
Method Article
AB Initio分子動力学シミュレーションからの溶融物と流体の分析
要約
溶融物や流体は、自然界における大量輸送のユビキタスベクターです。我々は、このようなシステムのab initio分子動力学シミュレーションを分析するためのオープンソースパッケージを開発しました。構造(接合、クラスター化、化学種分化)、輸送(拡散、粘度)、熱力学的特性(振動スペクトル)を計算します。
要約
我々は、流体のab initio分子動力学シミュレーションに起因する結果を分析するために、Pythonベースのオープンソースパッケージを開発しました。このパッケージは、ケイ酸塩や酸化物の溶融物、水性流体、およびさまざまな超臨界流体などの自然システムでの用途に最適です。パッケージは、ファイル形式と結晶学を扱う2つの主要なライブラリを含むPythonスクリプトのコレクションです。すべてのスクリプトは、コマンド ラインで実行されます。我々は、ユニバーサル分子動力学のために立って、UMDファイルに保存されるシミュレーションの原子軌道および関連する熱力学情報を格納するための単純化された形式を提案する。UMD パッケージは、一連の構造、輸送、熱力学的特性の計算を可能にします。対分布関数から始めて、結合長を定義し、原子間接続行列を構築し、最終的に化学種分化を決定します。化学種の寿命を決定すると、完全な統計分析を実行できます。次に、専用のスクリプトは、原子と化学種の平均二乗変位を計算します。原子速度の自己相関解析を実施すると、拡散係数と振動スペクトルが得られます。応力に適用される同じ分析によって、粘度が得られます。パッケージは、GitHubのウェブサイトを介して、オープンアクセスパッケージとしてERC IMPACTプロジェクトの専用ページから入手できます。
概要
流体と溶融物は、自然環境における活性化学および物理的輸送ベクトルです。原子拡散率の上昇は、化学交換や反応を好み、低粘度と様々な浮力を組み合わせることで大きな大量移送を好み、結晶溶融密度関係は惑星体内に重ね合わせる。周期格子の欠如、溶融状態に達するために必要な典型的な高温、および焼入れの難しさは、密度、拡散、粘度などの一連の明白な特性の実験的決定を極めて困難にします。これらの困難は、代替計算方法を強力で有用なツールにして、このクラスの材料を調査する。
計算能力の出現とスーパーコンピュータの利用可能性により、非結晶原子論系モンテカルロ1と分子動力学(MD)1,2の動的状態を研究するために、2つの主要な数値原子論的シミュレーション技術が現在採用されています。モンテカルロシミュレーションでは、構成空間はランダムにサンプリングされます。すべてのサンプリング観測値が互いに独立している場合、モンテカルロ法は平行法で線形スケーリングを示します。結果の品質は、乱数発生器の品質とサンプリングの代表性によって異なります。モンテカルロ法は、サンプリングが互いに独立している場合、平行化で線形スケーリングを示します。分子動力学(MD)では、構成空間は時間依存性原子軌道によってサンプリングされる。特定の構成から始めて、原子軌道はニュートン運動方程式を統合することによって計算されます。原子間の力はモデル間の潜在的な(古典的なMD)か第一原理法(ab initio、または第一原理、MD)を使用して計算することができる。結果の質は、軌道の長さと、局所的なミニマに引き付けされない能力に依存する。
分子動力学シミュレーションには、システムの動的挙動に関連する情報が多く含まれています。熱力学的平均特性は、内部エネルギー、温度、圧力など、計算に適した標準です。これらは、シミュレーションの出力ファイルから抽出して平均化できますが、原子の動きと相互関係に直接関連する量は、原子の位置と速度を抽出した後に計算する必要があります。
結果の視覚化に多くの努力が捧げられており、さまざまなパッケージが、オープンソースの異なるプラットフォームで利用可能かどうか[Ovito3、VMD4、Vesta5、Travis6など]。これらの可視化ツールはすべて、相互距離を効率的に処理するため、ペア分布関数と拡散係数の効率的な計算が可能になります。大規模分子動力学シミュレーションを行う様々なグループは、時にはシェアウェアやコミュニティへのアクセス制限の他の形態で、時には範囲が限定され、いくつかの特定のパッケージに使用するシミュレーションから生じる様々な他の特性を分析するための独自のソフトウェアを持っています。これらのパッケージの中には、原子間結合、幾何学的パターン、熱力学に関する情報を抽出する高度なアルゴリズムが開発され、実装されています。
ここでは、分子動力学シミュレーションの出力を分析するためにPythonで書かれたオープンソースパッケージである UMDパッケージ を提案します。UMD パッケージでは、構造、動的、熱力学的特性の幅広い計算が可能です(図 1)。パッケージは、GitHub のウェブサイト (https://github.com/rcaracas/UMD_package) から、オープン アクセス パッケージとして ERC IMPACT プロジェクトの専用ページ (http://moonimpact.eu/umd-package/) を介して入手できます。
汎用的で扱いやすくするために、実際の分子動力学の出力ファイルから熱力学状態と原子軌道に関連するすべての情報をまず抽出するアプローチです。この情報は専用ファイルに保存され、その形式はシミュレーションが実行された元の MD パッケージとは無関係です。私たちは、ユニバーサル分子ダイナミクスの略で、これらのファイル"umd"ファイルの名前を付けます。このように、当社のUMDパッケージは、適応の最小限の労力で、任意のソフトウェアを持つ任意のab initioグループによって簡単に使用することができます。現在のパッケージを使用する唯一の要件は、特定の MD ソフトウェアの出力から適切なパーサーを umd ファイル形式に書き込むだけです (まだ存在しない場合)。現時点では、VASP8 および QBox9 パッケージにこのようなパーサーを提供します。

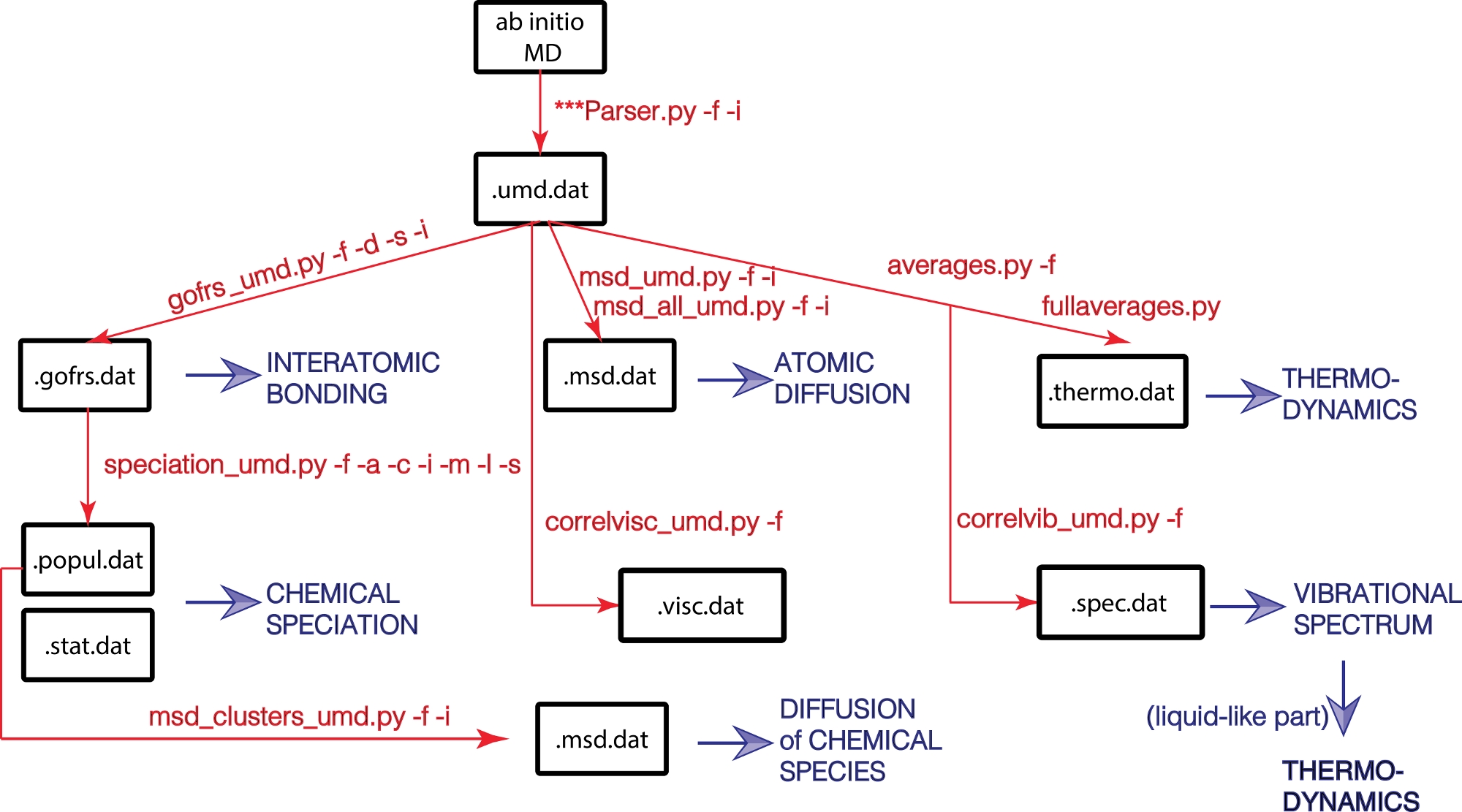
図 1: UMD ライブラリのフローチャート
物理プロパティは青で表示され、主要な Python スクリプトとそのオプションは赤で表示されます。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
umd ファイルは ASCII ファイルです。一般的な拡張子は"umd.dat"ですが、必須ではありません。すべての解析コンポーネントは、実際の名前拡張子に関係なく、umd 形式の ASCII ファイルを読み取ることができます。しかし、いくつかのシミュレーションで高速な大規模統計を実行するように設計された自動スクリプトの中には、特にumd.dat拡張子を持つファイルを検索するものもあります。各物理プロパティは 1 行で表されます。すべての行はキーワードで始まります。このように、フォーマットは非常に適応性が高く、バージョン全体で読みやすさを維持しながら、新しいプロパティをumdファイルに追加することができます。図 2に、4.6GPa及び3000Kにおけるパイロライトのシミュレーションのumdファイルの最初の30行を示す。

図2:4.6GPaおよび3000Kにおける液体火砕物のシミュレーションを記述するumdファイルの始まり。
ヘッダーの後には、各スナップショットの説明が続きます。各プロパティは、物理プロパティの名前、値、および単位をすべてスペースで区切って 1 行に書き込まれます。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
すべてのumdファイルには、シミュレーションセルの含有量を記述するヘッダ(原子数、電子、原子タイプ、原子の種類、化学記号、価電子数、質量など)の各原子の詳細が含まれています。空の行はヘッダーの終わりを示し、それを umd ファイルの主要部分から分離します。
次に、シミュレーションの各ステップが詳細になります。まず、瞬間的な熱力学パラメータがそれぞれ異なる線上に与えられ、(i)エネルギー、応力、同等の静水圧、密度、体積、格子パラメータなどのパラメータの名前を指定し、(ii)その値(s)、および(iii)その単位を指定します。次に原子を説明する表が来ます。ヘッダー行は、デカルト位置、速度、料金などの異なるメジャーとその単位を示します。その後、各原子は1行に詳細に記載されています。3つの x、y、z 軸に対応する3つのグループによって、エントリは、減少した位置、シミュレーションセルに折り畳まれたデカルト位置、デカルト位置(原子がシミュレーション中に複数のユニットセルを横断することができるという事実を適切に考慮する)、原子速度、原子力です。最後の2つのエントリはスカラーです:電荷と磁気モーメント。
2 つの主要なライブラリは、パッケージ全体の適切な機能を保証します。 umd_process.py ライブラリは、読み取りや印刷などの umd ファイルを扱います。 crystallography.py ライブラリは、実際の原子構造に関連するすべての情報を扱います。 crystallography.py ライブラリの根底にある考え方は、格子をベクトル空間として扱う事です。単位セルパラメータとその向きは、基底ベクトルを表します。「空間」は、一連のスカラー属性(特定の体積、密度、温度、および特定数の原子)、熱力学的特性(内部エネルギー、圧力、熱容量など)、および一連の緊張特性(応力と弾性)を有する。原子はこの空間に取り込みます。「ラティス」クラスは、このアンサンブルを定義し、特定の体積、密度、直接のラティスなどから相互格子を得るなど、いくつかの短い計算と一緒に定義します。「原子」クラスは原子を定義します。これらは、一連のスカラー特性(名前、記号、質量、電子の数など)と一連のベクトル特性(ラティスクラスに記述されたベクトル基に対する空間の位置、または普遍的なデカルト座標、速度、力などに対する相対)によって特徴付けられます。これら 2 つのクラスとは別に、 crystallography.py ライブラリには、原子距離やセルの乗算など、さまざまなテストや計算を実行する一連の関数が含まれています。要素の周期表も辞書として含まれています。
umd パッケージのさまざまなコンポーネントが、いくつかの出力ファイルを書き込みます。一般的な規則として、これらはすべて ASCII ファイルであり、すべてのエントリはタブで区切られ、可能な限り自明の説明として作成されます。たとえば、物理プロパティとその単位を常に明確に示します。 umd.dat ファイルは、この規則に完全に準拠しています。
プロトコル
1. 分子動力学の分析
注: このパッケージは、GitHub のウェブサイト (https://github.com/rcaracas/UMD_package) から、オープンアクセス パッケージとして ERC IMPACT プロジェクトの専用ページ (http://moonimpact.eu/umd-package/) を介して入手できます。
- パッケージから 1 つ以上の専用 Python スクリプトを使用して、特定の物理プロパティのセットを抽出します。コマンド ラインですべてのスクリプトを実行します。これらはすべて一連のフラグを採用しており、スクリプト間で可能な限り一貫性があります。フラグ、その意味、およびデフォルト値はすべて 表 1 にまとめられています。
| 旗 | 意味 | それを使用してスクリプト | 既定値 |
| -h | 短いヘルプ | すべての | |
| -f | UMD ファイル名 | すべての | |
| -i | 廃棄する熱化ステップ | すべての | 0 |
| -i | 原子間結合を含む入力ファイル | 種 分化 | 債券.インプット |
| -s | 周波数のサンプリング | msd, 種分化 | 1 (すべてのステップが考慮されます) |
| -a | 原子または陰イオンの一覧 | 種 分化 | |
| -c | カチの一覧 | 種 分化 | |
| -l | ボンド長さ | 種 分化 | 2 |
| -t | 温度 | 振動,レロジー | |
| -v | 平均二乗変位解析のための軌道のサンプリングウィンドウの幅の分離 | msd | 20 |
| -z | 平均二乗変位解析のための軌道のサンプリングウィンドウの開始の分離 | msd | 20 |
表 1: UMD パッケージで使用される最も一般的なフラグとその最も一般的な重要性。
- まず、VASP8 や QBox9 などの第一原理コードで実行される MD シミュレーションの出力を UMD ファイルに変換します。
- MD シミュレーションが VASP で実行された場合は、コマンド ラインで次のように入力します。
VaspParser.py -f -i <初期ステップ>
ここで–f フラグは VASP OUTCAR ファイルの名前を定義し、–i の熱化の長さを定義します。
注: –i で定義された最初のステップでは、シミュレーションの最初のステップを破棄できます。典型的な分子動力学の実行では、計算の最初の部分は、温度の温度化、すなわち、すべての原子が温度のガウス状分布を表すためにシステムを取り、平衡値の周りに温度、圧力、エネルギーなどの変動を示すためにシステム全体のためにかかる時間を表します。流体の統計的特性を解析する際には、シミュレーションのこの熱化部分を考慮しないでください。
- MD シミュレーションが VASP で実行された場合は、コマンド ラインで次のように入力します。
- を変換します。umd ファイルを に変換します。VMD4 や Vesta5 など、さまざまなパッケージで視覚化を容易にする xyz ファイル。コマンド ラインで次のように入力します。
umd2xyz.py -f -i -s
ここで –f は の名前を定義します。umd ファイル -i は破棄する熱化期間を定義し、–s に格納されている軌道のサンプリングの頻度を定義します。umd ファイル。既定値は –i 0 –s 1、つまり、破棄されずにシミュレーションのすべてのステップを考慮します。 - umd2poscar.py スクリプトを使用して 、umd ファイルを VASP タイプの POSCAR ファイルに逆にします。シミュレーションのスナップショットは、事前に定義された頻度で選択できます。コマンド ラインで次のように入力します。
umd2poscar.py -f -i -l -s
ここで –l は POSCAR ファイルに変換される最後のステップを表します。デフォルト値は -i 0 -l 1000000 -s 1 です。–l のこの値は、一般的な全体の軌道をカバーするのに十分な大きさです。
2. 構造解析を実行する
- gofrs_umd.pyスクリプトを実行して、アトミックタイプAとBのすべてのペアのペア分布関数 (PDF) g ᴀʙ(r) を計算します (図 3)。出力は、タブ区切りの 1 つの ASCII ファイルに、拡張 gofrs.dat 書き込まれます。コマンド ラインで次のように入力します。
gofrs_umd.py -f -s < Sampling_Frequency > -d <ディスレタイゼーション間隔> -i <初期ステップ>
注: デフォルトはSampling_Frequency(軌道をサンプリングする周波数)=1ステップです。分離間隔間隔 (g(r)) = 0.01 Å;InitialStep (破棄される軌道の先頭のステップ数) = 0。放射状PDF、g ᴀʙ(r)は、A型の原子を中心とした半径rと厚さdrの球状シェル内の距離d_ᴀʙ B型の平均数です(図3)。
ρでは、原子密度、NAおよびNBタイプAおよびBの原子数、およびδ(r-r ᴀʙ)は、原子AとBがrとr+drの間の距離にある場合に1に等しいデルタ関数を示します。最初の最大値の g ᴀʙ(r) の棄権は、A 型と B 型の原子の間で最も高い確率の結合長さを示します。最初の最小値は、最初の座標球の範囲を区切ります。したがって、最初の最小値までのPDF上の積分は、平均調整数を与えます。A型とB型のすべての組み合わせに対するg ᴀʙ(r)のフーリエ変換の合計は、回折計で実験的に得られたように、流体の回折パターンを生み出す。しかし実際には、高次の配位球がg ᴀʙ(r)から欠落している場合、回折パターン全体を得ることができない場合があります。

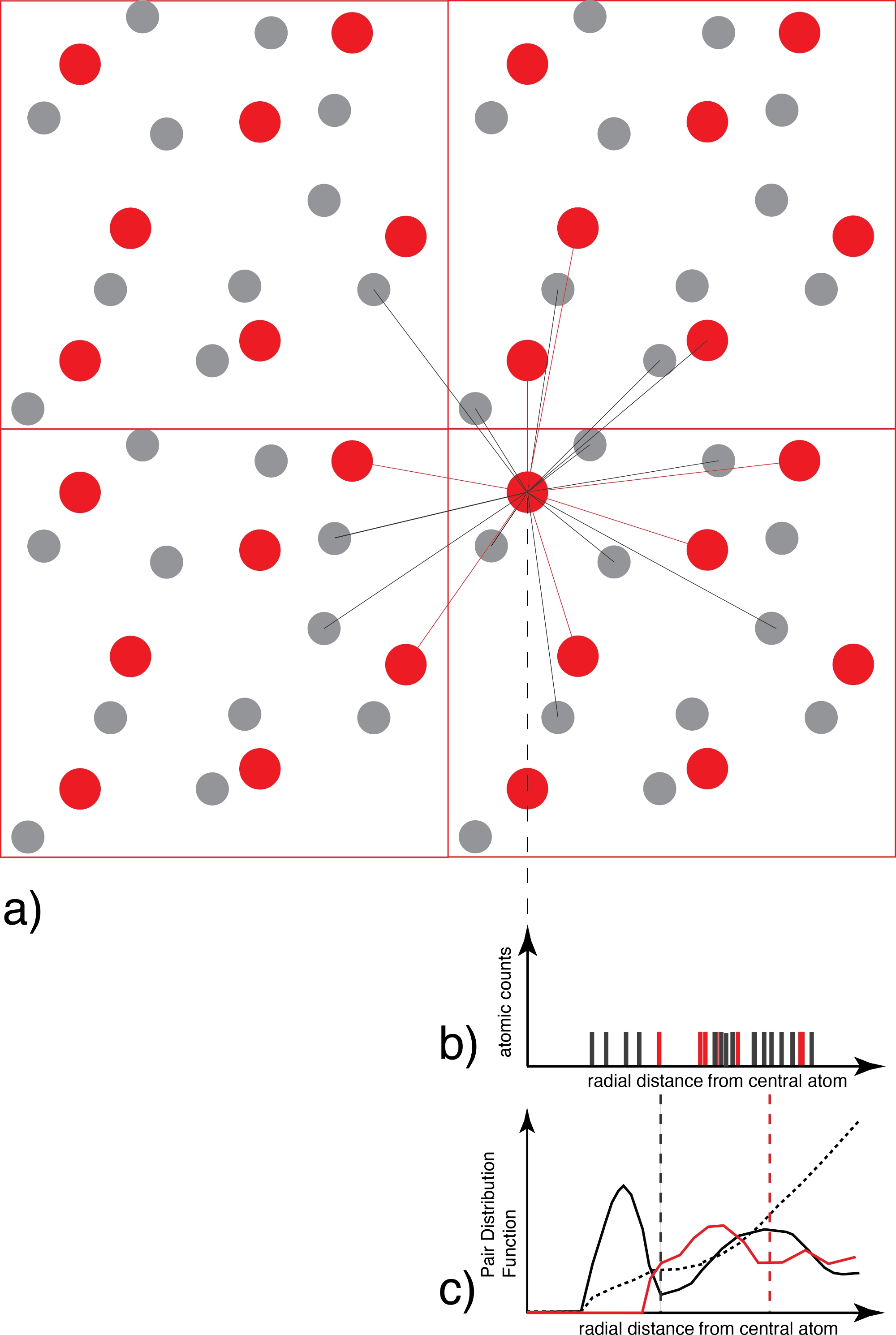
図3:ペア分布関数の決定
(a) 1つの種の各原子(例えば赤)について、配位種のすべての原子(例えば灰色および/または赤)は距離の関数として数えられる。(b) 各スナップショットの結果として得られる距離分布グラフは、この段階ではデルタ関数の集まりに過ぎず、すべての原子とすべてのスナップショットを平均化し、(c)連続するペア分布関数を生成する理想的なガス分布によって重み付けされる。 最初の最小値は 、最初の配位球の半径で、後で種分分析で使用します。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
- 最初の配位球の半径として平均原子間結合距離を抽出します。このためには、 g ᴀʙ(r)関数の最初の最大値の位置を特定します: gofrs.datファイルをスプレッドシートアプリケーションでプロットし、原子の各ペアの最大値と最小値を検索します。
- スプレッドシート ソフトウェアを使用して、最初の調整球の半径を PDF の最初の最小値である g ᴀʙ(r)として識別します。これは流体の構造解析全体の基礎となります。PDF は流体中の原子の平均結合状態を生成します。
- 最初のミニマ、すなわち、アブシッサの距離を抽出し、例えば bonds.inputと呼ばれる別のファイルに書き込みます。または、umD パッケージの analyze_gofr スクリプトのいずれかを実行して 、g ᴀʙ(r) 関数の最大値と最小値を識別します。コマンド ラインで次のように入力します。
analyze_gofr_semi_automatic.py - プログラムで開いたグラフに表示される g ᴀʙ(r) 関数の最大値と最小値の位置をクリックします。スクリプトは、現在のフォルダを自動的にスキャンし、すべての gofrs.datファイルを識別し、それぞれのファイルの分析を実行します。スクリプトが教育的な初期推測を必要とするたびに、ウィンドウの最大値と最小値をもう一度クリックします。
- 開いて、原子間の結合距離を含む bonds.input と呼ばれる自動的に生成されたファイルを見ます。
3. 種分分析を実行する
- グラフ理論内の接続性の概念を使用して、原子間の結合のトポロジを計算します:原子はノードであり、原子間結合はパスです。 speciation_umd.py スクリプトには bonds.input ファイルで定義された原子間結合距離が必要です。
注: 接続マトリックスは、各時間ステップで構築されます: 対応する最初の配位球の半径よりも小さい距離にある 2 つの原子は、結合されていると見なされます。つまり接続されています。さまざまな原子ネットワークは、この幾何学的基準によって接続が定義されたグラフ内のノードとして原子を扱うことによって構築されます。これらのネットワークは原子種であり、そのアンサンブルは、その特定の流体における原子種分化を定義する(図4)。

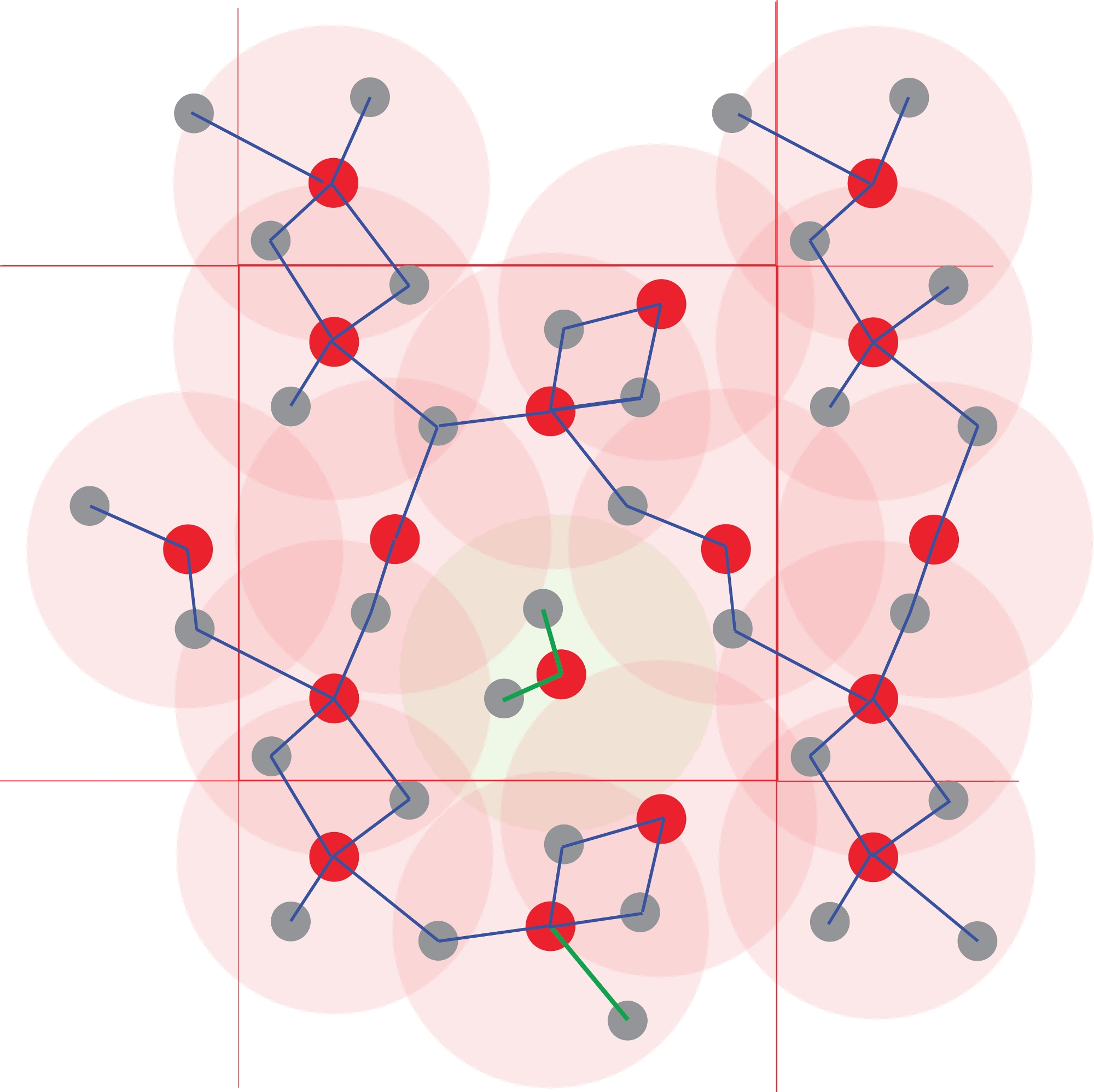
図4:原子クラスタの識別
配位多面体は、原子間距離を使用して定義されます。指定した半径よりも小さい距離にあるすべての原子は、結合されていると見なされます。ここでの閾値は、図1で定義された第1の配位球(明るい赤い円)に相当する。重合および従って化学種は、結合原子のネットワークから得られる。無限のポリマーを形成する他の原子から分離された中央のRed1Grey2クラスターに注意してください。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
- スペシエーション スクリプトを実行して接続行列を取得し、配位多面体または重合を取得します。コマンド ラインで次のように入力します。
speciation_umd.py -f -s -i <入力ファイル> -l -c <カチオン> -a <アニオンズ> -m -r <リング>
ここで-i フラグは、前のステップで生成された、例えば、原子間結合距離を持つファイルを示します。または、-l フラグで定義されたすべての結合に対して 1 つの長さのスクリプトを実行します。
注: -c フラグは、中心の原子を指定し、-a フラグをリガンドに指定します。中央原子とリガンドの両方が異なるタイプであることができます。この場合、カンマで区切る必要があります。m フラグは、分析で考慮されるために種が生きなければならない最小時間を示します。デフォルトでは、この最小時間はゼロで、最終的な分析でカウントされるすべてのオカレンスです。- 第 1 レベルで接続グラフをサンプリングして、コーディネーション多面体を識別するフラグ –r 0 を指定してspeciation_umd.pyスクリプトを実行します。例えば、カ チオン として示される中心原子は、1つ以上 の陰イオン で囲まれていてもよい(図4)。スペシレーションスクリプトは、配位多面体の一つ一つを識別します。すべての配位多面体の加重平均は、調整数を、PDFの統合から得られたものと同一に与える。コマンド ラインで次のように入力します。
speciation_umd.py -f -i <入力ファイル> -c <> -a <アニオン> -r 0
注: 流体の平均配位数は小数です。この分数は、コーディネートの平均特性から生まれます。種分化に基づく定義は、流体の構造をより直感的で有益な表現をもたらす。 - speciation_umd.py スクリプトを 実行するには、フラグ –r 1 を指定して、接続性グラフをすべての深度レベルでサンプリングして重合を取得します。原子グラフを通るネットワークは、原子が他の結合(例えば、交互のカチオンおよび陰イオンの配列)にさらに離れて結合されるので、ある深さを有する(図4)。
- 第 1 レベルで接続グラフをサンプリングして、コーディネーション多面体を識別するフラグ –r 0 を指定してspeciation_umd.pyスクリプトを実行します。例えば、カ チオン として示される中心原子は、1つ以上 の陰イオン で囲まれていてもよい(図4)。スペシレーションスクリプトは、配位多面体の一つ一つを識別します。すべての配位多面体の加重平均は、調整数を、PDFの統合から得られたものと同一に与える。コマンド ラインで次のように入力します。
- 2 つのファイルを開きます。ポパル.dat と.統計.dat 連続して;これらは、スペシテーション スクリプトの出力を構成します。各クラスターは、その化学式、それが形成された時間、それが死んだ時間、その寿命、このクラスターを形成する原子のリストを持つ行列を指定して、1行に書かれています。.popul.dat ファイルに示されているように、シミュレーションで見つかったすべての化学種の各原子クラスターの寿命をプロットします (図 5)。
- で見られる、各種の豊富な人口分析をプロットします。stat.dat ファイル。この分析は、絶対と相対の両方で、ケース -r 0 の座標多面体の実際の統計量に対応します。重合の場合、-r1では、原子の相対数に対する正規化を適用する必要がある場合があるため、慎重に扱う必要があります。豊富さは、生涯にわたって積分に対応します。ザ。stat.dat ファイルには、各クラスターのサイズ、すなわち、そのクラスターを形成する原子の数もリストされます。
4. 拡散係数の計算
- 自己拡散性を得るために時間の関数として原子の平均二乗変位(MSD)を抽出する。MSD の標準式は次のとおりです。

ここで、プリファクターは再正規化です。MSD ツールを使用すると、流体の動的な側面を分析するさまざまな方法があります。
注: T はシミュレーションの合計時間であり、N αはタイプ αの原子数です。初期時間 t0 は任意であり、シミュレーションの前半に及びます。ニニトは初期回数です。τ は、MSD が計算される時間間隔の幅です。最大値はシミュレーションの時間の長さの半分です。一般的な MSD 実装では、すべてのウィンドウは前のウィンドウの最後から開始されます。しかし、まばらなサンプリングは、MSDの傾きを変更することなく、MSDの計算を高速化することができます。このため、i 番目のウィンドウは時刻 t0(i) から始まりますが、(i+1)番目のウィンドウは時刻 t0(i) + τ + v で始まり、v の値はユーザー定義です。同様に、ウィンドウの幅は、ユーザーによって定義された個別のステップで増加します。z ("水平ステップ") と v ("垂直ステップ") の値は正またはゼロです。両方のデフォルトは 20 です。 - 一連の msd_umd スクリプトを使用して MSD を計算します。出力は.msd.dat ファイルでは、各原子タイプ、原子、またはクラスタの MSD が時間の関数として 1 つの列に出力されます。
- 各アトミックタイプの平均 MSD を計算します。MSD は各原子に対して計算され、その後、アトミックタイプごとに平均化されます。出力ファイルには、アトミック・タイプごとに 1 つの列が含まれます。コマンド ラインで次のように入力します。
msd_umd.py -f -z <水平ジャンプ> -v <バーティカルジャンプ> -b <弾道> - 各原子の MSD を計算します。MSD は各原子に対して計算され、その後、アトミックタイプごとに平均化されます。出力ファイルには、シミュレーション内のアトムごとに 1 つの列が含まれ、次にアトミックタイプごとに 1 つの列が含まれます。この機能により、液体や気体、または2つの液体など、2つの異なる環境で拡散する原子を識別できます。コマンド ラインで次のように入力します。
msd_all_umd.py -f -z <水平ジャンプ> -v <バーティカルジャンプ> -b <弾道> - 化学種の MSD を計算します。スペシテーション スクリプトで識別され、 で印刷されるクラスターの母集団を使用します。ポプル.dat ファイル。MSD は、個々のクラスタごとに計算されます。出力ファイルには、クラスターごとに 1 つの列が含まれます。大規模なポリマーの検討を避けるために、クラスターのサイズに制限を設けます。デフォルトは 20 個の原子です。コマンド ラインで次のように入力します。
msd_cluster_umd.py -f -p -s -b <弾道> -c <クラスタマックスサイズ>
注: デフォルト値は–b 100 –s 1 –c 20 です。
- 各アトミックタイプの平均 MSD を計算します。MSD は各原子に対して計算され、その後、アトミックタイプごとに平均化されます。出力ファイルには、アトミック・タイプごとに 1 つの列が含まれます。コマンド ラインで次のように入力します。
- スプレッドシートベースのソフトウェアを使用して MSD をプロットします(図 6)。MSD 対時間のログ ログ表現で、勾配の変化を特定します。弾道 体制を 表す最初の部分、通常は短い、すなわち衝突後の原子の速度の保全を分離する。2番目の長い部分は、 拡散 体制、すなわち衝突後の原子の速度の散乱を表します。
- MSD の傾きから拡散係数を次のように計算します。

ここで、Z は自由度の数(平面内の拡散の場合は Z = 2、空間での拡散の場合は Z = 3)、t は時間ステップです。
5. 時間相関関数
- 一般式を使用して、システムの慣性の尺度として時間相関関数を計算します。

A は、原子の位置、原子速度、応力、偏光などのさまざまな時間依存変数であり、それぞれの降伏は、緑久保関係12,13を介して、異なる物理的特性を介して、時にはさらに変換された後に起きます。 - 原子速度を解析し、液体の振動スペクトルと原子自己拡散係数の代替式を得る。
- vibr_spectrum_umd.pyスクリプトを実行して、各アトミックタイプの原子速度自動相関(VAC)関数を計算し、高速フーリエ変換を実行します。コマンド ラインで次のように入力します。
vibr_spectrum_umd.py -f -t <温度>
ここで –t は、ユーザーが定義する必要がある温度です。このスクリプトは、2 つのファイルを出力します。vels.scf.dat 各アトミックタイプの VAC 関数を持つファイルと.vibr.dat 各原子種で分解された振動スペクトルと合計値を持つファイルです。 - を開いて、 vels.scf.datを読みます。スプレッドシートのようなソフトウェアを使用して vels.scf.dat ファイルからVAC関数をプロットします。
- フーリエVACの本当の部分を維持します。これは、周波数の関数として、振動スペクトルを生成するものです。

ここで m は原子の質量です。 - スプレッドシートのようなソフトウェアを使用して 、vibr.dat ファイルから振動スペクトルをプロットします(図7)。流体のディフューシャブ特性と有限周波数のスペクトルのさまざまなピークに対応する 、ω=0の有限値を特定します。振動スペクトルへの各原子型の参加を特定します。
注: 原子タイプの分解は、異なる原子が拡散係数に対応する 異なるω=0寄与を有することを示しています。スペクトルの一般的な形状は、対応するソリッドよりも少ないフィーチャではるかに滑らかです。 - シェルでは、各原子種の拡散係数をもたらす振動スペクトルの積分を読み取ります。
注:熱力学的特性は、振動スペクトルからの統合によって得ることができますが、結果は2つの近似のために注意して使用する必要があります:統合は、必ずしも高温で保持されない準高調波近似の範囲内で有効です。拡散に対応するスペクトルの気体状部分を廃棄する必要があります。統合は、スペクトルの格子状の部分でのみ行われるべきです。ただし、この分離には通常、さらにいくつかの処理後の手順と計算が必要です14、これは現在の UMD パッケージではカバーされません。
- vibr_spectrum_umd.pyスクリプトを実行して、各アトミックタイプの原子速度自動相関(VAC)関数を計算し、高速フーリエ変換を実行します。コマンド ラインで次のように入力します。
- viscosity_umd.pyスクリプトを実行して、成分応力テンソルの自己相関を分析して、溶融の粘度を推定します。コマンド ラインで次のように入力します。
viscosity_umd.py -f -i -s -o -l
注: この機能は探索的なものであり、結果は慎重に行う必要があります。まず、シミュレーションの長さに対する粘度の収束を十分に確認します。- ストレステンソル15 の自己相関から流体の粘度を次のように導出します。

ここで、VとTはそれぞれ体積と温度で、κBはボルツマン定数であり、σ ijはデカルト座標で表される応力テンソルのij対角外成分である。 - より適切な適合度を使用して、粘度15,16のより強い推定値を得て、有限サイズとシミュレーションの有限期間から生じる可能性のある応力-テンソル自動相関関数のノイズを回避します。応力テンソルの自動相関関数の場合は、良好な結果をもたらす次の機能 form15,16 を使用します。

ここで、A、B、τ1、τ2、およびωは適合パラメータです。 統合後、粘度の式は次のようになります。
- ストレステンソル15 の自己相関から流体の粘度を次のように導出します。
6.シミュレーションに由来する熱力学パラメータ。
- averages.py 実行して、圧力、温度、密度、および内部エネルギーの平均値と広がり(標準偏差)を umd ファイルから抽出します。コマンド ラインで次のように入力します。
averages.py -f -s
の値は 、デフォルトとして –s 0 を使用します。 - ブロッキングメソッドを使用して、平均の統計的誤差を計算します。
注:この方法にはさまざまな種類があります。アレンとティルデスリー2の仕事に続いて、時間ブロックのシーケンスにわたって平均を平均し、ますます長い、および算術平均17に関して標準偏差を推定することが一般的です。サンプリングが相関していない場合、収束は、多くて長いブロック サイズの限界に達する可能性があります。収束の実際のしきい値は通常手動で選択する必要があります。- halving method18を使用する:最初のデータサンプルから始まり、各ステップ のκで、前のステップ のκ-1から2つの対応する連続したサンプルごとに平均化することによってサンプルの数を半分にします。

- fullaverages.py スクリプトを実行して、平均の誤差を含む完全な統計分析を実行します。コマンド ラインで次のように入力します。
fullaverages.py -s -u < ユニット>
注: スクリプトは、現在のディレクトリ内のすべての .umd.dat ファイルを検索し、それらのすべての分析を実行する時点に自動化されています。デフォルトは –s 0 –u 0 です。u 0 の場合、出力は最小限であり、-u 1 の出力はフルであり、いくつかの代替単位が出力されます。このスクリプトは、平均値の誤差を推定するために収束をチェックするためのグラフィカルイメージを作成するため、グラフィカルなサポートが必要です。
- halving method18を使用する:最初のデータサンプルから始まり、各ステップ のκで、前のステップ のκ-1から2つの対応する連続したサンプルごとに平均化することによってサンプルの数を半分にします。
結果
パイロライトは、鉄系コア19を除き、地化学的平均または地球全体のバルクケイ酸塩地球の組成に最もよく近似するモデル多成分ケイ酸塩メルト(0.5Na2O 2CaO 1.5Al2O3 4FeO 30MgO 24SiO2)です。初期の地球は一連の大規模な融解事象20によって支配され、最後の地球は原始月の円盤21の結露の後、惑星全体を巻き込んだかもしれない。パイロライトは、このような惑星規模のマグマ海洋の組成に最適な近似マントを表しています。その結果、3,000\u20125,000 K温度範囲におけるパイロライト融解物の物理的性質と、VASP実装におけるab initio分子動力学シミュレーションからの0\u2012150 GPa圧力範囲を広範囲に研究しました。これらの熱力学的条件は、地球の最も極端なマグマ海洋条件を完全に特徴付けます。我々の研究は、melts22の全体の詳細な分析のためのUMDパッケージの成功した使用例です。分布と平均結合長を計算し、カチオンと酸素の協調の変化を追跡し、その結果を様々な組成のアモルファスケイ酸塩に関する以前の実験および計算研究と比較した。私たちの詳細な分析は、標準的な配位数を基本的な構成要素に分解し、溶融物中のエキゾチックな配位多面体の存在を概説し、すべての配位多面体の寿命を抽出するのに役立ちました。また、軌道の長さと、モデル化されたシステム内に存在する原子の数の両方の観点から、シミュレーションにおけるサンプリングの重要性を概説した。後処理に関しては、UMD 分析はこれらの要因とは無関係ですが、UMD パッケージによって提供される結果を解釈する際には、これらの要因を考慮する必要があります。ここでは、溶融したピロライトへの応用により、UMDパッケージを使用して溶融物のいくつかの特徴的な特徴を抽出する方法の例をいくつか示します。
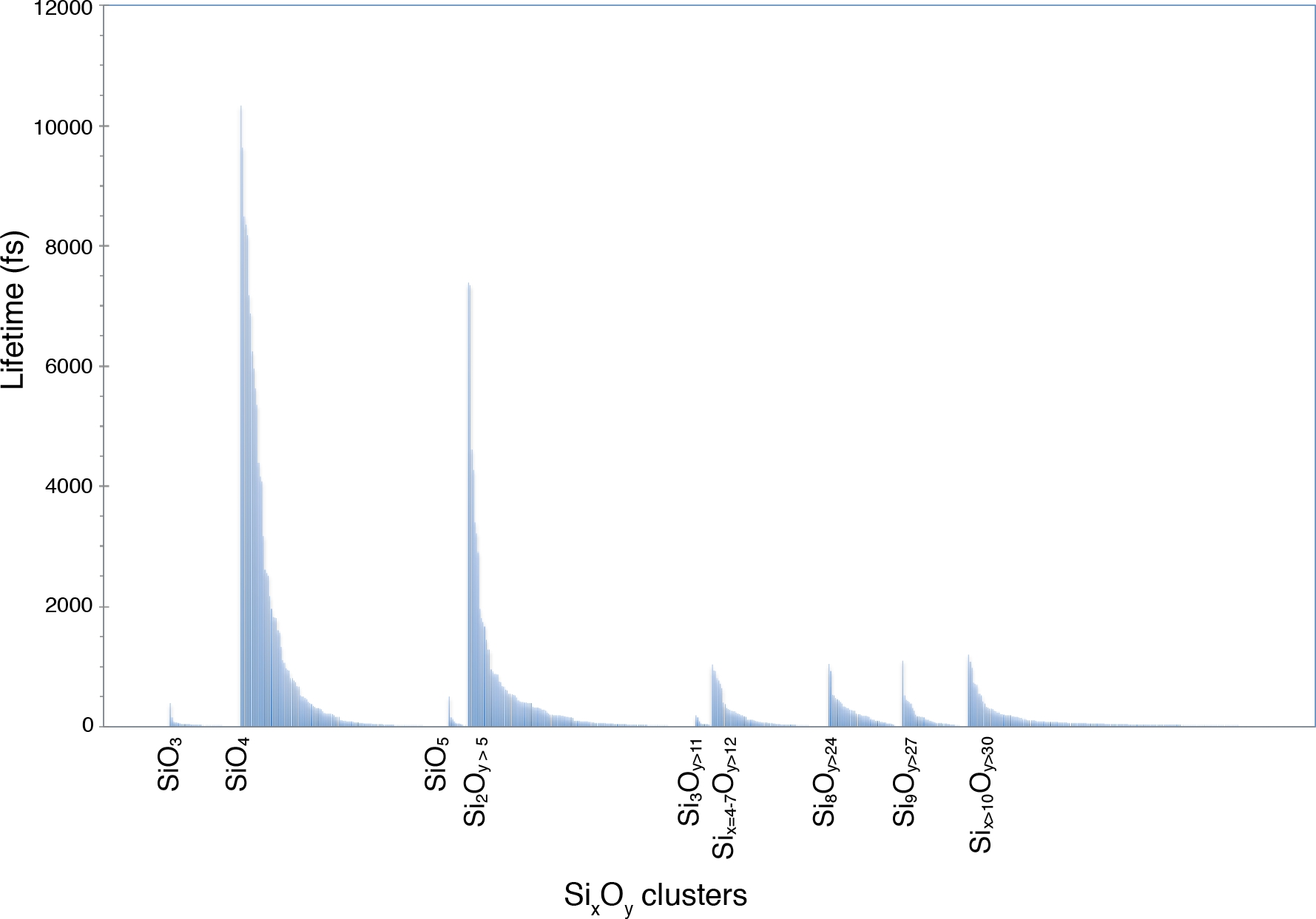
gofrs_umd.pyスクリプトから得られたSi-Oペア分布関数は、最初の配位球の半径(g(r)関数の最初の最小値)がT = 3000 KおよびP = 4.6 GPaで2.5オングストロームの周りにあることを示しています。g(r) の最大値は 1.635 Å にあり、これはベンド長に対する最適な近似値です。長い尾は温度によるものです。この限界をSi-O結合距離として使用すると、スペシテーション分析は、最大数ピコ秒まで持続できるSiO4単位が溶融物を支配することを示しています(図5)。Si2O7のようなダイマーやSi3Ox単位のような三量体の存在によって反映されるように、部分的な重合を示す溶融物の重要な部分があります。対応する寿命はピコ秒の順です。高次ポリマーはすべて、寿命がかなり短くなります。

図5:Si-O化学種の寿命
この種分化は、4.6 GPaおよび3000 Kの多成分融解で同定される。ラベルはSiO3、SiO4、SiO5モノマーと様々なSixOyポリマーを示しています。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
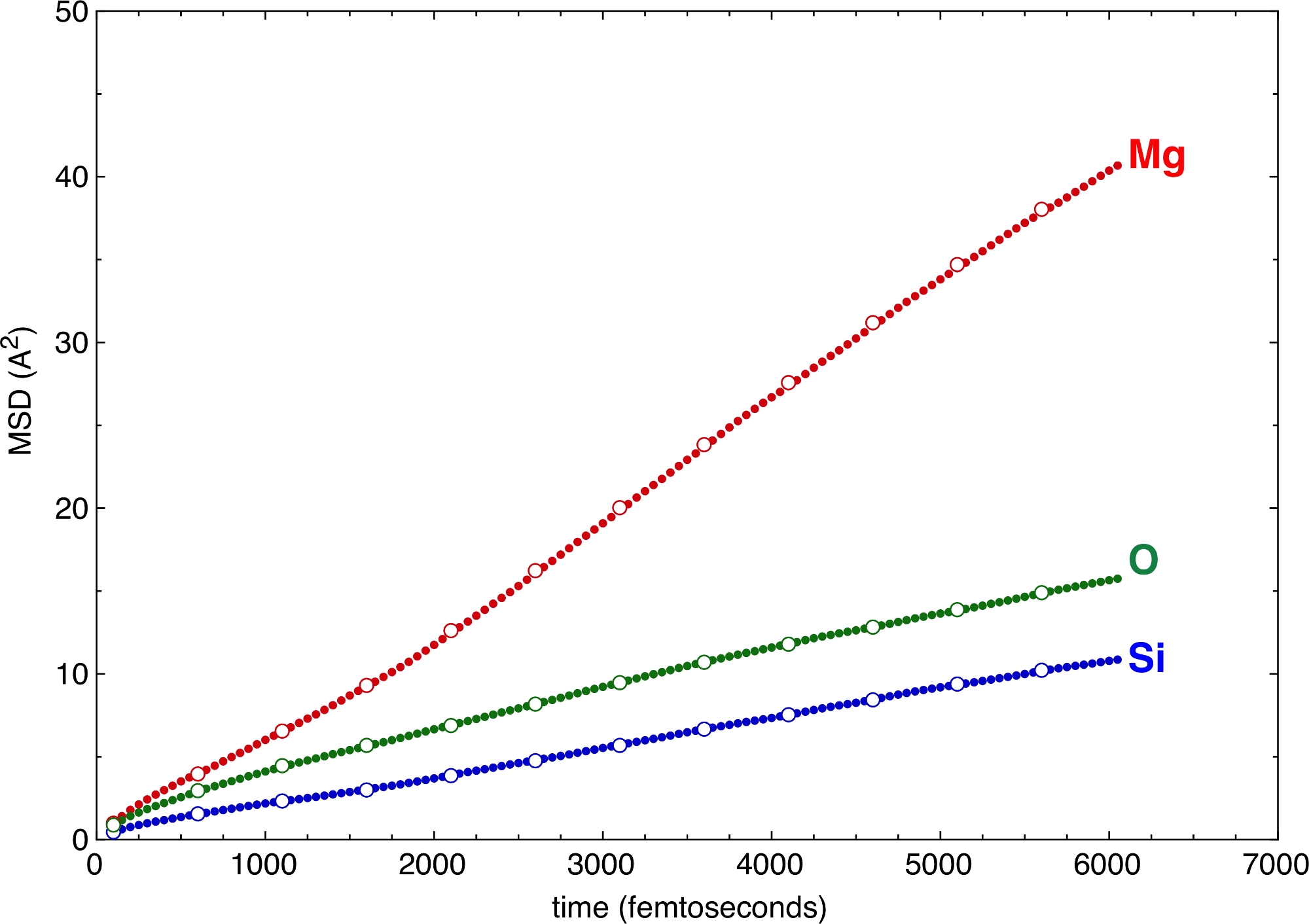
上の –z フラグと –v フラグで定義された垂直ステップと水平ステップの異なる値は、MSD のさまざまなサンプリングを生成します(図 6)。z と v の値が大きくても、傾きや異なる原子の拡散係数を定義するのに十分です。z と v の大きな値に行く場合、後処理の時間の増加は顕著です。MSDは、シミュレーションの品質に対して非常に強力な検証基準を提供します。MSD の拡散部分が十分に長くない場合、これはシミュレーションが短すぎるというサインであり、統計的な意味で流体状態に達しない。MSD のディフューシス部分の最小要件は、システムによって大きく異なります。1つは、すべての原子が流体10として考慮されるために、溶融物の構造において少なくとも一度は部位を変化させる必要がある。惑星科学の応用の優れた例は、その液体ライン11に近いか、あるいは下の高圧での複雑なケイ酸塩溶融物である。Si原子、主要なネットワーク形成カチオンは、2ダース以上のピコ秒の後にサイトを切り替えます。このしきい値より短いシミュレーションは、可能な構成空間をかなり低サンプリングします。しかし、協調陰イオンすなわちO原子は、中央Si原子よりも速く動くように、Siの移動が遅い部分を補うことができる。したがって、システム全体は、Si変位からのみ想定されるよりも、構成空間のより良いサンプリングを実際にカバーすることができます。

図 6: 平均二乗変位(MSD)
MSDは、多成分ケイ酸塩溶融物のいくつかの原子タイプについて示されている。さまざまな水平および垂直ステップを持つサンプリング、zとvは、一貫した結果を生成します。実円: -z 50 –v 50.開いた円: -z 250 -v 500. この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
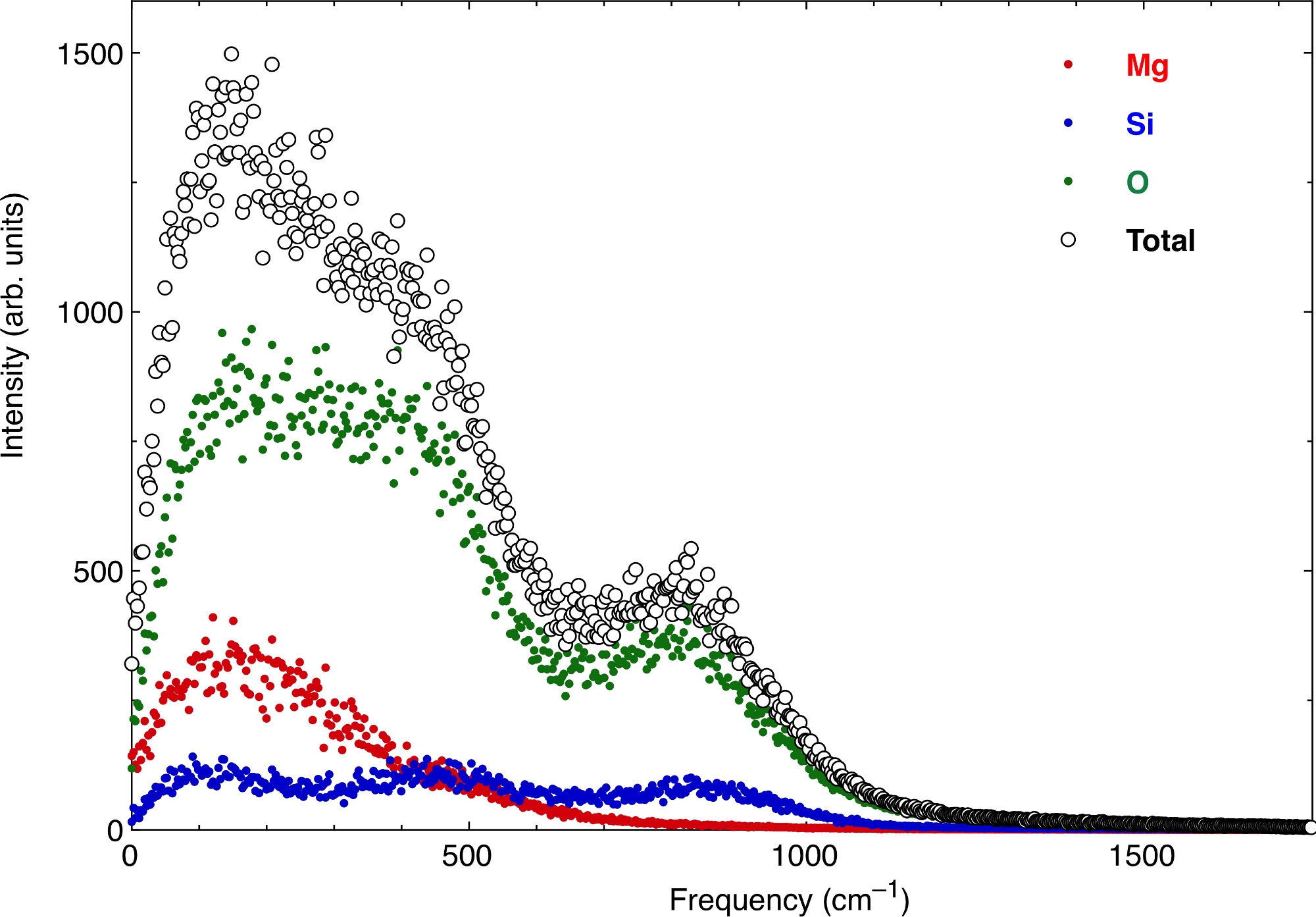
最後に、原子VAC関数は、溶融物の振動スペクトルを生み出す。 図7 は、上記と同じ圧力条件と温度条件でのスペクトルを示しています。Mg、Si、O原子の寄与と合計値を表します。ゼロ周波数では、融解の拡散特性に対応するスペクトルの有限値がある。振動スペクトルからの熱力学的特性の抽出は、このガス状の拡散特性をゼロから除去するだけでなく、より高い周波数での減衰を適切に考慮する必要があります。

図7:熱火物の振動スペクトルが溶ける。
原子速度自己相関関数のフーリエ変換の実部分は、振動スペクトルを生成します。 ここでスペクトルは、多成分ケイ酸塩溶融物について計算される。流体はゼロ周波数でゼロガス状の拡散特性を有する。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
UMD パッケージは、ab initio シミュレーションで、スナップショットの数が通常数十から数十万のスナップショットに制限され、ユニット セルあたり数百原子を使用する場合に、より適切に機能するように設計されています。ポストプロセスが実行されるマシンに十分なアクティブなメモリリソースがある場合、より大規模なシミュレーションも使用できます。コードは、計算できるさまざまなプロパティと、オープンソース ライセンスによって区別されます。
umd.dat ファイルは、シミュレーション全体で変更されないパーティクルの数を保持するアンサンブルに適しています。UMD パッケージは、シミュレーション ボックスの形状と体積が変化する計算に由来するファイルを読み取ることができます。これらは、NVT や NPT のような最も一般的な計算をカバーし、パーティクル、N、温度 T、体積、V、および/または圧力の数は一定に保たれています。
時間のペア分布関数だけでなく、種分化スクリプトのように、間の距離を推定する必要があるすべてのスクリプトは、軸間の角度が90°である3次、四角形、および直交性細胞を意味する直交単位セルでのみ動作します。
バージョン2.0の主な開発ラインは、距離の直交性制限の除去と、個々の化学結合の分析、原子間の角度の分析、第2の配位球の実装という、種分化スクリプトの機能の追加です。外部のコラボレーションの助けを借りて、我々は大規模なシステムで迅速な分析のためにGPUにコードを移植することに取り組んでいます。
開示事項
著者らは開示するものは何もない。
謝辞
この研究は、欧州連合ホライズン2020研究イノベーションプログラム(RCへのIMPACT 681818合意番号)、深炭素天文台のエクストリーム物理学化学局、およびノルウェー研究評議会がエクセレンスセンターの資金調達スキームを通じて、プロジェクト番号223272に支援しました。我々は、Stl2816シリーズのeDARIコンピューティング補助金を通じたGENCIスーパーコンピュータへのアクセス、PRACE RA4947プロジェクトを通じたアイリーンAMDスーパーコンピュータ、およびUNINETTシグマ2 NN9697Kを通じたフラムスーパーコンピュータへのアクセスを認める。FSはマリー・スクロトフスカ・キュリー・プロジェクト(補助金ABISSE No.750901)によって支援されました。
資料
| Name | Company | Catalog Number | Comments |
| getopt library | open-source | ||
| glob library | open-source | ||
| matplotlib library | open-source | ||
| numpy library | open-source | ||
| os library | open-source | ||
| Python software | The Python Software Foundation | Version 2 and 3 | open-source |
| random library | open-source | ||
| re library | open-source | ||
| scipy library | open-source | ||
| subprocess library | open-source | ||
| sys library | open-source |
参考文献
- Frenkel, D., Smit, B. . Understanding Molecular Simulation. From Algorithms to Applications. , (2001).
- Allen, M. P., Tildesley, D. J., Allen, T. . Computer Simulation of Liquids. , (1989).
- Zepeda-Ruiz, L. A., Stukowski, A., Oppelstrup, T., Bulatov, V. V. Probing the limits of metal plasticity with molecular-dynamics simulations. Nature Publishing Group. 550 (7677), 492-495 (2017).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modeling. 14 (1), 33-38 (1996).
- Momma, K., Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 44 (6), 1272-1276 (2011).
- Brehm, M., Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. Journal of Chemical Information and Modeling. 51 (8), 2007-2023 (2011).
- Stixrude, L. Visualization-based analysis of structural and dynamical properties of simulated hydrous silicate melt. Physics and Chemistry of Minerals. 37 (2), 103-117 (2009).
- Kresse, G., Hafner, J. Ab initio Molecular-Dynamics for Liquid-Metals. Physical Review B. 47 (1), 558-561 (1993).
- Gygi, F. Architecture of Qbox: A scalable first-principles molecular dynamics code. IBM Journal of Research and Development. 52 (1-2), 137-144 (2008).
- Harvey, J. P., Asimow, P. D. Current limitations of molecular dynamic simulations as probes of thermo-physical behavior of silicate melts. American Mineralogist. 100 (8-9), 1866-1882 (2015).
- Caracas, R., Hirose, K., Nomura, R., Ballmer, M. D. Melt-crystal density crossover in a deep magma ocean. Earth and Planetary Science Letters. 516, 202-211 (2019).
- Green, M. S. Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena. II. Irreversible Processes in Fluids. The Journal of Chemical Physics. 22 (3), 398-413 (1954).
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. Journal of the Physical Society of Japan. 12 (6), 570-586 (1957).
- Lin, S. T., Blanco, M., Goddard, W. A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. The Journal of Chemical Physics. 119 (22), 11792-11805 (2003).
- Meyer, E. R., Kress, J. D., Collins, L. A., Ticknor, C. Effect of correlation on viscosity and diffusion in molecular-dynamics simulations. Physical Review E. 90 (4), 1198-1212 (2014).
- Soubiran, F., Militzer, B., Driver, K. P., Zhang, S. Properties of hydrogen, helium, and silicon dioxide mixtures in giant planet interiors. Physics of Plasmas. 24 (4), 041401-041407 (2017).
- Flyvbjerg, H., Petersen, H. G. Error estimates on averages of correlated data. The Journal of Chemical Physics. 91 (1), 461-466 (1989).
- Tuckerman, M. E. . Statistical mechanics: theory and molecular simulation. , (2010).
- McDonough, W. F., Sun, S. S. The composition of the Earth. Chemical Geology. 120, 223-253 (1995).
- Elkins-Taton, L. T. Magma oceans in the inner solar system. Annual Review of Earth and Planetary Sciences. 40, 113-139 (2012).
- Lock, S. J., et al. The origin of the Moon within a terrestrial synestia. J. Geophysical Research: Planets. 123, 910-951 (2018).
- Solomatova, N. V., Caracas, R. Pressure-induced coordination changes in a pyrolitic silicate melt from ab initio molecular dynamics simulations. Journal of Geophysical Research: Solid Earth. 124, 11232-11250 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved