
Method Article
マウスを一回、二つの文化:個々のマウスの二つの神経原性のゾーンから成体神経幹細胞の単離および培養
Erratum Notice
要約
ここでは、脳室下帯と個々の成体マウスの歯状回から、付着性単層またはニューロスフェアのいずれかとして、神経前駆細胞培養を同時に生成するための詳細なプロトコルを記述します。
要約
ニューロスフェアアッセイおよび接着単層培養系は、in vitroで 、成体神経幹細胞の電位(増殖または分化)を決定するための貴重なツールである。これらのアッセイは、神経前駆細胞の増殖および分化に対する外因性因子の効果を決定するために、連続継代にわたってアッセイすることができる神経前駆細胞株を生成するために、遺伝的に異なる又は示差的処置動物から単離された細胞の前駆体の電位を比較するために使用することができる。ニューロスフェアアッセイは伝統的に、主として、それらが一次組織から単離し、脳組織内の前駆細胞数の迅速な推定値を与えるという大きな利点を有することが可能で胚葉マーカーの欠如のために、幹細胞の事後識別のために使用される個々の動物に由来する。接着単層培養物は、対照的に、伝統的に個々の動物間の増殖を比較するために使用されていない、それぞれの文化として、一般的に5〜8の間の動物の複合組織から開始されます。しかしながら、それらは、ニューロスフェアとは異なり、それらは前駆細胞の大部分が均一な集団からなり、単一細胞における分化プロセスを以下のために有用で大きな利点を有する。ここでは、初めて、個々の動物からの接着培養物を、詳細には、ニューロスフェア培養物の生成を説明し。これは脳室下帯(SVZ)および治療または遺伝的に異なるマウス系統の歯状回(DG)の両方での増殖および/または分化能力のペアの分析だけでなく、動物の使用量の大幅な削減など、多くの重要な意味を持っています。
概要
ニューロスフェアアッセイ1,2、付着単層培養3,4、どちらも1990年代初頭に開発されたが、まだ体外神経幹細胞アッセイにおけるゴールドスタンダードのまま。これらのアッセイでは、一次組織は、特定の脳領域から解剖マイクロあり、自由浮動のいずれかを形成するマイトジェンの上皮成長因子(EGF)及び線維芽細胞増殖因子-2(FGF2)の存在下での単一細胞懸濁液培養に解離クラスタ(ニューロスフェア)または付着性単層。どちらのシステムも、長所と短所と慎重に検討の数が1または他のシステムが選択される前に対処すべきである疑問に与えられるべきであります。
神経球前駆細胞数および電位の差の直接的な読み出しを可能にする。加えて、ニューロスフェアはまた彼らの正常な外部環境から取り外したときの細胞の本質的な仕様を研究するための有用なツールである。 Extrin原文の手がかりは、単に成長培地に、目的の因子を添加し、生成された神経球の数と大きさを定量化することによって研究することができる。ニューロスフェアの主な欠点は、しかし、それらが表面5上のものより分化した神経球である(特に、大規模なニューロスフェア)の中心の細胞と、自分自身のニッチを形成することである。ニューロスフェアは、幹細胞、前駆細胞、コミット、および分化した細胞の混合物を含有し、神経球内の細胞 - 細胞相互作用は、幹細胞の維持を打ち消す。ニューロスフェアは、真の幹細胞6-8のわずか数を含む理由である。
接着単層培養も生体内増殖をモデル化するための優れたin vitroの系を提供する。細胞は、より孤立し、均質たままで接着培養は、ニューロスフェアの異質性を排除することができます。これらの成長条件の下で前駆細胞を増殖さRAPIDLYとほとんど全ての細胞は、分裂し、特徴的な神経前駆マーカーネスチン、Sox2及びBLBPを表現している。ニューロスフェアアッセイと比較して、単層培養系の主な欠点は、個々の前駆体由来のクローンをモニターし、定量化することができないことである。
分離戦略の収率は、しばしば貧弱であったため、培養物の両方のタイプのほとんどのプロトコルの欠点は、動物の比較的大きな数字を使用する必要性であった。同時に、それは成体神経新生は、同様に個別化されたex vivoでのモデルの必要性をもたらす、脳9の個別化に寄与することが明らかとなった。この報告書に記載されるように、これらのニーズは、「ワンマウス - オン培養」プロトコルによって満たすことができる。
次の視覚的なプロトコルは、SVZとDGの個々の動物のいずれかとして、付着メートルの両方からの神経前駆体培養物の同時生成を説明しますonolayersまたはニューロスフェアとして。個々の動物からの培養物の生成は、個別に処理された動物または種々の個々のトランスジェニックまたは野生型マウスの間の比較が必要な場合に特に有用である。このプロトコルは、詳細な成体マウスからのSVZとDGの地域を同時にマイクロダイセクションのための命令、単一細胞懸濁液へのそれらの解離、接着単層培養またはニューロスフェアと多能および長期的な可能性の分析のいずれかとして、インビトロ培養、2枢機卿が含まれてい骨善意の幹細胞の特性。
プロトコル
1。基本的なセットアップと培養液の調製
- 実験を開始する前に、少なくとも2日間、接着単層培養のためのポリ-D-リジン(PDL)/ラミニンでコーティングしたプレートを準備します。調製するウェル/フラスコを被覆する表面(のdH 2 O中の10 mg / ml)を十分なPDLを追加し、室温で一晩インキュベートする。皿から溶液を除去およびDH 2 Oでお皿を3回洗浄空気乾燥させます。ラミニン(冷DMEM中の5 mg / mlの:F12)を追加し、37℃で一晩インキュベートする。ラミニンを削除し、直ちにプレートを使用することも、必要になるまで-20℃でラミニンで保管してください。
- エッジが丸くなるまで炎でガラスパスツールピペットを回転させることで「中」「小」の穴で火ポリッシュピペットを準備します。滅菌するオートクレーブ。
- 解剖の日に、2%B27、1×グルタ、2 mg / mlのヘパリンで神経基本培地を混合することにより、培地の適切な量を調製し、50単位/ mlペニシリン/ストレプトマイシン、20 ng / mlの精製マウスレセプターグレードの上皮成長因子(EGF)、および20 ng / mlの組換えウシ線維芽細胞増殖因子(FGF-2)。水浴中で37℃に培養液を温める。
- SVZ解離のために、0.05%トリプシン-EDTAを調製し、0.125 mg / mlのトリプシン、0.01 mg / mlのDNアーゼ阻害剤を含有する。 37℃にこれらのソリューションを平衡化
- 解剖顕微鏡を設定し、脳(はさみと小さなヘラ)を除去するために必要なツールを準備し、SVZとDG解剖(小刀、1ミリリットルのシリンジに取り付け、27G針、1×#7鉗子、1 X#5/45 70%エタノールに浸すことによって鉗子)。
2。大人のマウスの脳およびSVZ / DG Microdissectionsの収穫
- 適切な施設のガイドラインに従って、単一の成人(8週齢)マウスを麻酔。頸椎脱臼を実行します。
- 領域を滅菌するために毛皮の量を最小限に抑えるために70%エタノールでスプレーヘッドことはさみや脳にdheres。鋭いハサミを使用すると、脳幹の基部に動物を首を切る。
- 頭蓋底のヘッドを保持して、それぞれの目の空洞の中にはさみの小さなペアの1のブレードを配置し、冠状に切断することにより、2嗅球の間頭蓋骨を切った。次に、矢状縫合に沿って頭蓋骨を通して縦カットに続いて頭蓋底、2つの横方向の切断を行う注意 :ハサミの角度が、基礎となる脳の損傷を避けるために、可能な限り浅いことを確認してください。
- ハサミの刃や湾曲したピンセットのいずれかでスカルをバック剥離することにより脳を公開します。冷PBSに小さなヘラと場所を使って頭蓋骨から脳を解放します。
- 血液や毛皮を除去するためにPBSで脳を洗浄します。
- PBSを含有する10cmのプラスチック製ペトリ皿に脳を転送する
- 低倍率での解剖顕微鏡下で脳を含むペトリ皿を置き、BRAIを位置決めその腹側表面上にn。小脳によって所定の位置に頭を押さえながら細かい曲線状の鉗子を使用すると、嗅球を除去します。
- 背側面上に脳を回転させて、メスを使用すると、視交叉のレベルで脳の冠状切断を行う
- SVZを顕微解剖する(より詳細な手順については、アザリら 10も参照のこと)、カット冠状面を上に向くように、脳の吻側部分を配置し、高倍率に顕微鏡の焦点を合わせる。削除して、細かい湾曲したピンセットを用いてセプタムを捨てる。
- すぐに組織の中に、すぐに脳梁とその他が約1ミリメートルの下側脳室の左右の角にある細い曲線状ピンセットの1刃の先端を配置することによって、SVZ(心室の周囲の組織の薄い層)を解剖心室に隣接。お皿の底に向けてとventrの腹側面に向けた鉗子を押し下げるICLEは、組織の小さな三角形の部分を削除します。氷上のペトリ皿に解剖し、SVZを配置します。
- DGを顕微解剖する(より詳細な手順については、萩原ら 11も参照)、ペトリ皿とメスを用いて縦方向の亀裂に沿って切断し、脳の尾の部分を置く。
- 解剖顕微鏡下では、小脳や鉗子を使用して間脳を削除します。
- DGの周りの境界線が表示されるようになりましたように、顕微鏡に再び焦点を合わせる。歯状回を削除するには、DGとアンモン角の間の境界に沿って27のG針とスライドの先端を挿入します。細かい鉗子を使用して、周囲の組織からのDGを解放。
3。 SVZ組織解離
- 大きな片がなくなるまで約1分間、手術用メスの刃を用いて組織をミンチ。
- 温めておいた0.05%トリプシン-EDTAの1ミリリットルを使用して15mlチューブにみじん切りの組織を移し、7分間インキュベート37℃に設定した水浴
- 酵素反応を停止するには、DNアーゼIを含むトリプシンインヒビターの1ミリリットルを加え、チューブをフリックで内容物を混合。
- 5分間300×gで遠心分離することにより、サスペンションをペレットと上清を捨てる
- 増殖培地の1ミリリットル中にペレットを再懸濁し、P1000ピペットを用いて静かにピペッティングを約7〜10倍で解離注意:粉砕する以上は、細胞死の増加につながることができ負その後の細胞の成長に影響を与えます。
- 5ミリリットルの全体積に増殖培地を追加し、デブリと非解離組織塊を除去するための40mmの篩を通して細胞懸濁液を渡す。
- 5分間300×gで遠心し、上清を廃棄し、200ミリリットルの増殖培地中で得られたペレットを懸濁します。
4。 DGの組織解離
- 大きな片が残っていないと転送されるまで約1分間、手術用メスの刃を用いて組織をミンチ予め温めPDD酵素ミックス(パパイン2.5単位/ ml、ディスパーゼ1単位/ ml、DNアーゼI 250単位/ ml)に変換する。管ごとに3〜5分を反転することにより混合し、37℃で20分間インキュベートする。
- 機械的にそっと10倍をピペッティングによりミディアムボア、火災洗練された、パスツールピペットを用いて組織を解離する。
- 管ごとに3〜5分を反転することにより混合し、37℃でさらに10分間インキュベートする。
- さらに、機械的に小さな穴を使用して、組織を解離、火はそっと10倍をピペッティングによりパスツールピペットを磨いた。
- 5分間130×gで遠心分離する。
- 上清を除去し、1ミリリットルの緩衝液(1×HBSS、30 mMのグルコース、2のHEPES(pH7.4)を、26 mMの炭酸水素ナトリウム)でペレットを再懸濁します。緩衝液で10mlに構成しています。
- 5分間130×gで遠心分離する。
- 上清を除去し、20%パーコール5mlにペレットを再懸濁します。 (その後さらに20パーセントにこれを希釈して10倍のPBS 0.5mlに100パーセントパーコールの4.5ミリリットルを加え、90%パーコールを製造するために、3.9ミリリットル1X PBS)に1.1ミリリットル90%パーコールを追加することによって。
- 15分間遠心450 XG。
- 上清を除去し、10ミリリットルのバッファーでペレットを再懸濁します。
- 5分間130×gで遠心分離する。
- 200μlの増殖培地中でペレットを再懸濁。
5。接着単層培養の生成
- 96ウェルプレートのウェルコーティングされた単一のPDL /ラミニンに解離SVZまたはDG組織をプレートし、5%CO 2、37℃でインキュベートする。
- めっき後の約24時間後、細胞が被覆された表面に付着した後、さらに過剰な破片を除去する成長培地を交換する。
- 続く3〜4日ごとに、成長因子を補充する新鮮な培地で増殖培地の交換半。
- メッキと第一通路との間の時間は2〜3週間ほどかかる場合があります。細胞が約80%の集密度に達し、継代する準備が整うまで、 注意してください繰り返します。
- 培養物が約80%の集密度に達したときに井戸から培地を除去し、PBSで洗浄する。
注:これは、細胞の剥離およびニューロスフェアの形成につながり、さらに、細胞死のレベルを増加することができますように、細胞が90%の密集度を超えないようにしてください。 - (細胞は丸みを帯びたと切り離されているかどうかをチェックする)50ミリリットルをAccutaseを追加し、2〜3分間37℃でインキュベートする。
- 15mlチューブに細胞を除去し、ウェルのPBSで一度洗い、同じチューブに移す。
- PBSで5ミリリットルと5分間の遠心分離器300 XGに細胞を希釈する。
- 最初の通過のために、24ウェルプレートのウェルコーティングしたPDL /ラミニンに1ミリリットルとプレートに細胞を希釈する。
- その後の継代のために、200ミリリットルの増殖培地と数の再懸濁細胞は血球計数器を用いて。適切なサイズのコートされたウェルまたはフラスコ中の1×10 4細胞/ cm 2でプレート。 I>
7。接着単層培養の分化
- 20 ng / mlのEGFおよび10 ng / mlのbFGFを含有増殖培地中1×10 4細胞/ cm 2の密度で、PDL /ラミニンコートしたカバーガラス上に接着単層培養物を、プレートに増殖している細胞を分化する。
- 細胞が約80%の密集度(通常は2日間)に達したときに、5 ng / mlのbFGFおよび0 ng / mLのEGFを含む培地で増殖培地を交換してください。
- 5 ng / mlのbFGFを2日後に、さらに3日間両方のマイトジェンの非存在下で増殖培地で培地を交換注 :この期間に細胞死の有意な量が発生する。
- 5日間の合計した後、室温で20分間、4%パラホルムアルデヒド(PFA)で固定し、次いで任意死細胞を除去するためにPBSで分化した細胞を洗浄する。
- 4℃で1ミリリットルPBSにウェル内の任意のPFAや店舗カバーグラスを除去してPBSで再び洗浄します
- 培養培地20ml中に一匹の動物から解離SVZまたはDG組織を希釈し、10ml multidoserピペットを用いて、96ウェルプレートにおける200ミリリットル/ウエルプレート。
- DG由来のニューロスフェア用SVZ由来のニューロスフェア10〜12日間、6〜7日間で5%CO 2、37℃でインキュベートする。
注:これらの推奨インキュベーション時間よりも長い間成長が異常増殖をもたらすであろうし、ニューロスフェアおよび/ または、自発的な愛着と分化の中心に細胞死につながる。 - 数え、直立光顕微鏡に装着した接眼目盛を使用してニューロスフェアの直径を測定
9。ニューロスフェアを継代
一次ニューロスフェアをカウントされ、その大きさが記録された後、それらを単一の神経球またはバルク培養物のいずれかで始まるいくつかの継代にわたって拡張することができる。
- バルク培養ニューロスフェアの展開
- バルク文化としての通路を組み合わせニューロスフェアを、5分間300×gで15ミリリットルチューブと遠心分離機に移し、プレートからニューロスフェアを含む培地を除去します。
- 上澄み液を捨て、あらかじめ温めておいた0.05%トリプシン-EDTA 1mlにニューロスフェアを再懸濁し、3分間室温でインキュベートする。
- DNアーゼIを含むトリプシンインヒビターを等量加え、よく混ぜる。
- 300×gで5分間遠心、上清を除去し、増殖培地の1ミリリットルを加える。
- ニューロスフェアを解離P1000ピペットで上下に約10倍粉砕する。
- 細胞懸濁液10ミリリットルを削除し、トリパンブルー等容量の混在血球計数器を用いて生菌数を実行します。
- 適切なサイズの細胞培養ウェルまたはフラスコ中の1×10 4細胞/ cm 2の密度で細胞を再シードする。
- 37℃でインキュベート5%のCO 2を有する二次ニューロスフェアが形成されるまで。
- 単一のニューロスフェアの展開
- 通路に個々の神経球は、単一のニューロスフェアを含むウェルを選択し、慎重にニューロスフェアを乱すことなく、各ウェルから増殖培地を約160μLを取り外し、廃棄します。
- 各ウェルに継代するために、0.05%トリプシン-EDTAの100ミリリットルを加え、3分間室温でインキュベートする。
- 反応を停止させるのDNAseIを含む阻害剤トリプシンを100μlを加える。
- 上下にP200ピペットを用いて粉末化し、約10倍のニューロスフェアをバラバラにする。
- 増殖培地1.5 mlを含む24ウェルプレートの新しいウェルに解離したニューロスフェアを含有する200ミリリットルを転送する。二次ニューロスフェアが形成されるまで、5%CO 2、37℃でインキュベートする。
注:長期的な可能性、真の神経の基本的特性のいずれかを決定するTEMセル、ニューロスフェアは、少なくとも5〜10継代継代する必要があります。また、このような結果が12月14日の中の一部で物議を解釈に関する文献を参照してください。
10。ニューロスフェア培養の分化
プライマリまたは継代神経球を決定するために多能性分化させることができる。
- それらの培養プレートまたはフラスコから懸濁液中ニューロスフェアを削除し、10cmのプラスチックシャーレに転送。
- 解剖顕微鏡下でP20ピペットを用いて培地から約15から20のニューロスフェアを取り除き、成長因子およびPDL /ラミニンコーティングしたカバースリップすることなく、培地を含む24ウェルプレートに移す。
- 5%CO 2、37℃で約7日間分化。
- 室温で20分間、4%PFAで固定し、任意の死んだ細胞を除去するためにPBSで差別ニューロスフェアを洗ってください。
- RにPBSで再び洗浄します4℃で1ミリリットルPBS中ウェル内の任意のPFAや店舗カバースリップを残したまま削除
11。ニューロスフェアと接着培養の免疫染色
注意 :O4抗体で染色するためのブロッキングと染色の手順からトリトンを省略して、適切なIgM抗体二次抗体を使用してください。
- 室温で60分間(0.2%トリトンX-100を含有するPBS中10%正常ロバ血清)をブロッキング溶液中で分化した神経球または接着単層培養物を含有するカバースリップをインキュベートする。
- プライマリBIII-チューブリン、Map2a + bは、グリア線維性酸性タンパク質(GFAP)または室温で60分間O4抗体を含有する新鮮なブロッキング溶液中でインキュベートする。
- PBSで3回洗浄します。
- 暗所で室温で30分間、適切な蛍光結合二次抗体および4,6 - ジアミジノ-2 - フェニルインドール(DAPI 1:5,000)を含有する新鮮なブロッキング溶液中でインキュベートする。
- PBSで3回洗浄する。
- 暗い晩に蛍光封入し、空気乾燥を用いて顕微鏡スライド上にカバースリップをマウント
- 蛍光顕微鏡を使用してビューとイメージ。
結果
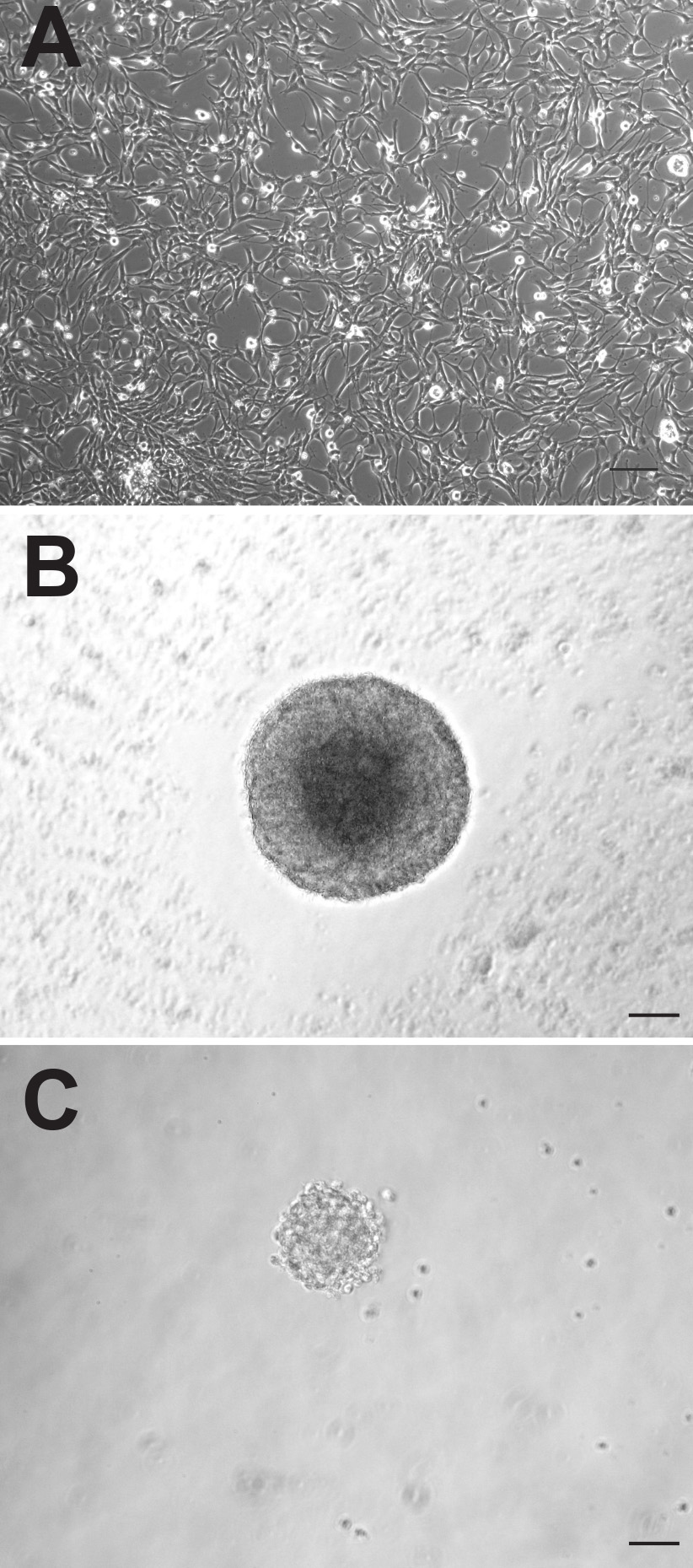
成体マウスの脳の神経原性の二領域の両方が、神経前駆細胞を含むが、これらの細胞は全く異なった場合に、インビトロで培養振る舞うことができる。両地域から生成された付着性単層培養は、しかし、SVZ由来の接着培養をより速く増殖し、1〜2日以前のDGから誘導されたものよりも、平均して、継代する必要があり、( 図1A)、形態学的に区別できないようである。ニューロスフェアのように、SVZ由来前駆細胞はまた、より速く増殖し、DG由来前駆細胞( 図1C)より大きいニューロスフェア( 図1B)を形成する。 SVZ由来のニューロスフェアは、一般的に、培養中の6-7日後にカウントされ、一方、DG由来のニューロスフェアは、通常10〜12日後に定量化される。また、神経前駆細胞の数は、はるかに大きな属であることができるニューロスフェアのほぼ10倍高い数によって証明されるように、DGと比較して、SVZ内に存在するこの領域からテッド(SVZ:DG対1173±74.9:145.3±26.4、P = <0.0001;群あたりn = 10動物; 図2A)。
研究は、SVZおよびDG内の前駆細胞は様々な刺激に応答することが示されている。 SVZ前駆細胞は、嗅覚学習および嗅覚富化によって活性化されるのに対し、DGにおける前駆細胞は、空間学習、特定の種類によって、そのような環境エンリッチメントや身体活動などの刺激により活性化される。これと一致して、我(TLW)のいずれかは、以前DGが神経興奮15-18によって活性化される潜在性幹細胞および前駆細胞の集団を含有することを実証した。対照的に、我々は、SVZ前駆細胞のKCl 17のレベルを脱分極に応答したニューロスフェアの数の減少と、まったく異なるこの刺激に反応することがわかった。ここでは、INDのSVZおよびDGに由来する単離された細胞の半分を、メッキ、この実験を繰り返してきたコントロールのKCl濃度でのKCl、残りの半分のレベルの脱分極中ividual動物。我々は、(DG前駆細胞が脱分極(101.2±17.4対184.8±12.5、p = 0.005、nは 5匹の動物)により活性化されるが、SVZ由来細胞の増殖は、実際には有意に減少していることを、以前に実証368.0±62.9対266.6±41.6、P = 0.02、N = 5動物; 図2B)。
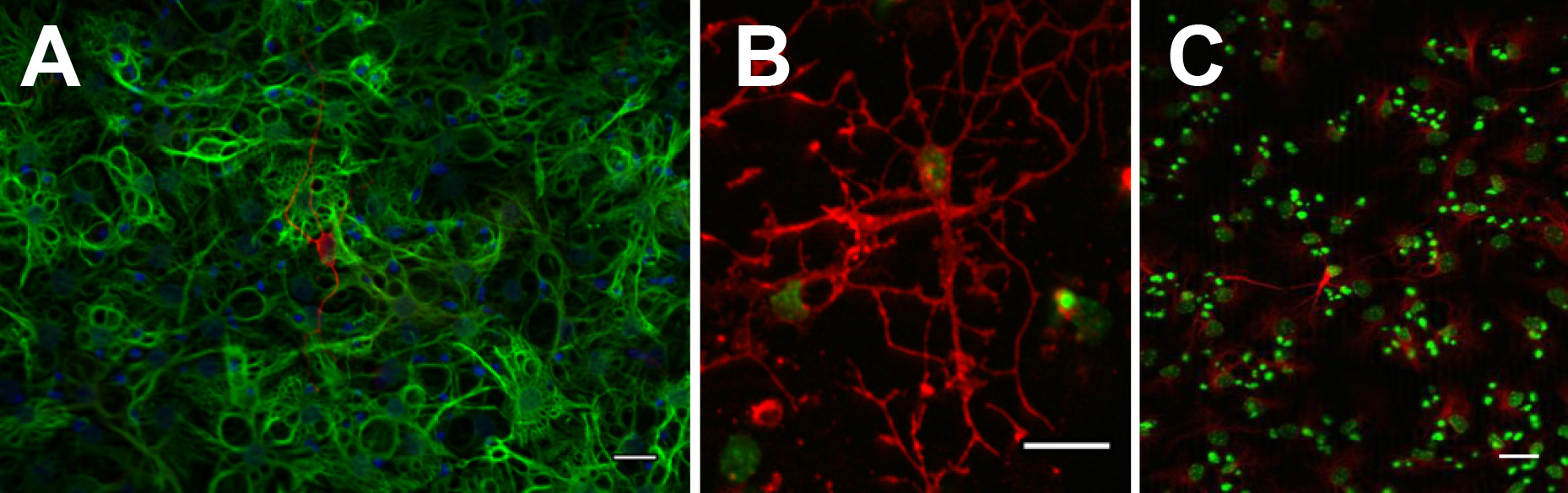
長期的な可能性、真の幹細胞は、単一の神経球または接着単層培養の基本的な機能の1を確認するためには、少なくとも10継代にわたってすなわち 、拡張、拡張することができなければならない。各継代において、単一細胞懸濁液の調製後、細胞数をカウントし、倍の増殖が算出される。理論上のセルの合計は、前の通路からの理論的合計することによって、その通過中折り膨張を乗じて算出される。これはDISPです継代数を有する線グラフとしてレイアウトさ(例えば、 図3を参照)の理論的全細胞数のlog10のに対してプロットした。多能性を確認するために、両方の単層培養および神経球をマイトジェン撤退によって区別することができ、両方の神経細胞を生じさせることを示すことがあり、グリア( 図4)。

図1成体マウスの前駆細胞が接着単層培養物(A)またはニューロスフェアとして培養することができる。(B:SVZ、C:DG)。スケールバーは50ミリメートルです。 拡大画像を表示するにはここをクリックしてください。
{kind=link}
load/51225/51225fig2highres.jpg "SRC =" / files/ftp_upload/51225/51225fig2.jpg "/>
図2より重要なことには、単一の神経球のマウスのDGと比較して、SVZから生成される(A)。 SVZとDGの前駆細胞は、in vitroでの脱分極(B)に対して異なる反応。

図3。長期増強を確認するためには、ニューロスフェアは、10以上の継代のために展開されます。

図4。ニューロスフェアBIII-tubuに分化させることができるLIN +ニューロン(A:赤)、GFAP +アストロサイト(A:緑)、O4 +オリゴデンドロサイト(B:赤)とMap2ab +ニューロン(C:赤)。 拡大画像を表示するにはここをクリックしてください。
{kind=link}
ディスカッション
本論文では、成体マウスの脳の二つの主要な神経性の領域からの神経前駆体培養物、などの付着性単層およびニューロスフェアの両方の開始のための詳細なプロトコルを提示します。これらのin vitro培養系のいずれかをしようとしたときに留意しなければならない重要なポイントがいくつかあります。まず、解離方法の選択は非常に重要であり、組織に依存する。我々の手では、0.05%トリプシン-EDTAは、SVZ組織の解離のために非常に有効であり、パパイン解離系技術を用いた場合よりも、より多数の神経球をもたらす。 DG組織の解離のためにしかし、我々は強く、パパインベースの解離方法をお勧めします。直接DG組織上の2つの解離方法を比較すると、我々は、生存細胞の有意に低い収量を観察し、トリプシンを用いたときより少ない神経球を、約10倍。解離のこの差は、組織compositioの差に起因する可能性が両地域間のn。 DGのコンパクトな組織は、大規模な神経網に囲まれ、細胞プロセスの甚大な被害は、解離の際に発生する可能性があります。
注意すべき第二の重要な点は、ニューロスフェアアッセイは、所与の組織試料中に存在する前駆細胞の数に関する定量的ステートメントを作成するのに有用であり得るが、いくつかの注意が、しかしながら、これらの絶対数の解釈に用いなければならない、ということである。ニューロスフェアの融合は、主要な交絡因子であり得る。いくつかの研究では、神経細胞は非常に運動性であってもおそらく「クローン性」の条件7,19は何かの下で、融合できることが示されている。生じたニューロスフェア周波数は培地成分、解剖手順と解離過程などの要因に大きく依存することができます。でも、経験豊富なハンドラとの間で、おそらく同一のサンプルから生成されたニューロスフェアの数はいくつかのバリエーションが明らかである( 図1Aを参照してください。。)より有用なノックアウト2与えられたサンプル間の前駆体頻度の直接比較( すなわち治療対対照または野生型対で)一回の実験ではなく、全体の定量的なステートメント内で同じ人が扱う前駆細胞数。
それは、これらの2つの培養システムが生成した細胞型の均一性が異なることに留意することが重要である特定の実験のために最も適している2つの培養法のどちらかを決定する。かなり均質な前駆細胞プール(〜細胞の98%がSox2の+で)を示し、接着性細胞培養、増殖中と比較して20、ニューロスフェアはより不均一であり、含まれているだけでなく、増殖している前駆細胞、分化したニューロンおよびアストロサイト21,22。これは、ニューロスフェアを大きくなどの通路の間に長時間培養されないことが重要で、それが彼らのコア中の分化細胞型を見つけることである可能性が高くなってニューロスフェア。
私たちは、伝統的に5〜8の間のマウスのDG組織からの接着単層神経前駆培養を開始。単一のマウスのDG又はSVZから接着単層培養物を確立しようとするとそのため、細心の注意は、組織摩砕にわたってによる過剰な細胞死を回避するために、組織解離手順の間に取られる必要があり、または拡張を服用解剖して、最終的な培養ステップ間の時間の期間。このプロトコルは、初めて、個々の動物のSVZとDGの両方から接着単層前駆体培養物の生成を説明しています。前駆体の増殖および分化の比較は単一の動物に基づいて行われる必要があるときに多くの例がある。これらは、直接対に統計を使用して個々の動物のDGとSVZを比較すると、個々の行動や生理的データ9で培養データをペアリングする機能があります。単一の動物培養もアル遺伝的関連研究のための文化当たり5-8ドナーの年齢に合致するプールが不可能な希少なトランスジェニック動物、だけでなく、ユニークな動物( 例えば 、F2交雑またはOUT飼育された動物)の低い使用。
開示事項
著者らは、開示することは何もありません。
謝辞
TLWはマリー·キュリー国際着信フェローシップによってサポートされていました。この作品は、基本的な制度の資金から融資された、BundesministeriumはBildungとForschung(BMBF)の資金調達、一部重点研究プログラム(SFB)、GKへの655の支援を受けて毛皮。著者らは、細胞培養および顕微鏡の支援のために本研究で用いたすべての動物の世話やメンテナンスのためのアンKarasinskyとオデットライター、スーザンRuhwald、ファニーボイーム、リチャードウェッツェルに感謝したいと思います。
資料
| Name | Company | Catalog Number | Comments |
| Poly-D-lysine | Sigma | P7280-5MG | |
| Laminin | Roche | 11243217001 | |
| Glass Pastuer pipettes | Volac | BS5732 | |
| DMEM:F12 (1:1) 1x | Life Technologies | 21331-020 | |
| Neural Basal Medium (1x) | Life Technologies | 21103-049 | |
| B27 supplements (50x) | Life Technologies | 17504-044 | |
| GlutaMAX | Life Technologies | 35050-038 | |
| Heparin | Sigma | H3393 | |
| Penacillin/Streptomycin | Life Technologies | 15140-122 | |
| EGF | PeproTech | AF-100-15 | |
| bFGF | PeproTech | 100-18B | |
| 0.05% Trypsin-EDTA | Life Technologies | 25300-054 | |
| Trypsin inhibitor | Sigma | T6522 | |
| DNaseI | Roche | 10104159001 | |
| Accutase | PAA | L11-007 | |
| Papain | Worthington | LS003120 | |
| Dispase | Life Technologies | 17105-041 | |
| Percoll | GE Healthcare | 17-0891-02 | |
| HBSS (with Calcium and Magnesium) | Life Technologies | 14025-050 | |
| Glucose | Roth | X997.2 | |
| HEPES | Sigma | H3375-500G | |
| NaHCO3 | Merck | K39347429847 | |
| 1 ml Syringes | Braun | 2016-10 | |
| 27 G Needles | Braun | 4657705 | |
| Scalpels (#22 disposable) | Braun | BA222 | |
| Dumont #7 forceps | FST | 11271-30 | |
| Dumont 5/45 forceps | FST | 11251-35 | |
| Scissors | FST | 14060-10 | |
| Iris spatula | FST | 10093-13 | |
| 70% Ethanol | |||
| PBS | Life Technologies | 14040-091 | |
| flasks/well plates | TPP | 92696 | |
| PFA (4%) | Sigma | P6148 | |
| Hemocytometer | Marienfeld | 650010 | |
| Trypan blue (0.4%) | Sigma | T8154 | |
| NDS | Millipore | 530 | |
| TritonX-100 | Sigma | T9284 | |
| Mouse monoclonal bIII-tubulin antibody | Promega | G712A | |
| Rabbit polyclonal glial fibrillary acidic protein antibody | Dako | 20334 | |
| O4 | R&D Systems | MAB1326 | |
| Map2a+b | Sigma | M1406 | |
| Donkey anti-mouse Cy3 antibody | Jackson ImmunoResearch | 715-505-151 | |
| Donkey anti-rabbit Alexa488 | Dianova | 711-545-152 | |
| 4,6-Diamidino-2-phenylindole (DAPI) | Invitrogen | 861405 | |
| Aqua Polymount | Polysciences Inc | 18606 | |
| 10 ml Combi tips | Eppendorf | 30089677 | |
| Plastic 10 ml and 25 ml serological pipettes | Corning | 4488/4489 | |
| EQUIPMENT | |||
| Pipetboy | Integra Biosciences | 521942 | |
| Multidoser pipette | Eppendorf | ||
| 37 °C waterbath | |||
| Dissecting microscope | |||
| 37 °C:5% CO2 incubator | |||
| Centrifuge | Eppendorf | 5810R |
参考文献
- Reynolds, B. A., Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 255, 1707-1710 (1992).
- Reynolds, B. A., Weiss, S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 175, 1-13 (1996).
- Palmer, T. D., Ray, J., Gage, F. H. FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol. Cell. Neurosci. 6, 474-486 (1995).
- Ray, J., Raymon, H. K., Gage, F. H. Generation and culturing of precursor cells and neuroblasts from embryonic and adult central nervous system. Methods Enzymol. 254, 20-37 (1995).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993, 18-29 (2003).
- Babu, H., Cheung, G., Kettenmann, H., Palmer, T. D., Kempermann, G. Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. PLoS One. 2, (2007).
- Jessberger, S., Clemenson, G. D., Gage, F. H. Spontaneous fusion and nonclonal growth of adult neural stem cells. Stem Cells. 25, 871-874 (2007).
- Reynolds, B. A., Tetzlaff, W., Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 12, 4565-4574 (1992).
- Freund, J., et al. Emergence of individuality in genetically identical mice. Science. 340, 756-759 (2013).
- Azari, H., Rahman, M., Sharififar, S., Reynolds, B. A. Isolation and expansion of the adult mouse neural stem cells using the neurosphere assay. J. Vis. Exp. , (2010).
- Hagihara, H., Toyama, K., Yamasaki, N., Miyakawa, T. Dissection of hippocampal dentate gyrus from adult mouse. J. Vis. Exp. , (2009).
- Jensen, J. B., Parmar, M. Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 34, 153-161 (2006).
- Pastrana, E., Silva-Vargas, V., Doetsch, F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell. Stem Cell. 8, 486-498 (2011).
- Reynolds, B. A., Rietze, R. L. Neural stem cells and neurospheres- re-evaluating the relationship. Nat. Methods. 2, 333-336 (2005).
- Walker, T. L., Turnbull, G. W., Mackay, E. W., Hannan, A. J., Bartlett, P. F. The latent stem cell population is retained in the hippocampus of transgenic Huntington's disease mice but not wild-type mice. PLoS One. 6, (2011).
- Walker, T. L., et al. Prolactin stimulates precursor cells in the adult mouse hippocampus. PLoS One. 7, (2012).
- Walker, T. L., et al. Latent stem and progenitor cells in the hippocampus are activated by neural excitation. J. Neurosci. 28, 5240-5247 (2008).
- Walker, T. L., et al. Prominin-1 allows prospective isolation of neural stem cells from the adult murine hippocampus. J. Neurosci. 33, (2013).
- Singec, I., et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods. 3, 801-806 (2006).
- Babu, H., et al. A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front. Neurosci. 5, 89 (2011).
- Parmar, M., Sjoberg, A., Bjorklund, A., Kokaia, Z. Phenotypic and molecular identity of cells in the adult subventricular zone in vivo and after expansion in vitro. Mol. Cell Neurosci. 24, 741-752 (2003).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99, 14506-14511 (2002).
Erratum
Formal Correction: Erratum: One Mouse, Two Cultures: Isolation and Culture of Adult Neural Stem Cells from the Two Neurogenic Zones of Individual Mice
Posted by JoVE Editors on 11/26/2014. Citeable Link.
A correction was made to One Mouse, Two Cultures: Isolation and Culture of Adult Neural Stem Cells from the Two Neurogenic Zones of Individual Mice. Many micro symbols were changed into milli symbols by accident: In the Protocols, sections 1.1, 1.3, 3.6, 3.7, 6.2, 6.6, 8.1, 9.1.6, 9.2.2, and 9.2.5 need to be fixed, as does Figure 1 description in the Results section.
Protocol section 1.1 was changed from:
At least two days prior to commencing the experiment, prepare Poly-D-lysine (PDL)/Laminin coated plates for adherent monolayer cultures. To prepare wells/flasks add enough PDL (10 mg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Allow to air dry. Add Laminin (5 mg/ml in cold DMEM:F12) and incubate at 37 °C overnight. Remove the Laminin and either use the plates immediately or store with the Laminin at -20 °C until required.
to:
At least two days prior to commencing the experiment, prepare Poly-D-lysine (PDL)/Laminin coated plates for adherent monolayer cultures. To prepare wells/flasks add enough PDL (10 µg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Allow to air dry. Add Laminin (5 µg/ml in cold DMEM:F12) and incubate at 37 °C overnight. Remove the Laminin and either use the plates immediately or store with the Laminin at -20 °C until required.
Protocol section 1.3 was changed from:
On the day of dissection, prepare the appropriate amount of culture medium by mixing Neural Basal Medium with 2% B27, 1x GlutaMAX, 2 µg/ml heparin, 50 units/ml Penicillin/Streptomycin, 20 ng/ml purified mouse receptor-grade epidermal growth factor (EGF), and 20 ng/ml recombinant bovine fibroblast growth factor (FGF-2). Warm the culture medium to 37 °C in a water bath.
to:
On the day of dissection, prepare the appropriate amount of culture medium by mixing Neural Basal Medium with 2% B27, 1x GlutaMAX, 2 mg/ml heparin, 50 units/ml Penicillin/Streptomycin, 20 ng/ml purified mouse receptor-grade epidermal growth factor (EGF), and 20 ng/ml recombinant bovine fibroblast growth factor (FGF-2). Warm the culture medium to 37 °C in a water bath.
Protocol section 3.6 was changed from:
Add growth medium to a total volume of 5 ml and pass the cell suspension through a 40 mm sieve to remove debris and undissociated tissue clumps.
to:
Add growth medium to a total volume of 5 ml and pass the cell suspension through a 40 µm sieve to remove debris and undissociated tissue clumps.
Protocol section 3.7 was changed from:
Centrifuge at 300 x g for 5 min, discard the supernatant and resuspend the resulting pellet in 200 ml growth medium.
to:
Centrifuge at 300 x g for 5 min, discard the supernatant and resuspend the resulting pellet in 200 µl growth medium.
Protocol section 6.2 was changed from:
Add 50 ml Accutase and incubate at 37 °C for 2-3 min (checking to see if the cells are rounded and detached).
to:
Add 50 µl Accutase and incubate at 37 °C for 2-3 min (checking to see if the cells are rounded and detached).
Protocol section 6.6 was changed from:
For subsequent passages, resuspend cells in 200 ml growth medium and count using a hemocytometer. Plate at 1 x 104 cells/cm2 in the appropriate sized coated well or flask.
to:
For subsequent passages, resuspend cells in 200 µl growth medium and count using a hemocytometer. Plate at 1 x 104 cells/cm2 in the appropriate sized coated well or flask.
Protocol section 8.1 was changed from:
Dilute the dissociated SVZ or DG tissue from one animal in 20 ml of culture medium and plate 200 ml/well across a 96-well plate using a 10 ml multidoser pipette.
to:
Dilute the dissociated SVZ or DG tissue from one animal in 20 ml of culture medium and plate 200 µl/well across a 96-well plate using a 10 ml multidoser pipette.
Protocol section 9.1.6 was changed from:
Remove 10 ml of the cell suspension and mix with an equal volume of trypan blue and perform a live cell count using a hemocytometer.
to:
Remove 10 µl of the cell suspension and mix with an equal volume of trypan blue and perform a live cell count using a hemocytometer.
Protocol section 9.2.2 was changed from:
Add 100 ml of 0.05% Trypsin-EDTA to each well to be passaged and incubate at room temperature for 3 min.
to:
Add 100 µl of 0.05% Trypsin-EDTA to each well to be passaged and incubate at room temperature for 3 min.
Protocol section 9.2.5 was changed from:
Transfer the 200 ml containing the dissociated neurosphere to a new well of a 24-well plate containing 1.5 ml of growth medium. Incubate at 37 °C with 5% CO2 until secondary neurospheres form.
to:
Transfer the 200 µl containing the dissociated neurosphere to a new well of a 24-well plate containing 1.5 ml of growth medium. Incubate at 37 °C with 5% CO2 until secondary neurospheres form.
Figure 1 description was updated from:
Figure 1. Adult mouse precursor cells can be cultured as adherent monolayer cultures (A) or as neurospheres (B: SVZ, C: DG). Scale bar is 50 mm.
to:
Figure 1. Adult mouse precursor cells can be cultured as adherent monolayer cultures (A) or as neurospheres (B: SVZ, C: DG). Scale bar is 50 µm.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved