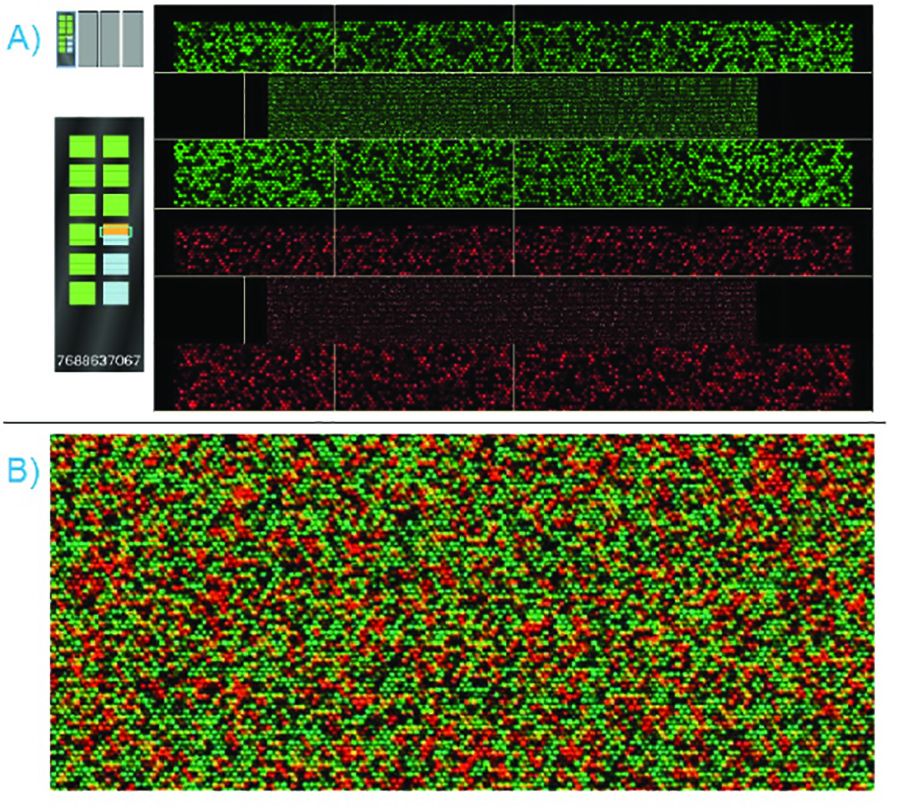
図6。成功したビーズチップスキャン - A)ビーズチップの両方赤と緑のレーザーでスキャンされ、スキャンソフトウェアの両方を同時に表示します。強度QCを渡すのセクションでは、左にビーズチップのディスプレイに緑の強調表示されます。強度QCを失敗のセクションでは、ビーズチップのディスプレイに赤くハイライト表示されます。B)のスキャンが完了すると、ソフトウェアは、赤と緑のディスプレイをオーバーレイします。ズームインした画像が表示されます。個々のビーズの色と強度はアレル存在を示している。 拡大画像を表示するには、ここをクリックしてください 。
{kind=link}

図7。 NORM-θクラスタのプロファイル対SNP NORM-R - AA AB、およびBB遺伝子型、赤、紫、青、B色)のSNPが必要な編集を表す3つの異なるクラスタとA)有効なSNP。ホモ接合ABであるべきミドルクラスタは、国連と呼ば残っている。 BBクラスタが誤ってA-B、C)悪いパフォーマンスのSNPと呼ばれています。明確なクラスターが存在しないように何の遺伝子型は、この強度プロットから得ることができない。 拡大画像を表示するには、ここをクリックしてください 。
{kind=link}
ディスカッション
大規模な遺伝子型決定アプリケーションは、より良好な多くのヒト疾患の根底にある遺伝的機構を理解するために使用されてきた。ゲノムワイド関連解析を介して任意の大幅な改変体の発見は、さらなる研究のための候補領域にフラグをすることができます。また、遺伝子型データは、配列決定プロジェクトでの品質管理のための優れたツールです。
サンプルスループットを最大化するために、複数のサンプルプレートを増幅し、その断片化され、再懸濁させた状態で保存することができる。 8枚のプレートには、複数のバッチのためのプロトコルの最初の24時間を組み合わせるとチップの処理〜2-8日間、十分な材料を提供し、一日で増幅することができる。増幅されたプレートをあらかじめ備蓄している場合は、新たなサンプルがスキャンは前回の実行から始まり直後にチップにハイブリダイズされる場合には、処理は追加のサンプル調製のために一時停止することなく、継続的に実行することができます。そのため、サンプルが完全に受けることが3日かかるでしょうがアッセイは、データは、毎日生成することができる。 Assuming24チップは毎日処理され、5日制は、12サンプルのビーズチップ上で実行される1,000以上のDNAサンプルを可能にします。いずれかのステップまたは試薬が失敗した場合は任意の補正を適用する前に、しかし、複数のバッチは、パフォーマンスの低下のリスクがあるかもしれません。配列がスキャンまたは分析されるまで、エラーが通知を免れるかもしれません。スループットが最大化されている場合、したがって、プロトコルの様々な段階におけるサンプルの数百人は、すでに発見に同じ欠陥の治療を受けている可能性があります。失われた試薬およびデータを復元することはできませんように、ユーザは、加速されたワークフローの必要性と、これらのリスクを比較検討する必要があります。
GenomeStudio解析ソフトウェアは、本当に遺伝子型判定プロセスの成功を測定する最初のチャンスである。 NORM-シータ強度プロット対NORM-Rが適切にクラスタ化されている場合、この値はSLI異なるが、サンプルの平均コールレートは、(全体のSNPの割合が正常に入力された)、99%に近づく必要がありますghtlyアレイタイプに応じ。 85から90パーセントよりも低いコールレートとの任意のサンプルからのデータが信頼できるものではなく、廃棄されるべきである。品質管理の目的のために、結果は、以前に知られている遺伝子型、可能な限りと比較すべきである。そのようなデータが存在しない場合は、意図的なサンプルの複製は、プレートまたはアレイの配置を確認するのに有用なツールである。これらの重複したペアは別個のチップ、プレート、バッチ、またはプロジェクトに配置する必要があり、その遺伝子型は生成時にチェックした。具体的な品質管理の制約が調査者の好みに応じて異なるが一般的なサンプル制約はコールレート、メンデルの矛盾、あるいは相互参照に基づいていますが、一般的なSNP制約は、症例と対照の間でサンプル呼出成功、ハーディ·ワインベルグ平衡、または欠測に基づいています臨床性別データへのX染色体のヘテロ接合性の[13]。
何らかの問題が発生した場合、分析スイートで見つかったコントロールダッシュボードは、、に提出することができます原因を特定するために、会社。これらのコントロールは、多くの場合、最有力ステップまたは試薬障害のために問題を絞り込むことができます。興味のある任意のSNPはインフィニウムジェノタイピング実験により発見された場合、さらなる研究が行われる前に、その強度プロットは、クラスタリングエラーをGenomeStudioをダブルチェックする必要があります。
失敗したのInfiniumジェノタイピング実験は、人間の処理エラーや低品質の投入DNAに起因する可能性が高い。サンプル定量化は、正確かつ精密である必要があります。最良の結果を得るには、任意のサンプルまたはチップに追加された試薬は、プロトコルが設定した音量で分配されなければなりません。ピペットは、適切に校正する必要があります。試薬が満了した後に実行すべきではないとRA1試薬保存、解凍した一回再凍結すべきではありません。可能な染色および伸長誤差を最小にするために、ホルムアミド/ EDTA混合物を毎月新たに調製すべきである。すべて-20℃の試薬は、手動霜取り冷凍庫に保管してください。 STで使用されるすべての実験器具aining、プロトコルの拡張、および洗浄の部分はすぐに廃止すると水と中性洗剤で十分に洗い流してください。 HYB室内の加湿貯水池を試験管ブラシと中性洗剤でこすりする必要があります。週に一度、自分のユーザーマニュアルの指示に従って、ガラススライドは、10%の漂白剤で洗浄する必要があります。
開示事項
この記事の著者は、開示することなく、競合する金融利害関係はありません。
謝辞
この仕事のための資金は、NIH P20 GM103456、NIH RC2 AR058959、およびNIH R56 AI063274によって提供されている
資料
| Name | Company | Catalog Number | Comments |
| Consumable or Equipment | Manufacturer | Part Number | Minimum Required for 96 Samples |
| 0.8 ml Deep Well Plate | Thermo Scientific | AB-0765 | 1 |
| Plate Mats | Thermo Scientific | AB-0674 | 2 |
| Reagent Basin | Fisher Scientific | 13-681-502 | 9 |
| Heat-seal Sheets | Thermo Scientific | AB-0559 | 1 |
| Flow-through Spacer | Fisher Scientific | NC9563984 | 6 |
| Pipette tips - 200 μl | Rainin | GP-L200F | 192 |
| Pipette tips - 10 μl | Rainin | GP-L10F | 16 |
| Pipette tips - 1,000 μl | Rainin | GP-L1000F | 16 |
| DNA Suspension Buffer | Teknova | T0220 | 0.5 ml |
| 0.1 N NaOH | Fisher Scientific | AC12419-0010 | 0.5 ml |
| Isopropanol (HPLC grade) | Fisher Scientific | A451 | 15 ml |
| Ethanol (200-proof) | Sigma-Aldrich | 459836 | 330 ml |
| Formamide (100%) | Thomas Scientific | C001K38 | 15 ml |
| EDTA (0.5 M) | Amresco | E177 | 0.2 ml |
| 10 μl 8-channel Pipette | Rainin | L8-10XLS | 1 |
| 200 μl 8-channel Pipette | Rainin | L8-200XLS | 2 |
| 1,000 μl Single-channel Pipette | Rainin | L-1000XLS | 1 |
| Microplate Shaker | VWR | 13500-890 | 1 |
| Refrigerated Microplate Centrifuge | VWR | BK369434 | 1 |
| Hybridization Oven | Illumina | SE-901-1001 | 1 |
| Hybex Microsample Incubator | SciGene | 1057-30-0 | 1 |
| Hybex MIDI Heat Block Insert | Illumina | BD-60-601 | 1 |
| Heat Sealer | Thermo Scientific | AB-0384 | 1 |
| Hyb Chamber w/ Insert and Mat | Illumina | BD-60-402 | 2 |
| Surgical Scissors | Fisher Scientific | 13-804-20 | 1 |
| Flow Through Assembly Parts | Illumina | WG-10-202 | 8 |
| Wash Rack and Dish | Illumina | BD-60-450 | 1 |
| Genepaint Chamber Rack | Tecan | 760-800 | 1 |
| Temperature Probe | Illumina | A1-99-109 | 1 |
| Staining Rack and Dish | Illumina | WG-10-207 | 1 |
| Self-Closing Tweezers | Ted Pella, Inc | 5374-NM | 1 |
| Vacuum Manifold | Ted Pella, Inc | 2240 | 1 |
| iScan or HiScan | Illumina | - | 1 |
参考文献
- Shi, M. M. Enabling large-scale pharmacogenetic studies by high-throughput mutation detection and genotyping technologies. Clin. Chem. 47 (2), 164-172 (2001).
- Livak, K. J. SNP genotyping by the 5'-nuclease reaction. Methods Mol. Biol. 212, 129-148 (2003).
- Seeb, J. E., Pascal, C. E., Ramakrishnan, R., Seeb, L. W. SNP genotyping by the 5'-nuclease reaction: advances in high throughput genotyping with non-model organisms. Methods in Mol. Biol. 578, 277-292 (2009).
- Bayés, M., Gut, I. G. Overview of Genotyping. Molecular Analysis and Genome Discovery. ed, , 2nd, John Wiley & Sons, Ltd. Chichester, UK. 1-23 (2011).
- Lee, Y., Seifert, S. N., Fornadel, C. M., Norris, D. E., Lanzaro, G. C. Single-Nucleotide Polymorphisms for High-Throughput Genotyping of Anopheles arabiensis in East and Southern Africa. J. Med. Entomol. 49 (2), 307-315 (2012).
- Liu, Z. SNP Genotyping Platforms. Next generation sequencing and whole genome selection in aquaculture. , Wiley-Blackwell. 123-132 (2011).
- Shen, R., Fan, J. B., et al. High-throughput SNP genotyping on universal bead arrays. Mutat. Res./Fundam and Mol. Mech. of Mutagenesis. 573 (1), 70-82 (2005).
- Ragoussis, J. Genotyping technologies for all. Drug Discov. Today: Technol. 3 (2), 115-122 (2006).
- Wu, C. C., Shete, S., Jo, E. J., et al. Whole-Genome Detection of Disease-Associated Deletions or Excess Homozygosity in a Case-Control Study of Rheumatoid Arthritis. Hum. Mol. Genet. , (2012).
- Green, E. K., Hamshere, M., Forty, L., et al. Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Mol. Psychiatry. , (2012).
- Shete, S., Hosking, F. J., Robertson, L. B., et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 41 (8), 899-904 (2009).
- Inc Illumina. Infinium HD Ultra User Guide 11328087 RevB-1. , 15-101 (2009).
- Turner, S., Armstrong, L. orenL., Yuki Bradford,, et al. Quality Control Procedures for Genome Wide Association Studies. Curr Protoc Hum Genet. , 1-19 (2011).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved