
Method Article
Analisi di fusioni e fluidi da Ab Initio Molecular Dynamics Simulations con il pacchetto UMD
In questo articolo
Riepilogo
Fusioni e fluidi sono vettori onnipresenti del trasporto di massa nei sistemi naturali. Abbiamo sviluppato un pacchetto open source per analizzare le simulazioni di dinamica molecolare ab initio di tali sistemi. Calcoliamo le proprietà strutturali (legame, clusterizzazione, speciazione chimica), trasporto (diffusione, viscosità) e termodinamiche (spettro vibrazionale).
Abstract
Abbiamo sviluppato un pacchetto open source basato su Python per analizzare i risultati derivanti dalle simulazioni di dinamica molecolare ab initio dei fluidi. Il pacchetto è più adatto per applicazioni su sistemi naturali, come silicati e fusioni di ossido, fluidi a base d'acqua e vari fluidi supercritici. Il pacchetto è una raccolta di script Python che includono due librerie principali che si occupano di formati di file e cristallografia. Tutti gli script vengono eseguiti dalla riga di comando. Proponiamo un formato semplificato per memorizzare le traiettorie atomiche e le informazioni termodinamiche rilevanti delle simulazioni, che vengono salvate in file UMD, che sta per Universal Molecular Dynamics. Il pacchetto UMD consente il calcolo di una serie di proprietà strutturali, di trasporto e termodinamiche. Partendo dalla funzione di distribuzione delle coppie, definisce le lunghezze dei legami, costruisce una matrice di connettività interatomica e infine determina la speciazione chimica. Determinare la durata della specie chimica consente di eseguire un'analisi statistica completa. Quindi gli script dedicati calcolano gli spostamenti quadrati medi per gli atomi e per le specie chimiche. L'analisi di auto-correlazione implementata delle velocità atomiche produce i coefficienti di diffusione e lo spettro vibrazionale. La stessa analisi applicata sulle sollecitazioni produce la viscosità. Il pacchetto è disponibile tramite il sito Web GitHub e tramite la propria pagina dedicata del progetto ERC IMPACT come pacchetto ad accesso aperto.
Introduzione
Fluidi e fusioni sono vettori di trasporto chimico e fisico attivi in ambienti naturali. Gli elevati tassi di diffusione atomica favoriscono gli scambi e le reazioni chimiche, la bassa viscosità unita alla variabile galleggiabilità favoriscono il trasferimento di massa di grandi dimensioni e le relazioni di densità cristallo-fusione favoriscono la stratificazione all'interno dei corpi planetari. L'assenza di un reticolo periodico, le tipiche alte temperature richieste per raggiungere lo stato fuso e la difficoltà di tempra rendono estremamente impegnativa la determinazione sperimentale di una serie di proprietà evidenti, come densità, diffusione e viscosità. Queste difficoltà rendono i metodi computazionali alternativi strumenti forti e utili per studiare questa classe di materiali.
Con l'avvento della potenza di calcolo e la disponibilità di supercomputer, due importanti tecniche di simulazione atomistica numerica sono attualmente impiegate per studiare lo stato dinamico di un sistema atomistico non cristallino, Monte Carlo1 e la dinamica molecolare (MD)1,2. Nelle simulazioni Monte Carlo lo spazio configurazionale viene campionato in modo casuale; I metodi Monte Carlo mostrano il ridimensionamento lineare in parallelizzazione se tutte le osservazioni di campionamento sono indipendenti l'una dall'altra. La qualità dei risultati dipende dalla qualità del generatore di numeri casuali e dalla rappresentatività del campionamento. I metodi Monte Carlo mostrano il ridimensionamento lineare in parallelizzazione se il campionamento è indipendente l'uno dall'altro. In dinamica molecolare (MD) lo spazio configurazionale è campionato da traiettorie atomiche dipendenti dal tempo. Partendo da una data configurazione, le traiettorie atomiche vengono calcolate integrando le equazioni newtoniane del moto. Le forze interatomiche possono essere calcolate usando i potenziali interatomici del modello (nella MD classica) o usando metodi di principi primi (in ab initio, o principi primi, MD). La qualità dei risultati dipende dalla lunghezza della traiettoria e dalla sua capacità di non essere attratta dai minimi locali.
Le simulazioni di dinamica molecolare contengono una pletora di informazioni, tutte relative al comportamento dinamico del sistema. Le proprietà medie termodinamiche, come l'energia interna, la temperatura e la pressione, sono piuttosto standard da calcolare. Possono essere estratti dai file di output delle simulazioni e mediati, mentre le quantità direttamente correlate al movimento degli atomi e alla loro relazione reciproca devono essere calcolate dopo l'estrazione delle posizioni e delle velocità atomiche.
Di conseguenza, un grande sforzo è stato dedicato alla visualizzazione dei risultati, e vari pacchetti sono disponibili oggi, su diverse piattaforme, open source o meno [Ovito3, VMD4, Vesta5, Travis6, ecc.]. Tutti questi strumenti di visualizzazione gestiscono in modo efficiente le distanze interatomiche e, come tali, consentono il calcolo efficiente delle funzioni di distribuzione delle coppie e dei coefficienti di diffusione. Vari gruppi che eseguono simulazioni di dinamica molecolare su larga scala hanno un software proprietario per analizzare varie altre proprietà risultanti dalle simulazioni, a volte in shareware o altre forme di accesso limitato alla comunità, e talvolta limitate nella portata e nell'uso di alcuni pacchetti specifici. Algoritmi sofisticati per estrarre informazioni sul legame interatomico, modelli geometrici e termodinamica sono sviluppati e implementati in alcuni di questi pacchetti3,4,5,6,7, ecc.
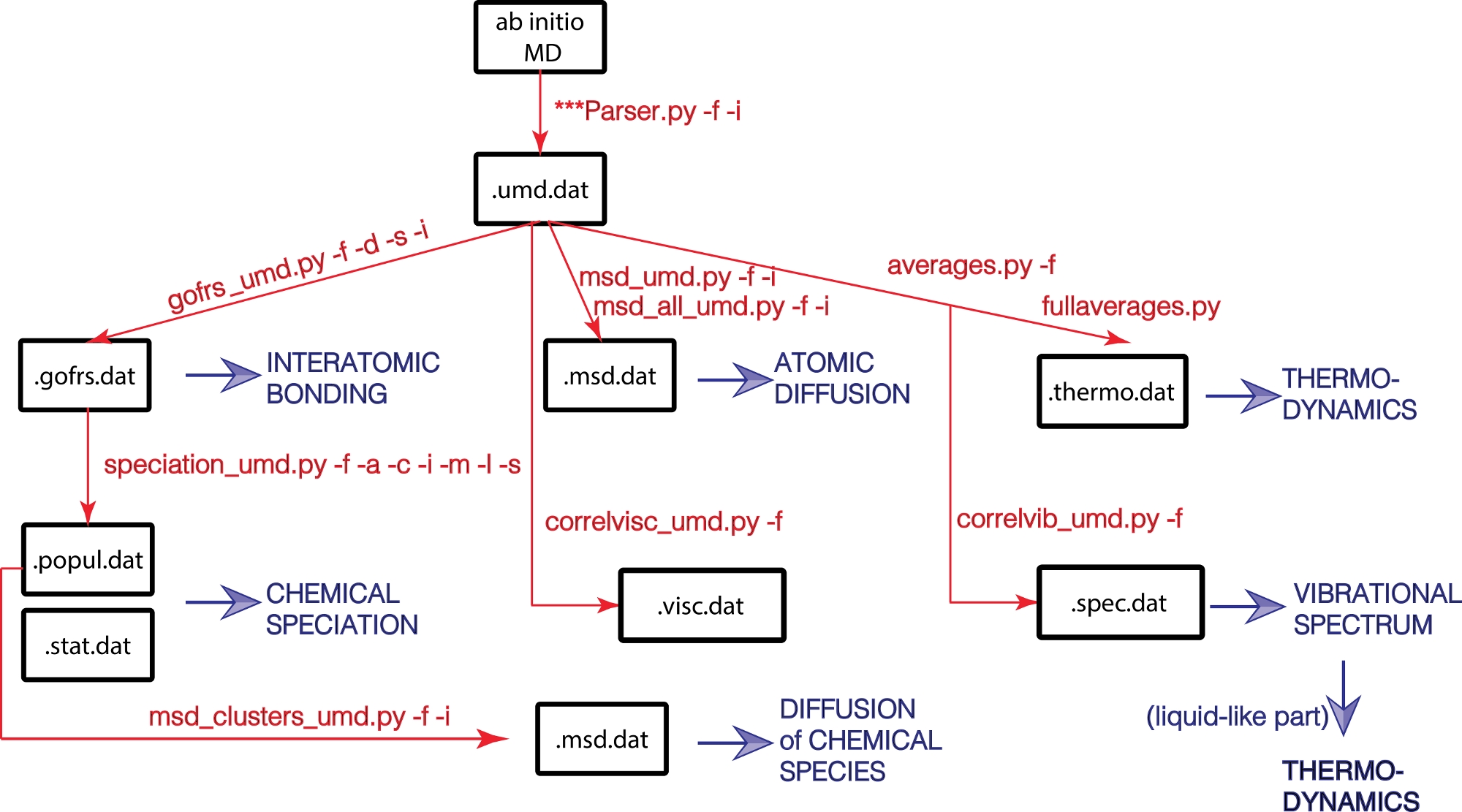
Qui proponiamo il pacchetto UMD - un pacchetto open source scritto in Python per analizzare l'output delle simulazioni di dinamica molecolare. Il pacchetto UMD consente il calcolo di un'ampia gamma di proprietà strutturali, dinamiche e termodinamiche (Figura 1). Il pacchetto è disponibile tramite il sito Web GitHub (https://github.com/rcaracas/UMD_package) e tramite una pagina dedicata (http://moonimpact.eu/umd-package/) del progetto ERC IMPACT come pacchetto ad accesso aperto.
Per renderlo universale e più facile da gestire, il nostro approccio è quello di estrarre prima tutte le informazioni relative allo stato termodinamico e alle traiettorie atomiche dal file di output dell'effettiva esecuzione di dinamica molecolare. Queste informazioni sono memorizzate in un file dedicato, il cui formato è indipendente dal pacchetto MD originale in cui è stata eseguita la simulazione. Chiamiamo questi file file "umd", che sta per Universal Molecular Dynamics. In questo modo, il nostro pacchetto UMD può essere facilmente utilizzato da qualsiasi gruppo ab initio con qualsiasi software, il tutto con un minimo sforzo di adattamento. L'unico requisito per utilizzare il pacchetto attuale è quello di scrivere il parser appropriato dall'output del particolare software MD nel formato di file umd, se questo non è ancora esistente. Per il momento, forniamo tali parser per i pacchetti VASP8 e QBox9 .

Figura 1: Diagramma di flusso della libreria UMD.
Le proprietà fisiche sono in blu e i principali script Python e le loro opzioni sono in rosso. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
I file umd sono file ASCII; l'estensione tipica è "umd.dat" ma non obbligatoria. Tutti i componenti di analisi possono leggere i file ASCII del formato umd, indipendentemente dall'estensione effettiva del nome. Tuttavia, alcuni degli script automatici progettati per eseguire statistiche veloci su larga scala su diverse simulazioni cercano specificamente i file con l'estensione umd.dat. Ogni proprietà fisica è espressa su una riga. Ogni riga inizia con una parola chiave. In questo modo il formato è altamente adattabile e consente di aggiungere nuove proprietà al file umd, preservandone la leggibilità in tutte le versioni. Le prime 30 righe del file umd della simulazione della pirolite a 4,6 GPa e 3000 K, utilizzate di seguito nella discussione, sono mostrate nella Figura 2.

Figura 2: L'inizio del file umd che descrive la simulazione della pirolite liquida a 4,6 GPa e 3000 K.
L'intestazione è seguita dalla descrizione di ogni istantanea. Ogni proprietà è scritta su una riga, contenente il nome della proprietà fisica, i valori e le unità, tutte separate da spazi. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tutti i file umd contengono un'intestazione che descrive il contenuto della cella di simulazione: il numero di atomi, elettroni e tipi atomici, nonché dettagli per ciascun atomo, come il suo tipo, simbolo chimico, numero di elettroni di valenza e la sua massa. Una riga vuota segna la fine dell'intestazione e la separa dalla parte principale del file umd.
Quindi ogni fase della simulazione è dettagliata. In primo luogo, vengono forniti i parametri termodinamici istantanei, ciascuno su una linea diversa, specificando (i) il nome del parametro, come energia, sollecitazioni, pressione idrostatica equivalente, densità, volume, parametri reticolari, ecc., (ii) i suoi valori e (iii) le sue unità. Viene dopo una tabella che descrive gli atomi. Una riga di intestazione fornisce le diverse misure, come le posizioni cartesiane, le velocità, le cariche, ecc. E le loro unità. Quindi ogni atomo è dettagliato su una riga. Per gruppi di tre, corrispondenti ai tre assi x, y, z , le voci sono: le posizioni ridotte, le posizioni cartesiane ripiegate nella cella di simulazione, le posizioni cartesiane (che tengono correttamente conto del fatto che gli atomi possono attraversare diverse celle unitarie durante una simulazione), le velocità atomiche e le forze atomiche. Le ultime due voci sono scalari: carica e momento magnetico.
Due librerie principali garantiscono il corretto funzionamento dell'intero pacchetto. La libreria umd_process.py si occupa dei file umd, come la lettura e la stampa. La biblioteca crystallography.py si occupa di tutte le informazioni relative all'effettiva struttura atomica. La filosofia alla base della biblioteca crystallography.py è quella di trattare il reticolo come uno spazio vettoriale. I parametri della cella unitaria insieme al loro orientamento rappresentano i vettori di base. Lo "Spazio" ha una serie di attributi scalari (volume specifico, densità, temperatura e numero specifico di atomi), proprietà termodinamiche (energia interna, pressione, capacità termica, ecc.) e una serie di proprietà tensoriali (stress ed elasticità). Gli atomi popolano questo spazio. La classe "Reticolo" definisce questo insieme, insieme a vari calcoli brevi, come volume specifico, densità, ottenendo il reticolo reciproco da quello diretto, ecc. La classe "Atomi" definisce gli atomi. Sono caratterizzati da una serie di proprietà scalari (nome, simbolo, massa, numero di elettroni, ecc.) e da una serie di proprietà vettoriali (la posizione nello spazio, sia relativa alla base vettoriale descritta nella classe Lattice, sia relativa alle coordinate cartesiane universali, velocità, forze, ecc.). Oltre a queste due classi, la libreria crystallography.py contiene una serie di funzioni per eseguire una varietà di test e calcoli, come le distanze atomiche o la moltiplicazione cellulare. La tavola periodica degli elementi è anche inclusa come dizionario.
I vari componenti del pacchetto umd scrivono diversi file di output. Come regola generale, sono tutti file ASCII, tutte le loro voci sono separate da schede e sono rese il più autoesplicative possibile. Ad esempio, indicano sempre chiaramente la proprietà fisica e le sue unità. I file umd.dat rispettano pienamente questa regola.
Protocollo
1. Analisi delle corse di dinamica molecolare
NOTA: Il pacchetto è disponibile tramite il sito Web GitHub (https://github.com/rcaracas/UMD_package) e tramite una pagina dedicata (http://moonimpact.eu/umd-package/) del progetto ERC IMPACT come pacchetto ad accesso aperto.
- Estrarre ogni set specifico di proprietà fisiche utilizzando uno o più script Python dedicati dal pacchetto. Eseguire tutti gli script dalla riga di comando; tutti impiegano una serie di bandiere, che sono il più coerenti possibile da uno script all'altro. I flag, il loro significato e i valori predefiniti sono tutti riepilogati nella Tabella 1.
| Bandiera | Significato | Script che lo utilizza | Valore predefinito |
| -h | Breve aiuto | tutto | |
| -f | Nome file UMD | tutto | |
| -i | Fasi di termalizzazione da scartare | tutto | 0 |
| -i | File di input contenente i legami interatomici | speciazione | bonds.input |
| -s | Campionamento della frequenza | msd, speciazione | 1 (ogni passo è considerato) |
| -a | Lista di atomi o anioni | speciazione | |
| -c | Elenco dei cationi | speciazione | |
| -l | Lunghezza del legame | speciazione | 2 |
| -t | Temperatura | vibrazioni, reologia | |
| -v | Discretizzazione della larghezza della finestra di campionamento della traiettoria per l'analisi dello spostamento medio-quadrato | Msd | 20 |
| -z | Discretizzazione dell'inizio della finestra di campionamento della traiettoria per l'analisi dello spostamento medio-quadrato | Msd | 20 |
Tabella 1: Flag più comuni utilizzati nel pacchetto UMD e loro significato più comune.
- Inizia trasformando l'output della simulazione MD eseguita in un codice di principi primi, come VASP8 o QBox9, in un file UMD.
- Se le simulazioni MD sono state eseguite in VASP, digitare nella riga di comando:
VaspParser.py -f -i
dove il flag –f definisce il nome del file VASP OUTCAR e la –i la lunghezza di termalizzazione.
NOTA: Il passaggio iniziale, definito da –i consente di scartare i primi passi delle simulazioni, che rappresentano la termalizzazione. In una tipica esecuzione di dinamica molecolare, la prima parte del calcolo rappresenta la termalizzazione, cioè il tempo impiegato dal sistema per tutti gli atomi per descrivere una distribuzione gaussiana della temperatura e per l'intero sistema per mostrare fluttuazioni di temperatura, pressione, energia, ecc. intorno ai valori di equilibrio. Questa parte di termalizzazione della simulazione non deve essere presa in considerazione quando si analizzano le proprietà statistiche del fluido.
- Se le simulazioni MD sono state eseguite in VASP, digitare nella riga di comando:
- Trasformare il file . file umd in . xyz per facilitare la visualizzazione su vari altri pacchetti, come VMD4 o Vesta5. Nella riga di comando digitare:
umd2xyz.py -f -i -s
dove –f definisce il nome di . umd file, –i definisce il periodo di termalizzazione da scartare e –s la frequenza del campionamento della traiettoria memorizzata nel file . umd . I valori predefiniti sono –i 0 –s 1, cioè considerando tutte le fasi della simulazione, senza che nessuno venga scartato. - Invertire il file umd in file POSCAR di tipo VASP utilizzando lo script umd2poscar.py; le istantanee delle simulazioni possono essere selezionate con una frequenza predefinita. Nella riga di comando digitare:
umd2poscar.py -f -i -l -s
dove –l rappresenta l'ultimo passaggio da trasformare in file POSCAR. I valori predefiniti sono -i 0 -l 10000000 -s 1. Questo valore di –l è abbastanza grande da coprire una tipica traiettoria intera.
2. Eseguire l'analisi strutturale
- Eseguire lo script gofrs_umd.py per calcolare la funzione di distribuzione delle coppie (PDF) gᴀʙ(r) per tutte le coppie di tipi atomici A e B (Figura 3). L'output è scritto in un file ASCII, separato da tabulazioni, con estensione gofrs.dat. Nella riga di comando digitare:
gofrs_umd.py -f -s < Sampling_Frequency > -d -i
NOTA: i valori predefiniti sono Sampling_Frequency (la frequenza per il campionamento della traiettoria) = 1 passo; DiscretizzazioneInterval (per tracciare il g(r)) = 0,01 Å; InitialStep (numero di passi all'inizio della traiettoria scartati) = 0. Il PDF radiale, gᴀʙ(r) è il numero medio di atomi di tipo B ad una distanza d_ᴀʙ all'interno di un guscio sferico di raggio r e spessore dr centrato sugli atomi di tipo A (Figura 3):
con ρ la densità atomica, NA e NB il numero di atomi di tipo A e B, e δ(r−rᴀʙ) la funzione delta che è uguale a 1 se gli atomi A e B si trovano ad una distanza tra r e r+dr. L'ascissa del primo massimo di gᴀʙ(r) dà la più alta lunghezza di legame di probabilità tra gli atomi di tipo A e B, che è la più vicina a una distanza media di legame che possiamo determinare. Il primo minimo delimita l'estensione della prima sfera di coordinamento. Quindi l'integrale sul PDF fino al primo minimo dà il numero medio di coordinamento. La somma delle trasformazioni di Fourier della gᴀʙ(r) per tutte le coppie di tipi atomici A e B produce il modello di diffrazione del fluido, ottenuto sperimentalmente con un diffrattometro. Tuttavia, in realtà, poiché spesso le sfere di coordinamento di alto ordine mancano dal gᴀʙ(r), il modello di diffrazione non può essere ottenuto nella sua interezza.

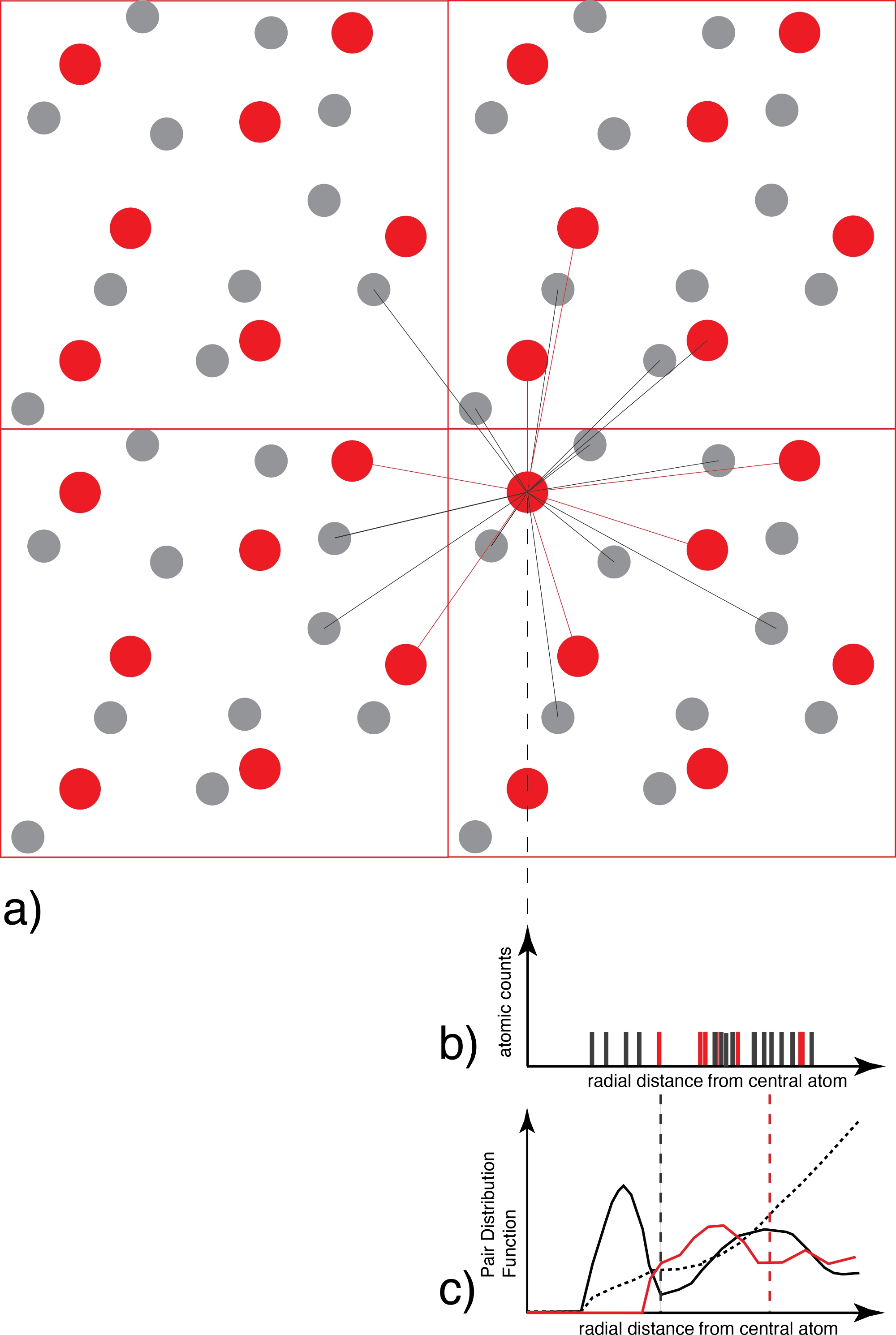
Figura 3: Determinazione della funzione di distribuzione delle coppie.
a) Per ogni atomo di una specie (ad esempio rosso), tutti gli atomi della specie coordinatrice (ad esempio grigio e/o rosso) sono conteggiati in funzione della distanza. (b) Il grafico di distribuzione della distanza risultante per ogni istantanea, che in questa fase è solo una raccolta di funzioni delta, viene quindi mediato su tutti gli atomi e tutte le istantanee e ponderato dalla distribuzione ideale del gas per generare (c) la funzione di distribuzione della coppia che è continua. Il primo minimo del g(r) è il raggio della prima sfera di coordinazione, utilizzato successivamente nell'analisi di speciazione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Estrarre le distanze medie di legame interatomico come raggi delle prime sfere di coordinazione. Per questo, identifica la posizione del primo massimo delle funzioni gᴀʙ(r): traccia il file gofrs.dat in un'applicazione per fogli di calcolo e cerca i massimi e i minimi per ogni coppia di atomi.
- Identificare il raggio della prima sfera di coordinamento, come primo minimo del PDF, gᴀʙ(r), utilizzando un software per fogli di calcolo. Questa è la base per l'intera analisi strutturale del fluido; il PDF restituisce lo stato medio di legame degli atomi nel fluido.
- Estrai le distanze dei primi minimi, cioè l'ascissa, e scrivili in un file separato, chiamato, ad esempio, bonds.input. In alternativa, eseguire uno degli script analyze_gofr del pacchetto UMD per identificare i massimi e i minimi delle funzioni gᴀʙ(r). Nella riga di comando digitare:
analyze_gofr_semi_automatic.py - Fare clic sulla posizione del massimo e del minimo della funzione gᴀʙ(r) visualizzata nel grafico che viene aperto dal programma. Lo script esegue automaticamente la scansione della cartella corrente, identifica tutti i file .dat e esegue l'analisi per ciascuno di essi. Fai di nuovo clic sul massimo e sul minimo nella finestra ogni volta che lo script ha bisogno di un'ipotesi iniziale istruita.
- Apri e guarda il file generato automaticamente chiamato bonds.input che contiene le distanze di legame interatomico.
3. Eseguire l'analisi di speciazione
- Calcola la topologia del legame tra gli atomi, usando il concetto di connettività all'interno della teoria dei grafi: gli atomi sono i nodi e i legami interatomici sono i percorsi. Lo script speciation_umd.py richiede le distanze di legame interatomico definite nel file bonds.input .
NOTA: La matrice di connettività è costruita ad ogni passo temporale: due atomi che si trovano a una distanza inferiore al raggio della loro corrispondente prima sfera di coordinazione sono considerati legati, cioè collegati. Varie reti atomiche sono costruite trattando gli atomi come nodi in un grafo le cui connessioni sono definite da questo criterio geometrico. Queste reti sono le specie atomiche e il loro insieme definisce la speciazione atomica in quel particolare fluido (Figura 4).

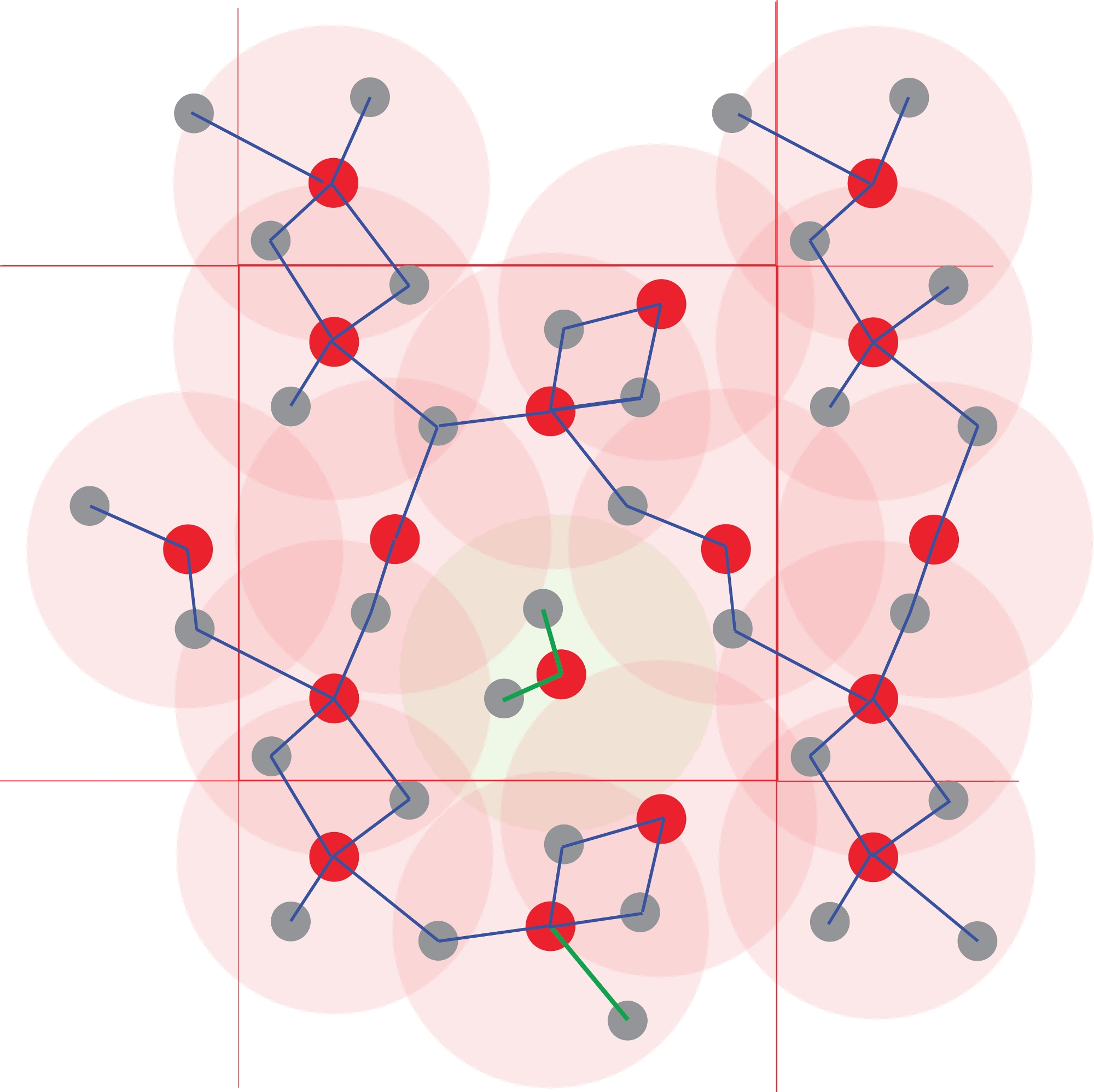
Figura 4: Identificazione degli ammassi atomici.
I poliedri di coordinazione sono definiti utilizzando distanze interatomiche. Tutti gli atomi a una distanza inferiore a un raggio specificato sono considerati legati. Qui la soglia corrisponde alla prima sfera di coordinazione (i cerchi rossi chiari), definita in Figura 1. La polimerizzazione e quindi le specie chimiche sono ottenute dalle reti degli atomi legati. Si noti il cluster centrale Red1Grey2, che è isolato dagli altri atomi, che formano un polimero infinito. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Eseguire lo script di speciazione per ottenere la matrice di connettività e ottenere il poliedro di coordinazione o la polimerizzazione. Nella riga di comando digitare:
speciation_umd.py -f -s -i -l -c -a -m -r
dove il flag -i fornisce al file le distanze di legame interatomico, che è stato prodotto ad esempio nel passaggio precedente. In alternativa, eseguire lo script con una sola lunghezza per tutti i vincoli definiti dal flag -l.
NOTA: il flag -c specifica gli atomi centrali e il flag -a i ligandi. Sia gli atomi centrali che i ligandi possono essere di diverso tipo; in questo caso, devono essere separati da virgole. La bandiera -m indica il tempo minimo di vita di una specie per essere considerata nell'analisi. Per impostazione predefinita, questo tempo minimo è zero, tutte le occorrenze vengono conteggiate in ultima analisi.- Eseguire lo script speciation_umd.py con il flag –r 0, che campiona il grafico di connettività al primo livello per identificare i poliedri di coordinazione. Ad esempio, un atomo centrale, indicato come catione , può essere circondato da uno o più anioni (Figura 4). Lo script di speciazione identifica ogni singolo poliedro di coordinazione. La media ponderata di tutti i poliedri di coordinazione dà il numero di coordinazione, identico a quello ottenuto dall'integrazione del PDF. Nella riga di comando digitare:
speciation_umd.py -f -i -c -a -r 0
NOTA: i numeri medi di coordinazione nei fluidi sono numeri frazionari. Questa frazionalità deriva dalla caratteristica media della coordinazione. La definizione basata sulla speciazione produce una rappresentazione più intuitiva e informativa della struttura del fluido, dove vengono quantificate le proporzioni relative delle diverse specie, cioè le coordinazioni. - Eseguire lo script speciation_umd.py con il flag –r 1, che campiona il grafico di connettività a tutti i livelli di profondità per ottenere la polimerizzazione. La rete attraverso il grafo atomico ha una certa profondità, poiché gli atomi sono legati più lontano ad altri legami (ad esempio, in sequenze di cationi e anioni alternati) (Figura 4).
- Eseguire lo script speciation_umd.py con il flag –r 0, che campiona il grafico di connettività al primo livello per identificare i poliedri di coordinazione. Ad esempio, un atomo centrale, indicato come catione , può essere circondato da uno o più anioni (Figura 4). Lo script di speciazione identifica ogni singolo poliedro di coordinazione. La media ponderata di tutti i poliedri di coordinazione dà il numero di coordinazione, identico a quello ottenuto dall'integrazione del PDF. Nella riga di comando digitare:
- Aprire i due file . popul.dat e . stat.dat consecutivamente; questi costituiscono l'output dello script di speciazione. Ogni ammasso è scritto su una riga, specificando la sua formula chimica, il momento in cui si è formato, il momento in cui è morto, la sua vita, una matrice con l'elenco degli atomi che formano questo ammasso. Traccia la durata di ogni cluster atomico di tutte le specie chimiche trovate nella simulazione come trovato nel file .popul.dat (Figura 5).
- Traccia l'analisi della popolazione con l'abbondanza di ciascuna specie, come si trova nel file . stat.dat file. Questa analisi, sia assoluta che relativa, corrisponde alle statistiche effettive dei poliedri di coordinazione per il caso -r 0; nel caso della polimerizzazione, con -r 1 questo deve essere trattato con attenzione in quanto potrebbe essere necessario applicare una normalizzazione sul numero relativo di atomi. L'abbondanza corrisponde all'integrale nel corso delle vite. Le. stat.dat file elenca anche la dimensione di ogni cluster, cioè quanti atomi lo formano.
4. Calcola i coefficienti di diffusione
- Estrarre gli spostamenti quadrati medi (MSD) degli atomi in funzione del tempo per ottenere l'auto-diffusività. La formula standard del MSD è:

dove i prefattori sono rinormalizzazioni. Con lo strumento MSD, ci sono diversi modi per analizzare gli aspetti dinamici dei fluidi.
NOTA: T è il tempo totale della simulazione e N α è il numero di atomi di tipo α. Il tempo iniziale t0 è arbitrario e copre la prima metà della simulazione. Ninit è il numero di volte iniziali. τ è la larghezza dell'intervallo di tempo durante il quale vengono calcolati i DMS; il suo valore massimo è la metà della durata della simulazione. Nelle tipiche implementazioni MSD, ogni finestra inizia alla fine della precedente. Ma un campionamento più scarso può accelerare il calcolo del MSD, senza alterare la pendenza del MSD. Per questo, l'i-esima finestra inizia al tempo t0(i), ma la finestra (i+1)-esima inizia al tempo t0(i) + τ + v, dove il valore di v è definito dall'utente. Allo stesso modo, la larghezza della finestra viene aumentata in passi discreti definiti dall'utente, come tali: τ(i) = τ(i-1) + z. I valori di z ("passo orizzontale") e v ("passo verticale") sono positivi o zero; il valore predefinito per entrambi è 20. - Calcolare l'MSD utilizzando la serie di script msd_umd . Il loro output è stampato in un file . msd.dat file, in cui il MSD di ogni tipo atomico, atomo o cluster viene stampato su una colonna in funzione del tempo.
- Calcolare il MSD medio di ciascun tipo atomico. I MSD vengono calcolati per ogni atomo e quindi mediati per ogni tipo atomico. Il file di output contiene una colonna per ogni tipo atomico. Nella riga di comando digitare:
msd_umd.py -f -z -v -b - Calcolare il MSD di ogni atomo. I MSD vengono calcolati per ogni atomo e quindi mediati per ogni tipo atomico. Il file di output contiene una colonna per ogni atomo nella simulazione e quindi una colonna per ogni tipo atomico. Questa caratteristica consente di identificare gli atomi che si diffondono in due ambienti diversi, come liquido e gas, o due liquidi. Nella riga di comando digitare:
msd_all_umd.py -f -z -v -b - Calcolare il MSD delle specie chimiche. Utilizzare la popolazione di cluster identificati con lo script di speciazione e stampati in . popul.dat file. I codici MSD vengono calcolati per ogni singolo cluster. Il file di output contiene una colonna per ogni cluster. Per evitare di considerare polimeri su larga scala, porre un limite alla dimensione del cluster; il suo valore predefinito è 20 atomi. Nella riga di comando digitare:
msd_cluster_umd.py -f -p -s -b -c
NOTA: i valori predefiniti sono: –b 100 –s 1 –c 20.
- Calcolare il MSD medio di ciascun tipo atomico. I MSD vengono calcolati per ogni atomo e quindi mediati per ogni tipo atomico. Il file di output contiene una colonna per ogni tipo atomico. Nella riga di comando digitare:
- Tracciare il CODICE MSD utilizzando un software basato su fogli di calcolo (Figura 6). In una rappresentazione log-log dell'MSD rispetto al tempo, identificare la variazione di pendenza. Separare la prima parte, solitamente breve, che rappresenta il regime balistico , cioè la conservazione della velocità degli atomi dopo le collisioni. La seconda parte più lunga rappresenta il regime diffusivo , cioè la dispersione della velocità degli atomi dopo le collisioni.
- Calcolare i coefficienti di diffusione dalla pendenza del MSD come:

dove Z è il numero di gradi di libertà (Z = 2 per la diffusione nel piano, Z = 3 per la diffusione nello spazio), e t è il passo temporale.
5. Funzioni di correlazione temporale
- Calcola le funzioni di correlazione temporale come misura dell'inerzia del sistema usando la formula generale:

A può essere una varietà di variabili dipendenti dal tempo, come le posizioni atomiche, le velocità atomiche, le sollecitazioni, la polarizzazione, ecc., Ognuna delle quali produce - attraverso le relazioni di Green-Kubo12,13 - diverse proprietà fisiche, a volte dopo un'ulteriore trasformazione. - Analizzare le velocità atomiche per ottenere lo spettro vibrazionale del liquido e l'espressione alternativa dei coefficienti di autodisponsazione atomica.
- Eseguire lo script vibr_spectrum_umd.py per calcolare la funzione di auto-correlazione (VAC) velocità atomica per ogni tipo atomico ed eseguire la sua trasformata di Fourier veloce. Nella riga di comando digitare:
vibr_spectrum_umd.py -f -t
dove –t è la temperatura che deve essere definita dall'utente. Lo script stampa due file: . vels.scf.dat file con la funzione VAC per ogni tipo atomico e . vibr.dat file con lo spettro vibrazionale decomposto su ogni specie atomica e il valore totale. - Aprire e leggere il file vels.scf.dat. Traccia la funzione VAC dal file vels.scf.dat utilizzando un software simile a un foglio di calcolo.
- Mantieni la parte reale del Fourier VAC. Questo è ciò che produce lo spettro vibrazionale, in funzione della frequenza:

dove m sono le masse atomiche. - Traccia lo spettro vibrazionale dal file vibr.dat utilizzando un software simile a un foglio di calcolo (Figura 7). Identificare il valore finito a ω=0 che corrisponde al carattere diffusivo del fluido e ai vari picchi dello spettro a frequenza finita. Identificare la partecipazione di ciascun tipo atomico allo spettro vibrazionale.
NOTA: La decomposizione sui tipi atomici mostra che atomi diversi hanno diversi contributi ω=0, corrispondenti ai loro coefficienti di diffusione. La forma generale dello spettro è molto più liscia con meno caratteristiche rispetto a un solido corrispondente. - Al guscio, leggi l'integrale sullo spettro vibrazionale, che produce i coefficienti di diffusione per ogni specie atomica.
NOTA: Le proprietà termodinamiche possono essere ottenute mediante integrazione dallo spettro vibrazionale, ma i risultati devono essere usati con cautela a causa di due approssimazioni: l'integrazione è valida all'interno dell'approssimazione quasi-armonica, che non necessariamente si mantiene ad alte temperature; e la parte gassosa dello spettro corrispondente alla diffusione deve essere scartata. L'integrazione dovrebbe quindi essere fatta solo sulla parte reticolare dello spettro. Ma questa separazione di solito richiede diverse ulteriori fasi e calcoli di post-elaborazione14, che non sono coperti dall'attuale pacchetto UMD.
- Eseguire lo script vibr_spectrum_umd.py per calcolare la funzione di auto-correlazione (VAC) velocità atomica per ogni tipo atomico ed eseguire la sua trasformata di Fourier veloce. Nella riga di comando digitare:
- Eseguire lo script viscosity_umd.py per analizzare l'auto-correlazione del tensore di sollecitazione dei componenti per stimare la viscosità del fuso. Nella riga di comando digitare:
viscosity_umd.py -f -i -s -o -l
NOTA: questa funzione è esplorativa e qualsiasi risultato deve essere preso con cautela. In primo luogo, controllare accuratamente la convergenza della viscosità rispetto alla lunghezza della simulazione.- Derivare la viscosità del fluido dall'autocorrelazione del tensore di sollecitazione15 come:

dove V e T sono rispettivamente il volume e la temperatura, κB è la costante di Boltzmann e σ ij la componente ij fuori diagonale del tensore di sollecitazione, espressa in coordinate cartesiane. - Utilizzare un adattamento più adeguato per ottenere una stima più robusta della viscosità15,16 ed evitare il rumore della funzione di auto-correlazione stress-tensore che potrebbe derivare dalla dimensione finita e dalla durata finita delle simulazioni. Per la funzione di auto-correlazione del tensore di sollecitazione, utilizzare la seguente forma funzionale15,16 che produce buoni risultati:

dove A, B, τ1, τ2 e ω sono parametri di adattamento. Dopo l'integrazione, l'espressione per la viscosità diventa:
- Derivare la viscosità del fluido dall'autocorrelazione del tensore di sollecitazione15 come:
6. Parametri termodinamici derivanti dalle simulazioni.
- Eseguire averages.py per estrarre i valori medi e lo spread (come deviazione standard) per pressione, temperatura, densità ed energia interna dai file umd . Nella riga di comando digitare:
averages.py -f -s
con –s 0 come valore predefinito. - Calcola l'errore statistico della media, utilizzando metodi di blocco.
NOTA: Ci sono vari sapori di questo metodo. Seguendo il lavoro di Allen e Tildesley2, è comune fare la media su sequenze di blocchi temporali, di lunghezza sempre più lunga, e stimare la deviazione standard rispetto alla media aritmetica17. La convergenza può essere raggiunta nel limite di molte e abbastanza lunghe dimensioni di blocco, quando il campionamento non è correlato. Anche se il valore di soglia effettivo per la convergenza di solito deve essere scelto manualmente.- Utilizzare il metodo di dimezzamento18: a partire dal campione di dati iniziale, ad ogni fase κ, dimezzare il numero dei campioni facendo la media su ogni due campioni consecutivi corrispondenti dal passaggio precedente κ−1:

- Eseguire lo script fullaverages.py per eseguire l'analisi statistica completa, incluso l'errore della media. Nella riga di comando digitare:
fullaverages.py -s -u
Nota : lo script viene automatizzato al punto di cercare tutti i file .umd.dat nella directory corrente ed eseguire l'analisi per tutti loro. I valori predefiniti sono –s 0 –u 0. Per -u 0 l'output è minimo, e per -u 1 l'output è completo, con diverse unità alternative stampate. Questo script richiede supporto grafico, in quanto crea un'immagine grafica per verificare la convergenza per stimare l'errore sulla media.
- Utilizzare il metodo di dimezzamento18: a partire dal campione di dati iniziale, ad ogni fase κ, dimezzare il numero dei campioni facendo la media su ogni due campioni consecutivi corrispondenti dal passaggio precedente κ−1:
Risultati
La pirolite è un modello di silicato fuso multicomponente (0,5Na2O 2CaO 1,5Al2O3 4FeO 30 MgO 24SiO2) che meglio si avvicina alla composizione della Terra di silicato di massa, la media geochimica o l'intero pianeta, ad eccezione del suo nucleo a base di ferro19. La Terra primordiale era dominata da una serie di eventi di fusione su larga scala20, l'ultimo potrebbe aver inghiottito l'intero pianeta, dopo la sua condensazione per il disco protolunare21. La pirolite rappresenta la migliore approssimante alla composizione di tali oceani di magma su scala planetaria. Di conseguenza, abbiamo studiato ampiamente le proprietà fisiche della fusione di pirolite nell'intervallo di temperatura 3.000\u20125.000 K e nell'intervallo di pressione 0\u2012150 GPa dalle simulazioni di dinamica molecolare ab initio nell'implementazione VASP. Queste condizioni termodinamiche caratterizzano interamente le condizioni oceaniche di magma più estreme della Terra. Il nostro studio è un ottimo esempio di un uso riuscito del pacchetto UMD per l'intera analisi approfondita dei fusi22. Abbiamo calcolato la distribuzione e le lunghezze medie dei legami, tracciato i cambiamenti nella coordinazione catione-ossigeno e confrontato i nostri risultati con precedenti studi sperimentali e computazionali su silicati amorfi di varie composizioni. La nostra analisi approfondita ha aiutato a scomporre i numeri di coordinazione standard nei loro costituenti di base, delineare la presenza di poliedri di coordinazione esotici nella fusione ed estrarre la durata di vita per tutti i poliedri di coordinazione. Ha anche sottolineato l'importanza del campionamento nelle simulazioni in termini sia di lunghezza della traiettoria che di numero di atomi presenti nel sistema modellato. Per quanto riguarda la post-elaborazione, l'analisi UMD è indipendente da questi fattori, tuttavia, dovrebbero essere presi in considerazione quando si interpretano i risultati forniti dal pacchetto UMD. Qui, mostriamo alcuni esempi di come il pacchetto UMD può essere utilizzato per estrarre diverse caratteristiche dei fusi, con un'applicazione alla pirolite fusa.
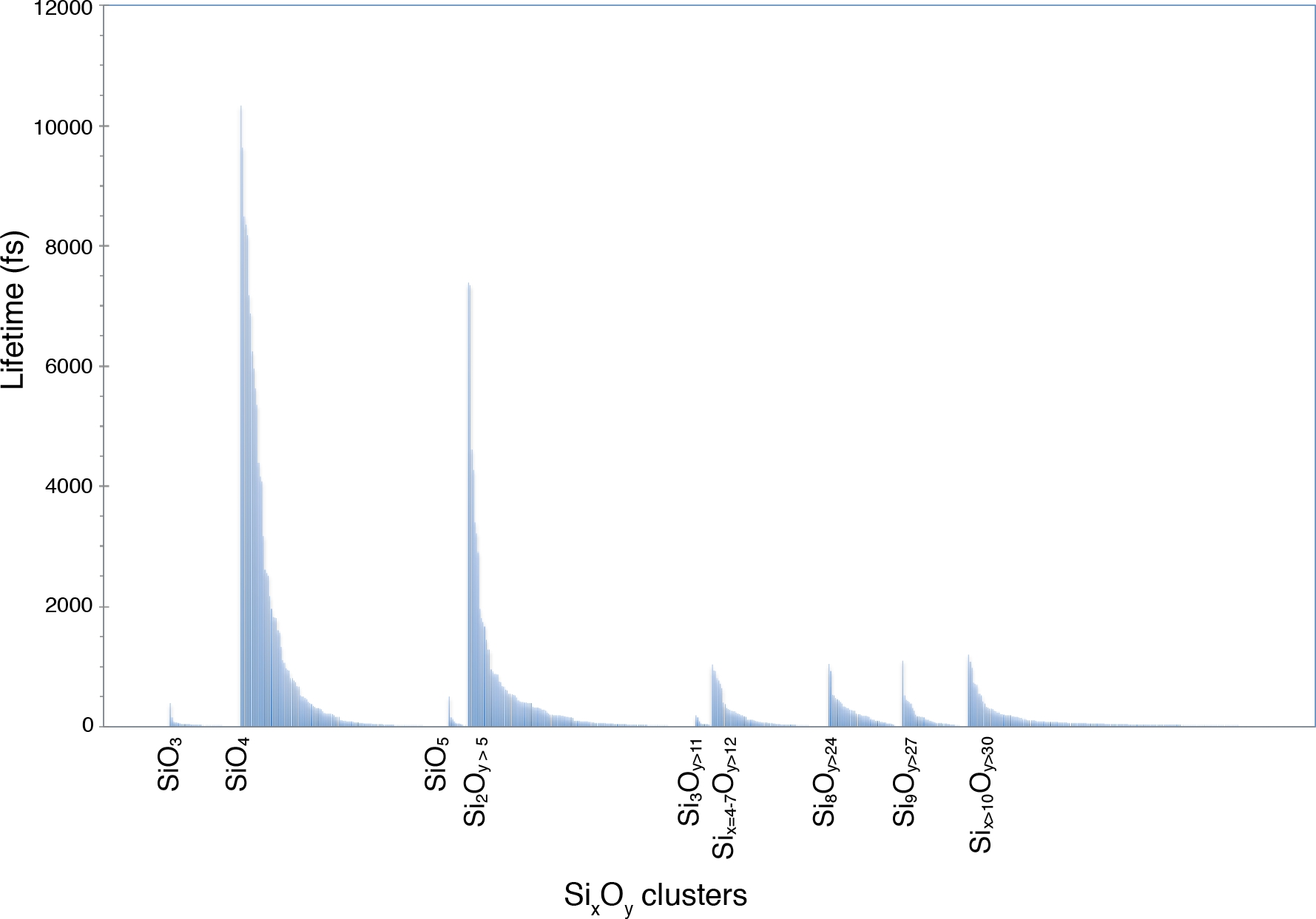
La funzione di distribuzione della coppia Si-O ottenuta dallo script gofrs_umd.py mostra che il raggio della prima sfera di coordinazione, che è il primo minimo della funzione g(r), si trova intorno a 2,5 angstrom a T = 3000 K e P = 4,6 GPa. Il massimo di g(r) si trova a 1,635 Å, la migliore approssimazione alla lunghezza della piegatura. La lunga coda è dovuta alla temperatura. Usando questo limite come distanza di legame Si-O, l'analisi di speciazione mostra che le unità SiO4, che possono durare fino a pochi picosecondi, dominano la fusione (Figura 5). C'è una parte importante del fuso che mostra una polimerizzazione parziale, come riflesso dalla presenza di dimeri come Si2O7 e trimeri come le unità Si3Ox. La loro durata corrispondente è nell'ordine del picosecondo. I polimeri di ordine superiore hanno tutti una durata considerevolmente più breve.

Figura 5: Durata della vita della specie chimica Si-O.
La speciazione è identificata in un fuso multicomponente a 4,6 GPa e 3000 K. Le etichette contrassegnano i monomeri SiO3, SiO4 e SiO5 e i vari polimeri SixOy. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
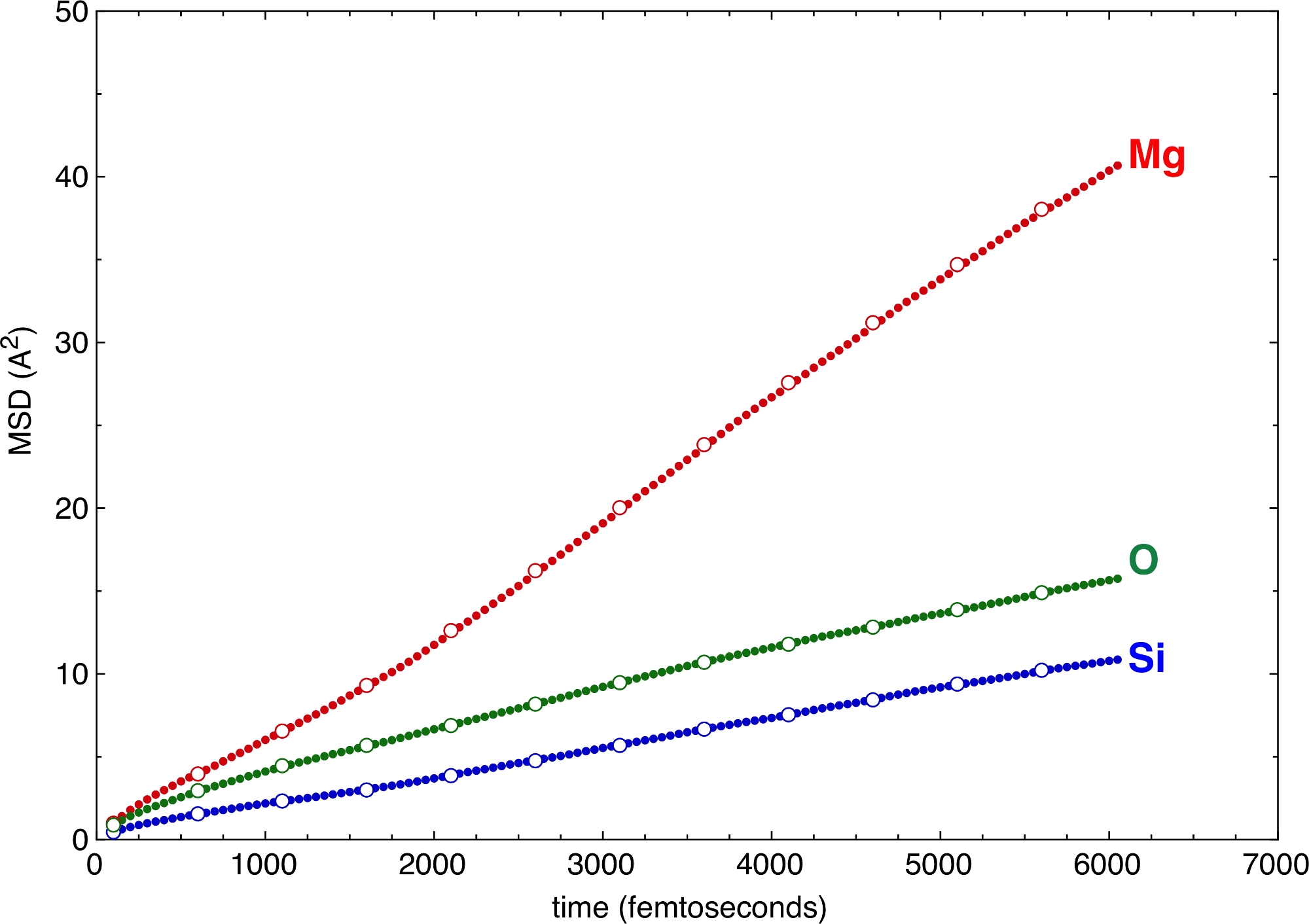
I diversi valori dei gradini verticali e orizzontali, definiti dai flag –z e –v sopra, producono vari campionamenti del MSD (Figura 6). Anche grandi valori di z e v sono sufficienti per definire le pendenze e quindi i coefficienti di diffusione dei diversi atomi. Il guadagno in tempo per la post-elaborazione è notevole quando si passa a grandi valori di z e v. Il MSD offre un criterio di convalida molto forte per la qualità delle simulazioni. Se la parte di diffusione del MSD non è sufficientemente lunga, ciò è un segno che la simulazione è troppo breve e non riesce a raggiungere lo stato fluido in senso statistico. Il requisito minimo per la parte diffusiva del MSD dipende in larga misura dal sistema. Si può richiedere che tutti gli atomi cambino il loro sito almeno una volta nella struttura del fuso in modo che possa essere considerato come un fluido10. Un eccellente esempio con applicazioni nelle scienze planetarie è la fusione di silicati complessi ad alte pressioni vicino o anche al di sotto della loro linea liquida11. Gli atomi di Si, i principali cationi che formano la rete, cambiano sito dopo più di due dozzine di picosecondi. Le simulazioni più brevi di questa soglia sarebbero considerevolmente sottocampionate del possibile spazio configurazionale. Tuttavia, poiché gli anioni coordinati, vale a dire gli atomi O, si muovono più velocemente degli atomi centrali di Si, possono compensare parte della lenta mobilità di Si. Come tale, l'intero sistema potrebbe effettivamente coprire un campionamento migliore dello spazio configurazionale rispetto a quanto ipotizzato solo dagli spostamenti di Si.

Figura 6: Spostamenti quadrati medi (MSD).
I MSD sono illustrati per alcuni tipi atomici di un silicato multicomponente fuso. Il campionamento con vari passaggi orizzontali e verticali, z e v, produce risultati coerenti. Cerchi solidi: -z 50 –v 50. Cerchi aperti: -z 250 –v 500. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
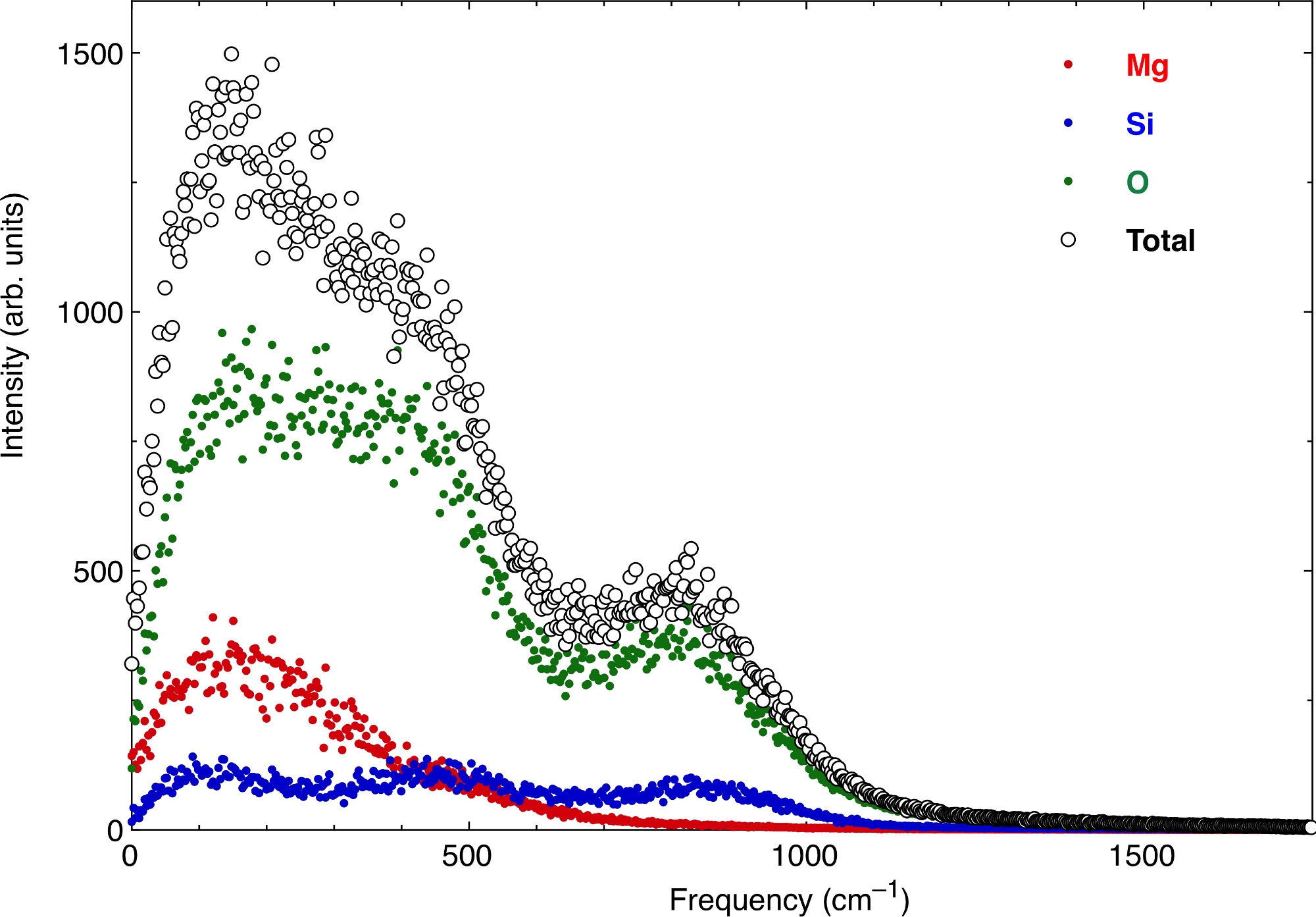
Infine, le funzioni VAC atomiche producono lo spettro vibrazionale della fusione. La Figura 7 mostra lo spettro alle stesse condizioni di pressione e temperatura di cui sopra. Rappresentiamo i contributi degli atomi Mg, Si e O, nonché il valore totale. A frequenza zero c'è un valore finito dello spettro, che corrisponde al carattere di diffusione del fuso. L'estrazione delle proprietà termodinamiche dallo spettro vibrazionale deve rimuovere questo carattere diffusivo gassoso da zero, ma anche tenere adeguatamente conto del suo decadimento a frequenze più elevate.

Figura 7: Lo spettro vibrazionale della fusione della pirolite.
La parte reale della trasformata di Fourier della funzione di auto-correlazione velocità-velocità atomica produce lo spettro vibrazionale. Qui lo spettro viene calcolato per un silicato multicomponente fuso. I fluidi hanno un carattere diffusivo simile a un gas diverso da zero a frequenza zero. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Il pacchetto UMD è stato progettato per funzionare meglio con le simulazioni ab initio, in cui il numero di istantanee è in genere limitato a decine o centinaia di migliaia di istantanee, con poche centinaia di atomi per unità di cella. Sono inoltre trattabili simulazioni più grandi a condizione che la macchina su cui viene eseguita la post-elaborazione disponga di risorse di memoria attive sufficienti. Il codice si distingue per la varietà di proprietà che può calcolare e per la sua licenza open source.
I file umd.dat sono appropriati per gli insiemi che mantengono invariato il numero di particelle durante la simulazione. Il pacchetto UMD è in grado di leggere i file derivanti da calcoli in cui la forma e il volume della scatola di simulazione variano. Questi coprono i calcoli più comuni, come NVT e NPT, in cui il numero di particelle, N, temperatura T, volume, V e / o pressione, P, sono mantenuti costanti.
Per il tempo inizia la funzione di distribuzione della coppia così come tutti gli script che devono stimare le distanze interatomiche, come gli script di speciazione, funzionano solo per le celle unitarie ortogonali, cioè per le celle cubiche, tetragonali e ortorombiche, dove gli angoli tra gli assi sono di 90°.
Le principali linee di sviluppo per la versione 2.0 sono la rimozione della restrizione di ortogonalità per le distanze e l'aggiunta di ulteriori funzionalità per gli script di speciazione: per analizzare i singoli legami chimici, per analizzare gli angoli interatomici e per implementare la seconda sfera di coordinazione. Con l'aiuto di una collaborazione esterna, stiamo lavorando al porting del codice su una GPU per un'analisi più rapida in sistemi più grandi.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo lavoro è stato sostenuto dal Consiglio europeo della ricerca (CER) nell'ambito del programma di ricerca e innovazione Horizon 2020 dell'Unione europea (numero di accordo di sovvenzione 681818 IMPACT a RC), dalla direzione estrema di fisica e chimica dell'Osservatorio Deep Carbon e dal Consiglio di ricerca della Norvegia attraverso il suo schema di finanziamento dei Centri di eccellenza, il numero di progetto 223272. Riconosciamo l'accesso ai supercomputer GENCI attraverso la serie stl2816 di sovvenzioni per l'elaborazione eDARI, al supercomputer Irene AMD attraverso il progetto PRACE RA4947 e al supercomputer Fram attraverso UNINETT Sigma2 NN9697K. FS è stato sostenuto da un progetto Marie Skłodowska-Curie (convenzione di sovvenzione ABISSE No.750901).
Materiali
| Name | Company | Catalog Number | Comments |
| getopt library | open-source | ||
| glob library | open-source | ||
| matplotlib library | open-source | ||
| numpy library | open-source | ||
| os library | open-source | ||
| Python software | The Python Software Foundation | Version 2 and 3 | open-source |
| random library | open-source | ||
| re library | open-source | ||
| scipy library | open-source | ||
| subprocess library | open-source | ||
| sys library | open-source |
Riferimenti
- Frenkel, D., Smit, B. . Understanding Molecular Simulation. From Algorithms to Applications. , (2001).
- Allen, M. P., Tildesley, D. J., Allen, T. . Computer Simulation of Liquids. , (1989).
- Zepeda-Ruiz, L. A., Stukowski, A., Oppelstrup, T., Bulatov, V. V. Probing the limits of metal plasticity with molecular-dynamics simulations. Nature Publishing Group. 550 (7677), 492-495 (2017).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modeling. 14 (1), 33-38 (1996).
- Momma, K., Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 44 (6), 1272-1276 (2011).
- Brehm, M., Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. Journal of Chemical Information and Modeling. 51 (8), 2007-2023 (2011).
- Stixrude, L. Visualization-based analysis of structural and dynamical properties of simulated hydrous silicate melt. Physics and Chemistry of Minerals. 37 (2), 103-117 (2009).
- Kresse, G., Hafner, J. Ab initio Molecular-Dynamics for Liquid-Metals. Physical Review B. 47 (1), 558-561 (1993).
- Gygi, F. Architecture of Qbox: A scalable first-principles molecular dynamics code. IBM Journal of Research and Development. 52 (1-2), 137-144 (2008).
- Harvey, J. P., Asimow, P. D. Current limitations of molecular dynamic simulations as probes of thermo-physical behavior of silicate melts. American Mineralogist. 100 (8-9), 1866-1882 (2015).
- Caracas, R., Hirose, K., Nomura, R., Ballmer, M. D. Melt-crystal density crossover in a deep magma ocean. Earth and Planetary Science Letters. 516, 202-211 (2019).
- Green, M. S. Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena. II. Irreversible Processes in Fluids. The Journal of Chemical Physics. 22 (3), 398-413 (1954).
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. Journal of the Physical Society of Japan. 12 (6), 570-586 (1957).
- Lin, S. T., Blanco, M., Goddard, W. A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. The Journal of Chemical Physics. 119 (22), 11792-11805 (2003).
- Meyer, E. R., Kress, J. D., Collins, L. A., Ticknor, C. Effect of correlation on viscosity and diffusion in molecular-dynamics simulations. Physical Review E. 90 (4), 1198-1212 (2014).
- Soubiran, F., Militzer, B., Driver, K. P., Zhang, S. Properties of hydrogen, helium, and silicon dioxide mixtures in giant planet interiors. Physics of Plasmas. 24 (4), 041401-041407 (2017).
- Flyvbjerg, H., Petersen, H. G. Error estimates on averages of correlated data. The Journal of Chemical Physics. 91 (1), 461-466 (1989).
- Tuckerman, M. E. . Statistical mechanics: theory and molecular simulation. , (2010).
- McDonough, W. F., Sun, S. S. The composition of the Earth. Chemical Geology. 120, 223-253 (1995).
- Elkins-Taton, L. T. Magma oceans in the inner solar system. Annual Review of Earth and Planetary Sciences. 40, 113-139 (2012).
- Lock, S. J., et al. The origin of the Moon within a terrestrial synestia. J. Geophysical Research: Planets. 123, 910-951 (2018).
- Solomatova, N. V., Caracas, R. Pressure-induced coordination changes in a pyrolitic silicate melt from ab initio molecular dynamics simulations. Journal of Geophysical Research: Solid Earth. 124, 11232-11250 (2019).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati