
Method Article
Analisando derretimentos e fluidos de simulações de dinâmica molecular da Ab Initio com o pacote UMD
Neste Artigo
Resumo
Derretimentos e fluidos são vetores onipresentes do transporte em massa em sistemas naturais. Desenvolvemos um pacote de código aberto para analisar simulações de dinâmica molecular ab initio de tais sistemas. Calculamos propriedades estruturais (ligação, clusterização, especiação química), transporte (difusão, viscosidade) e propriedades termodinâmicas (espectro vibracional).
Resumo
Desenvolvemos um pacote de código aberto baseado em Python para analisar os resultados decorrentes de simulações de dinâmica molecular ab initio de fluidos. O pacote é mais adequado para aplicações em sistemas naturais, como derretimento de silicato e óxido, fluidos à base de água e vários fluidos supercríticos. O pacote é uma coleção de scripts Python que incluem duas grandes bibliotecas que lidam com formatos de arquivo e com cristalografia. Todos os scripts são executados na linha de comando. Propomos um formato simplificado para armazenar as trajetórias atômicas e informações termodinâmicas relevantes das simulações, que são salvas em arquivos UMD, em posição de Universal Molecular Dynamics. O pacote UMD permite a computação de uma série de propriedades estruturais, de transporte e termodinâmicas. A partir da função de distribuição de pares, ele define os comprimentos dos títulos, constrói uma matriz de conectividade interatômica e, eventualmente, determina a especiação química. Determinar a vida útil das espécies químicas permite executar uma análise estatística completa. Em seguida, scripts dedicados computam os deslocamentos médios quadrados para os átomos, bem como para as espécies químicas. A análise de auto correlação implementada das velocidades atômicas produz os coeficientes de difusão e o espectro vibracional. A mesma análise aplicada nos estresses rende a viscosidade. O pacote está disponível através do site do GitHub e através de sua própria página dedicada do projeto ERC IMPACT como pacote de acesso aberto.
Introdução
Fluidos e derretimentos são vetores ativos de transporte químico e físico em ambientes naturais. As taxas elevadas de difusão atômica favorecem trocas químicas e reações, a baixa viscosidade aliada à flutuação variada favorece uma grande transferência de massa, e as relações de densidade cristalina favorecem camadas dentro de corpos planetários. A ausência de uma rede periódica, temperaturas típicas de alta temperatura necessárias para atingir o estado derretido, e a dificuldade para saciar tornam a determinação experimental de uma série de propriedades óbvias, como densidade, difusão e viscosidade, extremamente desafiadoras. Essas dificuldades tornam os métodos computacionais alternativos ferramentas fortes e úteis para investigar essa classe de materiais.
Com o advento do poder computacional e a disponibilidade de supercomputadores, duas grandes técnicas numéricas de simulações atomísticas são atualmente empregadas para estudar o estado dinâmico de um sistema atomista não cristalino, Monte Carlo1 e dinâmica molecular (MD)1,2. Em Monte Carlo, o espaço configuracional é amostrado aleatoriamente; Os métodos de Monte Carlo mostram escala linear na paraleloização se todas as observações amostrais forem independentes umas das outras. A qualidade dos resultados depende da qualidade do gerador de números aleatórios e da representatividade da amostragem. Os métodos de Monte Carlo mostram escala linear na paraleloização se a amostragem for independente uma da outra. Na dinâmica molecular (MD) o espaço configuracional é amostrado por trajetórias atômicas dependentes do tempo. A partir de uma determinada configuração, as trajetórias atômicas são computadas pela integração das equações newtonianas de movimento. As forças interatômicas podem ser computadas usando potenciais interatômicos de modelo (em MD clássico) ou usando métodos de princípios iniciais (em ab initio, ou primeiros princípios, MD). A qualidade dos resultados depende do comprimento da trajetória e sua capacidade de não ser atraído para a minima local.
Simulações de dinâmica molecular contêm uma infinidade de informações, todas relacionadas ao comportamento dinâmico do sistema. Propriedades médias termodinâmicas, como energia interna, temperatura e pressão, são bastante padrão para calcular. Eles podem ser extraídos dos arquivos de saída das simulações e mediados, enquanto as quantidades relacionadas diretamente ao movimento dos átomos, bem como à sua relação mútua precisam ser computadas após a extração das posições atômicas e velocidades.
Consequentemente, muito esforço tem sido dedicado para visualizar os resultados, e vários pacotes estão disponíveis hoje, em diferentes plataformas, código aberto ou não [Ovito3, VMD4, Vesta5, Travis6, etc.]. Todas essas ferramentas de visualização lidam eficientemente com distâncias interatômicas e, como tal, permitem a computação eficiente de funções de distribuição de pares e coeficientes de difusão. Vários grupos que realizam simulações de dinâmica molecular em larga escala têm software proprietário para analisar várias outras propriedades resultantes das simulações, às vezes em shareware ou outras formas de acesso limitado à comunidade, e às vezes limitados em escopo e uso para alguns pacotes específicos. Algoritmos sofisticados para extrair informações sobre ligação interatômica, padrões geométricos e termodinâmica são desenvolvidos e implementados em alguns desses pacotes3,4,5,6,7, etc.
Aqui propomos o pacote UMD - um pacote de código aberto escrito em Python para analisar a saída de simulações de dinâmica molecular. O pacote UMD permite a computação de uma ampla gama de propriedades estruturais, dinâmicas e termodinâmicas (Figura 1). O pacote está disponível através do site do GitHub (https://github.com/rcaracas/UMD_package) e através de uma página dedicada (http://moonimpact.eu/umd-package/) do projeto ERC IMPACT como um pacote de acesso aberto.
Para torná-lo universal e mais fácil de manusear, nossa abordagem é primeiro extrair todas as informações relacionadas ao estado termodinâmico e as trajetórias atômicas do arquivo de saída da dinâmica molecular real. Essas informações são armazenadas em um arquivo dedicado, cujo formato é independente do pacote MD original onde a simulação foi executada. Nós nomeamos esses arquivos "umd", que significa Universal Molecular Dynamics. Desta forma, nosso pacote UMD pode ser facilmente usado por qualquer grupo ab initio com qualquer software, tudo com um esforço mínimo de adaptação. O único requisito para usar o pacote atual é escrever o analisador apropriado da saída do software MD em particular para o formato de arquivo umd, se isso ainda não existir. Por enquanto, fornecemos tais analisadores para os pacotes VASP8 e QBox9 .

Figura 1: Fluxograma da biblioteca umd.
As propriedades físicas estão em azul, e os principais scripts python e suas opções estão em vermelho. Clique aqui para ver uma versão maior desta figura.
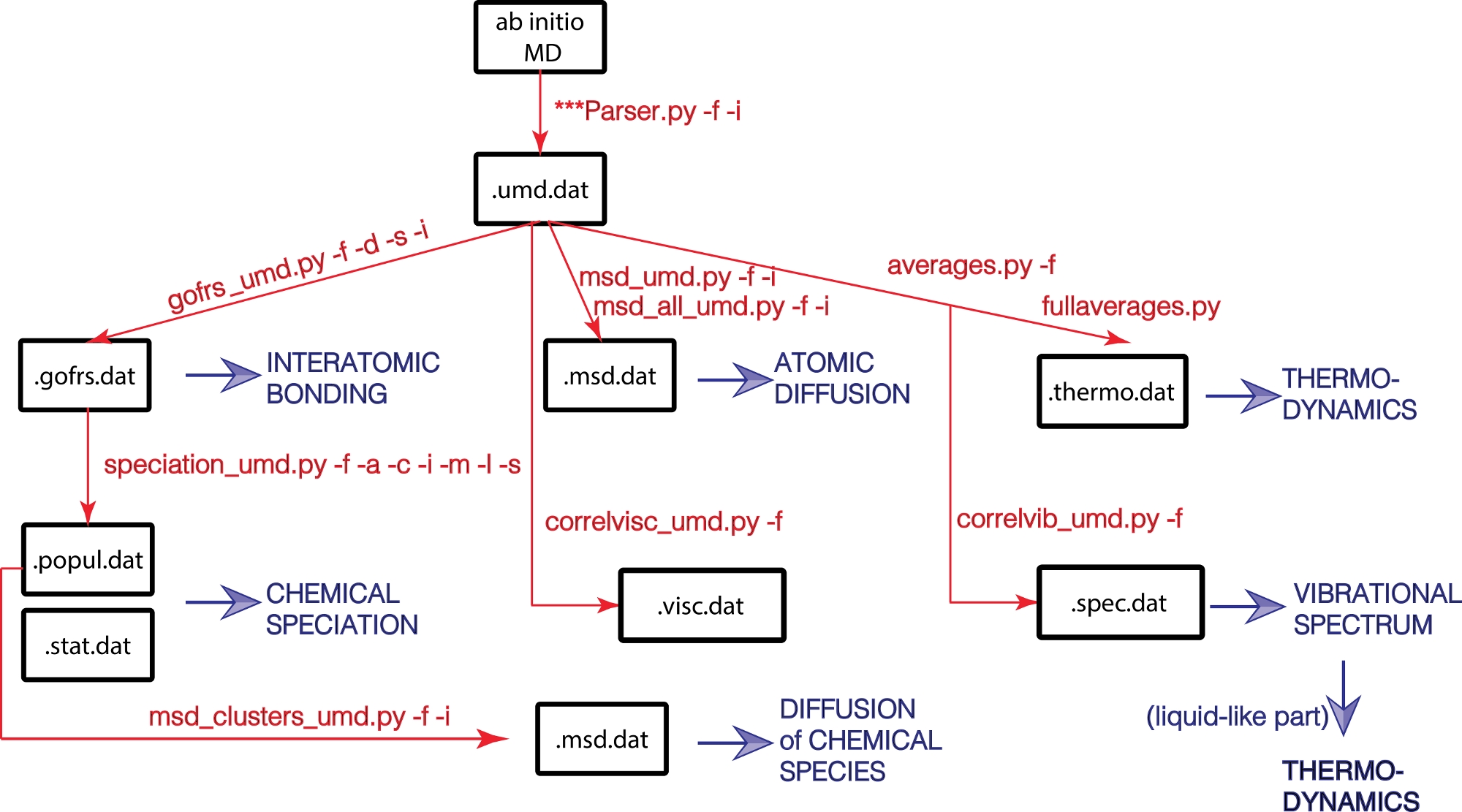{kind=link}
Os arquivos umd são arquivos ASCII; extensão típica é "umd.dat", mas não obrigatória. Todos os componentes de análise podem ler arquivos ASCII do formato umd, independentemente da extensão real do nome. No entanto, alguns dos scripts automáticos projetados para executar estatísticas rápidas em grande escala ao longo de várias simulações procuram especificamente arquivos com a extensão umd.dat. Cada propriedade física é expressa em uma linha. Cada linha começa com uma palavra-chave. Desta forma, o formato é altamente adaptável e permite que novas propriedades sejam adicionadas ao arquivo umd, preservando toda a sua legibilidade ao longo das versões. As primeiras 30 linhas do arquivo umd da simulação de pirolito a 4,6 GPa e 3000 K, usadas abaixo na discussão, são mostradas na Figura 2.

Figura 2: O início do arquivo umd descrevendo a simulação de pirolito líquido a 4,6 GPa e 3000 K.
O cabeçalho é seguido pela descrição de cada instantâneo. Cada imóvel está escrito em uma linha, contendo o nome da propriedade física, o valor(s) e as unidades, todas separadas por espaços. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Todos os arquivos de umd contêm um cabeçalho descrevendo o conteúdo da célula de simulação: o número de átomos, elétrons e tipos atômicos, bem como detalhes para cada átomo, como seu tipo, símbolo químico, número de elétrons de valência, e sua massa. Uma linha vazia marca a extremidade do cabeçalho e a separa da parte principal do arquivo umd.
Em seguida, cada passo da simulação é detalhado. Primeiro, são dados os parâmetros termodinâmicos instantâneos, cada um em uma linha diferente, especificando (i) o nome do parâmetro, como energia, tensões, pressão hidrostática equivalente, densidade, volume, parâmetros de rede, etc., (ii) seu valor(s) e (iii) suas unidades. Uma tabela descrevendo os átomos vem em seguida. Uma linha de cabeçalho dá as diferentes medidas, como posições cartesianas, velocidades, cargas, etc., e suas unidades. Então cada átomo é detalhado em uma linha. Por grupos de três, correspondentes aos eixos três x, y, z , as entradas são: as posições reduzidas, as posições cartesianas dobradas na célula de simulação, as posições cartesianas (que levam em conta o fato de que os átomos podem atravessar várias células unitárias durante uma simulação), as velocidades atômicas e as forças atômicas. As duas últimas entradas são escalares: carga e momento magnético.
Duas grandes bibliotecas garantem o bom funcionamento de todo o pacote. A biblioteca umd_process.py lida com os arquivos umd, como leitura e impressão. A biblioteca crystallography.py trata de todas as informações relacionadas à estrutura atômica real. A filosofia subjacente da biblioteca crystallography.py é tratar a rede como um espaço vetorial. Os parâmetros da célula unitária, juntamente com sua orientação, representam os vetores de base. O "Espaço" tem uma série de atributos escalares (volume específico, densidade, temperatura e número específico de átomos), propriedades termodinâmicas (energia interna, pressão, capacidade térmica, etc.), e uma série de propriedades tensores (estresse e elasticidade). Átomos povoam este espaço. A classe "Rede" define este conjunto, ao lado de vários poucos cálculos curtos, como volume específico, densidade, obtenção da rede recíproca do direto, etc. A classe "Átomos" define os átomos. Caracterizam-se por uma série de propriedades escalares (nome, símbolo, massa, número de elétrons, etc.) e uma série de propriedades vetoriais (a posição no espaço, seja em relação à base vectorial descrita na classe Rettice, ou relativa às coordenadas cartesianas universais, velocidades, forças, etc.). Além dessas duas classes, a biblioteca crystallography.py contém uma série de funções para realizar uma variedade de testes e cálculos, como distâncias atômicas ou multiplicação celular. A tabela periódica dos elementos também é incluída como dicionário.
Os vários componentes do pacote umd escrevem vários arquivos de saída. Como regra geral, são todos arquivos ASCII, todas as suas entradas são separadas por guias, e são feitas o mais autoexplicativo possível. Por exemplo, eles sempre indicam claramente a propriedade física e suas unidades. Os arquivos umd.dat cumprem totalmente esta regra.
Protocolo
1. A análise da dinâmica molecular corre
NOTA: O pacote está disponível através do site do GitHub (https://github.com/rcaracas/UMD_package) e através de uma página dedicada (http://moonimpact.eu/umd-package/) do projeto ERC IMPACT como um pacote de acesso aberto.
- Extrair cada conjunto específico de propriedades físicas usando um ou mais scripts Python dedicados do pacote. Execute todos os scripts na linha de comando; todos eles empregam uma série de bandeiras, que são o mais consistentes possível de um script para outro. As bandeiras, seu significado e valores padrão estão todos resumidos na Tabela 1.
| Bandeira | Significado | Script usando-o | Valor padrão |
| -h | Ajuda curta | todo | |
| -f | Nome do arquivo UMD | todo | |
| -i | Etapas de termoqueização a serem descartadas | todo | 0 |
| -i | Arquivo de entrada contendo as ligações interatômicas | especiação | bonds.input |
| -s | Amostragem da frequência | msd, especiação | 1 (cada passo é considerado) |
| -a | Lista de átomos ou ânions | especiação | |
| -c | Lista de cations | especiação | |
| -l | Comprimento do vínculo | especiação | 2 |
| -t | Temperatura | vibrações, reologia | |
| -v | Discretização da largura da janela amostral da trajetória para a análise de deslocamento médio-quadrado | Msd | 20 |
| -z | Discretização do início da janela amostral da trajetória para a análise de deslocamento médio-quadrado | Msd | 20 |
Tabela 1: Bandeiras mais comuns usadas no pacote UMD e seu significado mais comum.
- Comece com a transformação da saída da simulação MD realizada em um código de princípios inicialmente, como VASP8 ou QBox9, em um arquivo UMD.
- Se as simulações de MD foram feitas em VASP, então no tipo de linha de comando:
VaspParser.py -f -i
onde –f bandeira define o nome do arquivo VASP OUTCAR, e o comprimento de termelétrica .
NOTA: A etapa inicial, definida por –i permite descartar os primeiros passos das simulações, que representam a termequeização. Em uma dinâmica molecular típica, a primeira parte do cálculo representa a termeterização, ou seja, o tempo que leva o sistema para que todos os átomos descrevam uma distribuição gaussiana da temperatura, e para todo o sistema apresentar flutuações da temperatura, pressão, energia, etc. em torno dos valores de equilíbrio. Esta parte de termebrição da simulação não deve ser levada em conta ao analisar as propriedades estatísticas do fluido.
- Se as simulações de MD foram feitas em VASP, então no tipo de linha de comando:
- Transforme o . arquivos humd em . arquivos xyz para facilitar a visualização em vários outros pacotes, como VMD4 ou Vesta5. No tipo de linha de comando:
umd2xyz.py -f -i -s
onde –f define o nome do . umd file, –i define o período de termequeização a ser descartado, e –s a frequência da amostragem da trajetória armazenada no . arquivo umd . Os valores padrão são -i 0 –s 1, ou seja, considerando todas as etapas da simulação, sem que nenhum seja descartado. - Inverta o arquivo umd em arquivos POSCAR do tipo VASP usando o script umd2poscar.py; instantâneos das simulações podem ser selecionados com uma frequência predefinida. No tipo de linha de comando:
umd2poscar.py -f -i -l -s
onde – l representa o último passo a ser transformado em arquivo POSCAR. Os valores padrão são -i 0 -l 10000000 -s 1. Esse valor de - l é grande o suficiente para cobrir uma trajetória típica inteira.
2. Realizar a análise estrutural
- Execute o script gofrs_umd.py para calcular a função de distribuição de pares (PDF) g(r) para todos os pares dos tipos atômicos A e B (Figura 3). A saída é escrita em um arquivo ASCII, separado por guias, com os gofrs de extensão.dat. No tipo de linha de comando:
gofrs_umd.py -f -s < Sampling_Frequency > -d -i
NOTA: Os padrões são Sampling_Frequency (a frequência para amostragem da trajetória) = 1 passo; DiscretizaçãoInterval (para plotagem do g(r)) = 0,01 Å; InitialStep (número de passos no início da trajetória que são descartadas) = 0. O PDF radial, g(r) é o número médio de átomos do tipo B a uma distância d_não de uma camada esférica de raio r e espessura dr centrada nos átomos do tipo A (Figura 3):
com a densidade atômica, NA e NB o número de átomos dos tipos A e B, e δ(r−r)) a função delta que é igual a 1 se os átomos A e B se encontram a uma distância entre r e r+dr. A abscissa do primeiro máximo de g(r) dá o maior comprimento de ligação de probabilidade entre os átomos dos tipos A e B, que é o mais próximo de uma distância média de ligação que podemos determinar. O primeiro mínimo delimita a extensão da primeira esfera de coordenação. Assim, a integral sobre o PDF até o primeiro mínimo dá o número médio de coordenação. A soma das transformações de Fourier do g(r) para todos os pares dos tipos atômicos A e B produz o padrão de difração do fluido, como obtido experimentalmente com um difratómetro. No entanto, na realidade, como muitas vezes faltam as esferas de coordenação de alta ordem do g(r), o padrão de difração não pode ser obtido em sua totalidade.

Figura 3: Determinação da função de distribuição do par.
a Para cada átomo de uma espécie (por exemplo vermelho), todos os átomos das espécies coordenadoras (por exemplo cinza e/ou vermelho) são contados em função da distância. (b) O gráfico de distribuição de distância resultante para cada instantâneo, que nesta fase é apenas uma coleção de funções delta, é então mediado sobre todos os átomos e todos os instantâneos e ponderados pela distribuição ideal de gás para gerar (c) a função de distribuição do par que é contínua. O primeiro mínimo do g(r) é o raio da primeira esfera de coordenação, utilizado posteriormente na análise de especiação. Clique aqui para ver uma versão maior desta figura.
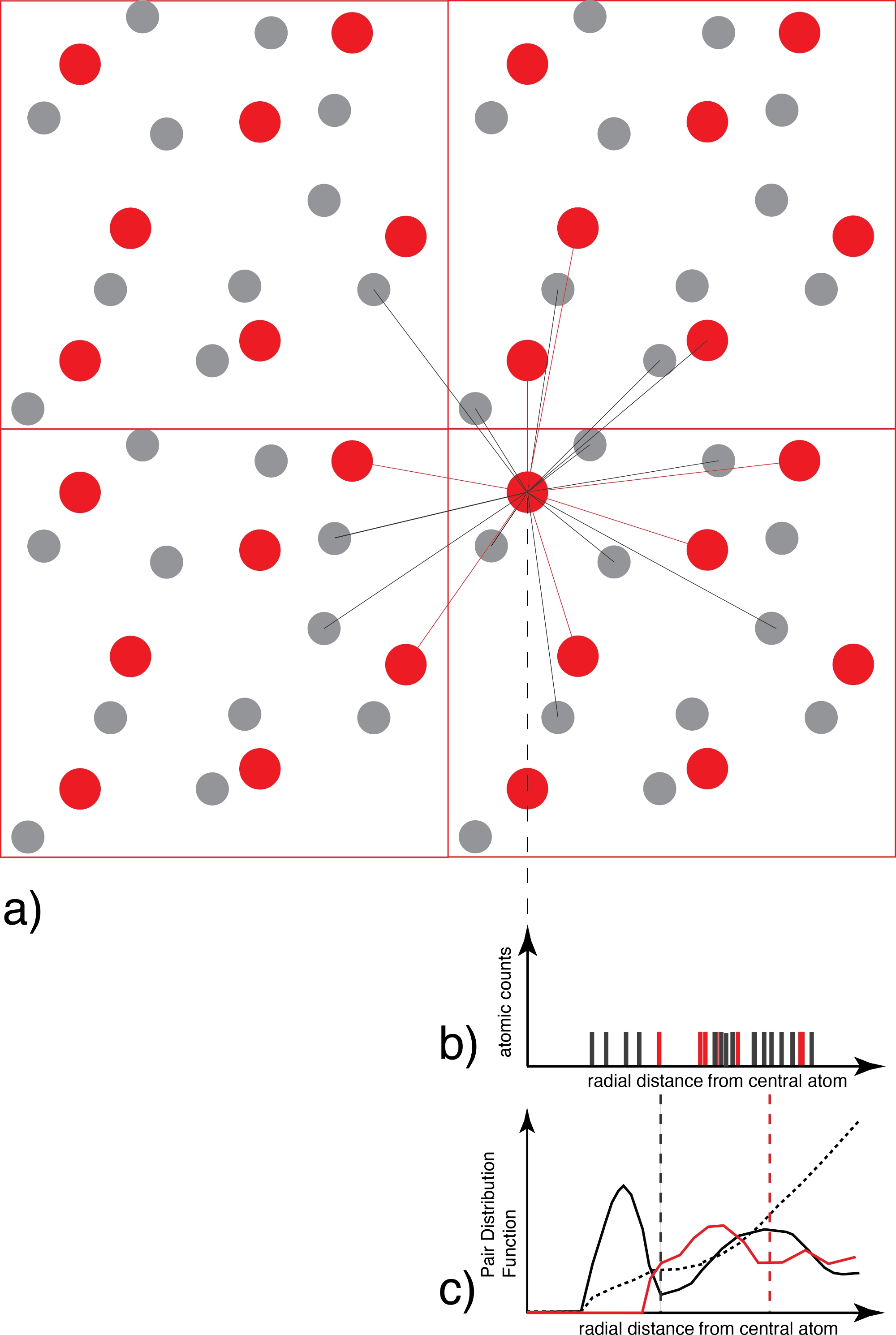{kind=link}
- Extrair as distâncias médias de ligação interatômicas como os raios das primeiras esferas de coordenação. Para isso, identifique a posição do primeiro máximo das funções g(r): plote os gofrs.dat arquivo em um aplicativo de planilha e procure o máximo e o minima para cada par de átomos.
- Identifique o raio da primeira esfera de coordenação, como o primeiro mínimo do PDF, g(r), utilizando software de planilha. Esta é a base para toda a análise estrutural do fluido; o PDF produz o status médio de ligação dos átomos no fluido.
- Extrair as distâncias do primeiro minima, ou seja, a abscissa, e escrevê-las em um arquivo separado, chamado, por exemplo, bonds.input. Alternativamente, execute um dos scripts analyze_gofr do pacote UMD para identificar a maxima e o minima das funções g(r). No tipo de linha de comando:
analyze_gofr_semi_automatic.py - Clique na posição do máximo e do mínimo da função g(r) exibida no gráfico que é aberto pelo programa. O script verifica automaticamente a pasta atual, identifica todos os arquivos .dat e executa a análise para cada um deles. Clique novamente no máximo e no mínimo na janela toda vez que o script precisar de um palpite inicial educado.
- Abra e olhe para o arquivo gerado automaticamente chamado bonds.input que contém as distâncias de títulos interatômicos.
3. Realize a análise de especiação
- Calcular a topologia da ligação entre os átomos, usando o conceito de conectividade dentro da teoria dos gráficos: os átomos são os nós e as ligações interatômicas são os caminhos. O script speciation_umd.py precisa das distâncias de títulos interatômicos definidas no arquivo bonds.input .
NOTA: A matriz de conectividade é construída a cada passo: dois átomos que ficam a uma distância menor do que o raio de sua esfera de primeira coordenação correspondente são considerados ligados, ou seja, conectados. Várias redes atômicas são construídas tratando os átomos como nós em um gráfico cujas conexões são definidas por este critério geométrico. Estas redes são as espécies atômicas, e seu conjunto define a especiação atômica nesse fluido particular (Figura 4).

Figura 4: Identificação dos aglomerados atômicos.
A coordenação poliedra é definida utilizando distâncias interatômicas. Todos os átomos a uma distância menor do que um raio especificado são considerados ligados. Aqui o limiar corresponde à primeira esfera de coordenação (os círculos vermelhos claros), definido na Figura 1. A polimerização e, portanto, espécies químicas são obtidas das redes dos átomos ligados. Observe o aglomerado central Red1Grey2, que está isolado dos outros átomos, que formam um polímero infinito. Clique aqui para ver uma versão maior desta figura.
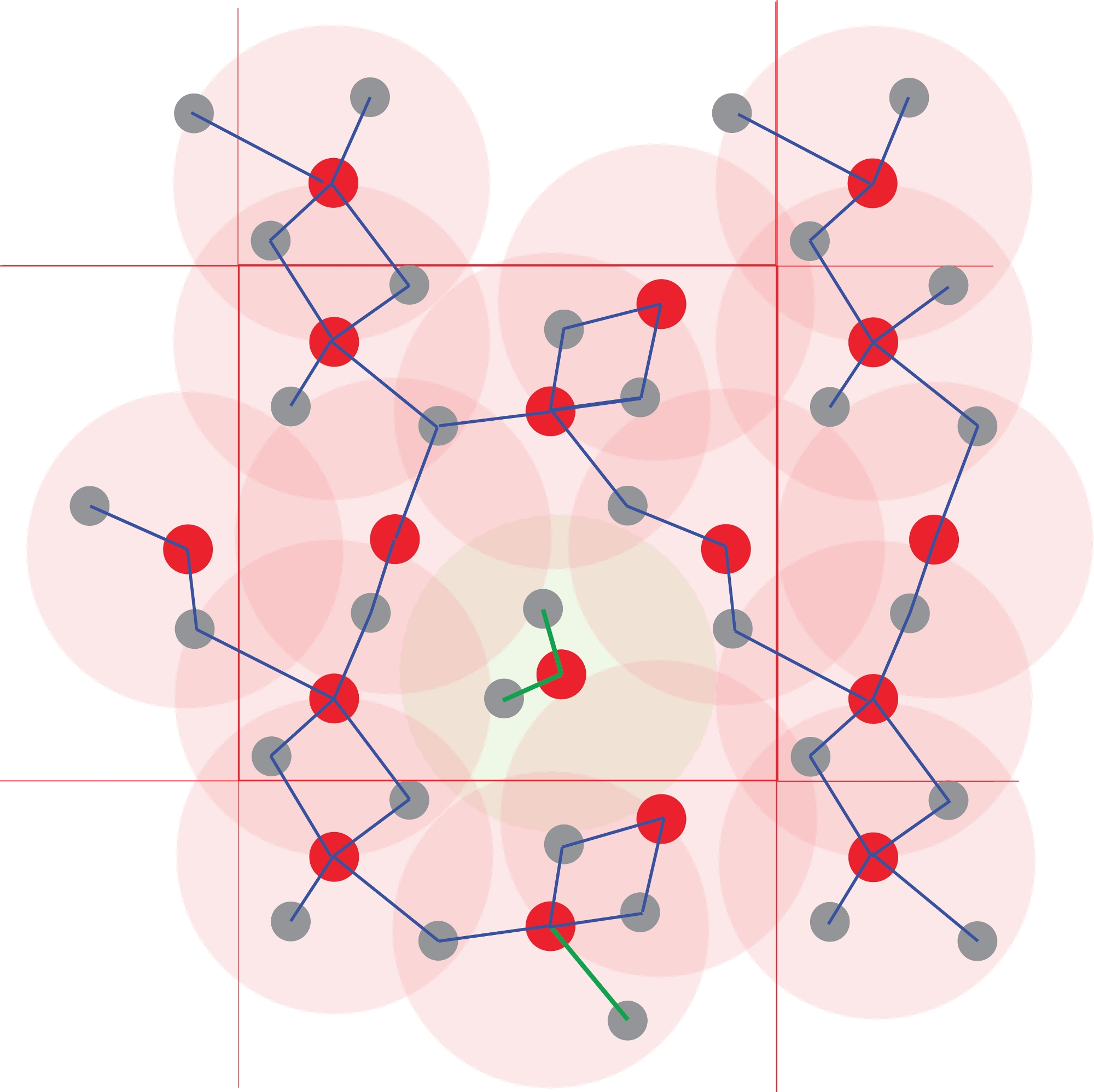{kind=link}
- Execute o script de especiação para obter a matriz de conectividade e obter a coordenação poliedra ou a polimerização. No tipo de linha de comando:
speciation_umd.py -f -s -i -l -c -a -m -r
onde a bandeira -i dá o arquivo com as distâncias de ligação interatômica, que foi produzida, por exemplo, na etapa anterior. Alternativamente, execute o script com um único comprimento para todos os títulos definidos pela bandeira -l.
NOTA: A bandeira -c especifica os átomos centrais e a bandeira -a os ligantes. Tanto átomos centrais quanto ligantes podem ser de diferentes tipos; neste caso, eles devem ser separados por írgulas. A bandeira -m dá o tempo mínimo que uma espécie deve viver para ser considerada na análise. Por padrão este tempo mínimo é zero, todas as ocorrências sendo contadas na análise final.- Execute o script speciation_umd.py com a bandeira –r 0, que mostra o gráfico de conectividade no primeiro nível para identificar a coordenação poliédra. Por exemplo, um átomo central, denotado como um ceo pode ser cercado por um ou mais ânions (Figura 4). O script de especiação identifica cada um dos poliédes de coordenação. A média ponderada de toda a coordenação poliedra dá o número de coordenação, idêntico ao obtido a partir da integração do PDF. No tipo de linha de comando:
speciation_umd.py -f -i -c -a -r 0
NOTA: Os números médios de coordenação em fluidos são números fracionados. Essa fracionamento vem da característica média da coordenação. A definição baseada na especiação produz uma representação mais intuitiva e informativa da estrutura do fluido, onde as proporções relativas das diferentes espécies, ou seja, coordenações, são quantificadas. - Execute o script speciation_umd.py com a bandeira –r 1, que mostra o gráfico de conectividade em todos os níveis de profundidade para obter a polimerização. A rede através do gráfico atômico tem uma certa profundidade, à medida que os átomos são ligados mais longe a outras ligações (por exemplo, em sequências de cáras alternados e ânions) (Figura 4).
- Execute o script speciation_umd.py com a bandeira –r 0, que mostra o gráfico de conectividade no primeiro nível para identificar a coordenação poliédra. Por exemplo, um átomo central, denotado como um ceo pode ser cercado por um ou mais ânions (Figura 4). O script de especiação identifica cada um dos poliédes de coordenação. A média ponderada de toda a coordenação poliedra dá o número de coordenação, idêntico ao obtido a partir da integração do PDF. No tipo de linha de comando:
- Abra os dois arquivos. popul.dat e . estat.dat consecutivamente; estes constituem a saída do script de especiação. Cada aglomerado é escrito em uma linha, especificando sua fórmula química, o tempo em que se formou, o tempo em que morreu, sua vida, uma matriz com a lista dos átomos formando este aglomerado. Plote a vida útil de cada aglomerado atômico de todas as espécies químicas encontradas na simulação como encontrado no arquivo .popul.dat (Figura 5).
- Traçar a análise populacional com a abundância de cada espécie, como encontrado no . .dat arquivo . Esta análise, tanto absoluta quanto relativa, corresponde às estatísticas reais da coordenação poliedra para o caso -r 0; para o caso da polimerização, com -r 1 isso precisa ser tratado cuidadosamente, pois alguma normalização sobre o número relativo de átomos pode precisar ser aplicada. A abundância corresponde à integral ao longo da vida. O. stat.dat arquivo também lista o tamanho de cada cluster, ou seja, quantos átomos o formam.
4. Coeficientes de difusão computacional
- Extrair os deslocamentos quadrados médios (MSD) dos átomos em função do tempo para obter a auto-difusividade. A fórmula padrão do MSD é:

onde os pré-fatores são renormalizações. Com a ferramenta MSD, existem diferentes maneiras de analisar os aspectos dinâmicos dos fluidos.
NOTA: T é o tempo total da simulação e N α é o número de átomos de α tipo. O tempo inicial t0 é arbitrário e abrange a primeira metade da simulação. Ninit é o número de vezes iniciais. τ é a largura do intervalo de tempo sobre o qual o MSD é computado; seu valor máximo é metade do tempo da simulação. Em implementações típicas de MSD, cada janela começa no final do anterior. Mas uma amostragem mais esparsa pode acelerar o cálculo do MSD, sem alterar a inclinação do MSD. Para isso, a janela i-th começa no tempo t0(i), mas a janela (i+1)-th começa no tempo t0(i) + τ + v, onde o valor de v é definido pelo usuário. Da mesma forma, a largura da janela é aumentada em passos discretos definidos pelo usuário, como tal: τ(i) = τ(i-1) + z. Os valores de z ("passo horizontal") e v ("passo vertical") são positivos ou zero; o padrão para ambos é 20. - Calcule o MSD usando a série de scripts msd_umd . Sua saída é impressa em um . msd.dat arquivo, onde o MSD de cada tipo atômico, átomo ou cluster é impresso em uma coluna em função do tempo.
- Calcule o MSD médio de cada tipo atômico. O MSD é computado para cada átomo e, em seguida, mediado para cada tipo atômico. O arquivo de saída contém uma coluna para cada tipo atômico. No tipo de linha de comando:
msd_umd.py -f -z -v -b - Calcule o MSD de cada átomo. O MSD é computado para cada átomo e, em seguida, mediado para cada tipo atômico. O arquivo de saída contém uma coluna para cada átomo na simulação e, em seguida, uma coluna para cada tipo atômico. Este recurso permite identificar átomos que difundem em dois ambientes diferentes, como líquido e gás, ou dois líquidos. No tipo de linha de comando:
msd_all_umd.py -f -z -v -b - Calcular o MSD da espécie química. Use a população de clusters identificados com o script de especiação e impressos no . arquivo popul.dat . O MSD é computado para cada cluster individual. O arquivo de saída contém uma coluna para cada cluster. Para evitar considerar polímeros de grande escala, coloque um limite no tamanho do cluster; seu padrão é de 20 átomos. No tipo de linha de comando:
msd_cluster_umd.py -f -p -s -b -c
NOTA: Os valores padrão são: –b 100 –s 1 –c 20.
- Calcule o MSD médio de cada tipo atômico. O MSD é computado para cada átomo e, em seguida, mediado para cada tipo atômico. O arquivo de saída contém uma coluna para cada tipo atômico. No tipo de linha de comando:
- Plote o MSD usando um software baseado em planilha (Figura 6). Em uma representação de log-log do MSD versus tempo, identifique a alteração de inclinação. Separe a primeira parte, geralmente curta, que representa o regime balístico , ou seja, a conservação da velocidade dos átomos após colisões. A segunda parte mais longa representa o regime difuso , ou seja, a dispersão da velocidade dos átomos após colisões.
- Calcule os coeficientes de difusão da inclinação do MSD como:

onde Z é o número de graus de liberdade (Z = 2 para difusão em plano, Z = 3 para difusão no espaço), e t é o passo do tempo.
5. Funções de correlação de tempo
- Calcular as funções de correlação de tempo como uma medida da inércia do sistema usando a fórmula geral:

A pode ser uma variedade de variáveis dependentes do tempo, como as posições atômicas, velocidades atômicas, tensões, polarização, etc., cada uma rendendo — através das relações Verde-Kubo12,13 — diferentes propriedades físicas, às vezes após uma nova transformação. - Analise as velocidades atômicas para obter o espectro vibracional do líquido e expressão alternativa dos coeficientes atômicos de auto-difusão.
- Execute o script vibr_spectrum_umd.py para calcular a função de auto-correlação de velocidade at atômica (VAC) para cada tipo atômico e realize sua transformação rápida-Fourier. No tipo de linha de comando:
vibr_spectrum_umd.py -f -t
onde – t é a temperatura que deve ser definida pelo usuário. O script imprime dois arquivos: o . vels.scf.dat arquivo com a função VAC para cada tipo atômico, e o . vibr.dat arquivo com o espectro vibracional decomposto em cada espécie atômica e o valor total. - Abra e leia o vels.scf.dat. Plote a função VAC a partir do arquivo vels.scf.dat usando software semelhante a planilha.
- Mantenha a parte real do VAC Fourier. Isto é o que produz o espectro vibracional, em função da frequência:

onde m estão as massas atômicas. - Plote o espectro vibracional a partir do arquivo vibr.dat usando software semelhante a planilha (Figura 7). Identifique o valor finito em ω=0 que corresponde ao caráter difuso do fluido e aos vários picos do espectro em frequência finita. Identifique a participação de cada tipo atômico ao espectro vibracional.
NOTA: A decomposição nos tipos atômicos mostra que diferentes átomos têm contribuições diferentes de ω=0, correspondendo aos seus coeficientes de difusão. A forma geral do espectro é muito mais suave com menos características do que para um sólido correspondente. - Na concha, leia a integral sobre o espectro vibracional, que produz os coeficientes de difusão para cada espécie atômica.
NOTA: As propriedades termodinâmicas podem ser obtidas por integração a partir do espectro vibracional, mas os resultados devem ser usados com cautela devido a duas aproximações: a integração é válida dentro da aproximação quase harmônica, que não necessariamente se mantém em altas temperaturas; e a parte semelhante a gás do espectro correspondente à difusão precisa ser descartada. A integração deve então ser feita apenas sobre a parte de rede do espectro. Mas essa separação geralmente requer várias outras etapas pós-processamento e cálculos14, que não são cobertos pelo atual pacote UMD.
- Execute o script vibr_spectrum_umd.py para calcular a função de auto-correlação de velocidade at atômica (VAC) para cada tipo atômico e realize sua transformação rápida-Fourier. No tipo de linha de comando:
- Execute o script viscosity_umd.py para analisar a auto-correlação dos componentes tensor de estresse para estimar a viscosidade do derretimento. No tipo de linha de comando:
viscosity_umd.py -f -i -s -o -l
NOTA: Este recurso é exploratório e qualquer resultado deve ser tomado com cautela. Em primeiro lugar, verifique minuciosamente a convergência da viscosidade em relação ao comprimento da simulação.- Derivar a viscosidade do fluido da auto-correlação do tensor de estresse15 como:

onde V e T são o volume e a temperatura, respectivamente, κB é a constante Boltzmann e σ ij o componente ij off-diagonal do tensor de estresse, expresso em coordenadas cartesianas. - Use um ajuste mais adequado para obter uma estimativa mais robusta da viscosidade15,16 e evite o ruído da função de auto-correlação de tensão de estresse que pode surgir do tamanho finito e da duração finita das simulações. Para a função de correlação automática do tensor de estresse, utilize a seguinte forma funcional15,16 que produz bons resultados:

onde A, B, τ1, τ2 e ω são parâmetros de ajuste. Após a integração, a expressão para a viscosidade torna-se:
- Derivar a viscosidade do fluido da auto-correlação do tensor de estresse15 como:
6. Parâmetros termodinâmicos decorrentes das simulações.
- Execute averages.py para extrair os valores médios e o spread (como desvio padrão) para pressão, temperatura, densidade e energia interna dos arquivos umd . No tipo de linha de comando:
averages.py -f -s
com -s 0 como padrão. - Calcular o erro estatístico da média, utilizando métodos de bloqueio.
NOTA: Existem vários sabores deste método. Seguindo o trabalho de Allen e Tildesley2, é comum a média sobre sequências de blocos temporários, de comprimento cada vez mais longo, e estimar o desvio padrão em relação à média aritmética17. A convergência pode ser alcançada no limite de tamanhos de blocos longos e longos o suficiente, quando a amostragem não é corrigida. Embora o valor limite real para a convergência geralmente precise ser escolhido manualmente.- Use o método de halving18: começando com a amostra inicial de dados, a cada etapa κ, reduza pela metade o número de amostras, com uma média sobre cada duas amostras consecutivas correspondentes da etapa anterior κ−1:

- Execute o script fullaverages.py para realizar a análise estatística completa, incluindo o erro da média. No tipo de linha de comando:
fullaverages.py -s -u
NOTA: O script é automatizado a ponto de procurar todos os arquivos .umd.dat no diretório atual e realizar a análise para todos eles. Os padrões são -s 0 –u 0. Para -u 0 a saída é mínima, e para -u 1 a saída está na íntegra, com várias unidades alternativas impressas. Este script requer suporte gráfico, pois cria uma imagem gráfica para verificar a convergência para estimar o erro na média.
- Use o método de halving18: começando com a amostra inicial de dados, a cada etapa κ, reduza pela metade o número de amostras, com uma média sobre cada duas amostras consecutivas correspondentes da etapa anterior κ−1:
Resultados
Pirolite é um modelo de derretimento de silicato multicomponimento (0,5Na2O 2CaO 1.5Al2O3 4FeO 30MgO 24SiO2) que melhor aproxima a composição do silicato a granel da Terra — a média geoquímica ou todo o nosso planeta, exceto por seu núcleo baseado em ferro19. O início da Terra foi dominado por uma série de eventos de fusão em larga escala20, o último pode ter engolido todo o planeta, após sua condensação para o disco protolunar21. A pirolite representa a melhor aproximação à composição de tais oceanos de magma em escala planetária. Consequentemente, estudamos extensivamente as propriedades físicas do fusão pirolito na faixa de temperatura de 3.000\u20125.000 K e 0\u2012150 GPa de simulações de dinâmica molecular ab initio na implementação do VASP. Estas condições termodinâmicas caracterizam inteiramente as condições mais extremas do oceano magma da Terra. Nosso estudo é um excelente exemplo de um uso bem sucedido do pacote UMD para toda a análise aprofundada dos derretimentos22. Calculamos a distribuição e os comprimentos médios dos títulos, traçamos as mudanças na coordenação de cátion-oxigênio, e comparamos nossos resultados com estudos experimentais e computacionais anteriores sobre silicatos amorfos de várias composições. Nossa análise aprofundada ajudou a decompor os números de coordenação padrão em seus constituintes básicos, delinear a presença de poliedra de coordenação exótica no derretimento, e extrair vidas para toda a coordenação poliedra. Também destacou a importância da amostragem em simulações em termos tanto de duração da trajetória quanto do número de átomos presentes no sistema modelado. Quanto ao pós-processamento, a análise umd é independente desses fatores, no entanto, eles devem ser levados em conta ao interpretar os resultados fornecidos pelo pacote UMD. Aqui, mostramos alguns exemplos de como o pacote UMD pode ser usado para extrair várias características características dos derretimentos, com uma aplicação para pirolito derretido.
A função de distribuição do par Si-O obtida a partir do script gofrs_umd.py mostra que o raio da primeira esfera de coordenação, que é o primeiro mínimo da função g(r), fica em torno de 2,5 angstroms em T = 3000 K e P = 4,6 GPa. O máximo do g(r) está em 1.635 Å — esta é a melhor aproximação ao comprimento da curva. A cauda longa é devido à temperatura. Usando esse limite como distância de ligação Si-O, a análise de especiação mostra que as unidades SiO4, que podem durar até alguns picosegundos, dominam o derretimento (Figura 5). Há uma parte importante do derretimento que mostra polimerização parcial, como refletido pela presença de dimers como Si2O7, e aparadores como unidades Si3Ox. Sua vida correspondente é na ordem do picosegundo. Polímeros de ordem superior têm uma vida útil consideravelmente menor.

Figura 5: Vida útil da espécie química Si-O.
A especiação é identificada em um derretimento multicomponimento a 4,6 GPa e 3000 K. Os rótulos marcam os monômeros SiO3, SiO4 e SiO5 e os vários polímeros SixOy. Clique aqui para ver uma versão maior desta figura.
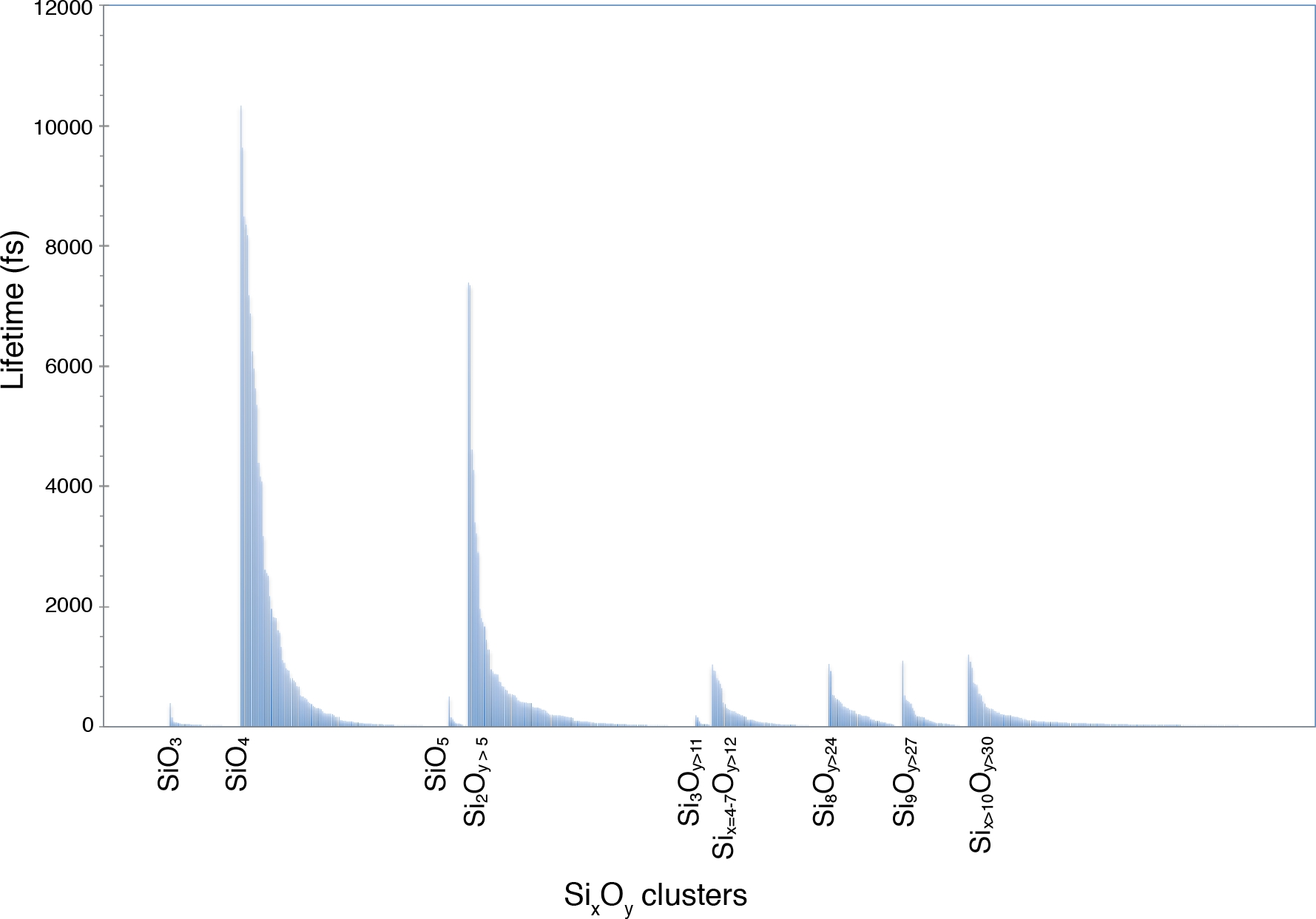{kind=link}
Os diferentes valores das etapas verticais e horizontais, definidos pelas bandeiras –z e v acima, produzem várias amostras do MSD (Figura 6). Mesmo grandes valores de z e v são suficientes para definir as encostas e, assim, os coeficientes de difusão dos diferentes átomos. O ganho de tempo para o pós-processamento é notável quando se vai a grandes valores de z e v. O MSD oferece um critério de validação muito forte para a qualidade das simulações. Se a parte de difusão do MSD não for suficientemente longa, isso é um sinal de que a simulação é muito curta, e não consegue atingir o estado fluido em um sentido estatístico. O requisito mínimo para a parte difusa do MSD depende muito do sistema. Pode-se exigir que todos os átomos mudem seu local pelo menos uma vez na estrutura do derretimento para que ele seja considerado como um fluido10. Um excelente exemplo com aplicações em ciências planetárias é o complexo derretimento do silicato em altas pressões próximas ou mesmo abaixo de sua linha liquidus11. Os átomos de Si, os principais cations formadores de rede, trocam de local após mais de duas dúzias de picosegundos. Simulações mais curtas que esse limiar seriam consideravelmente menos amostrais do possível espaço configuracional. No entanto, como os ânions coordenadores, ou seja, os átomos O, movem-se mais rápido que os átomos centrais do Si, eles podem compensar parte da mobilidade lenta de Si. Como tal, todo o sistema poderia de fato cobrir uma melhor amostra do espaço configuracional do que assumido apenas a partir dos deslocamentos si.

Figura 6: Deslocamentos quadrados médios (MSD).
O MSD é ilustrado para alguns tipos atômicos de um derretimento de silicato multicompontado. A amostragem com várias etapas horizontais e verticais, z e v, produz resultados consistentes. Círculos sólidos: -z 50 –v 50. Círculos abertos: -z 250 –v 500. Clique aqui para ver uma versão maior desta figura.
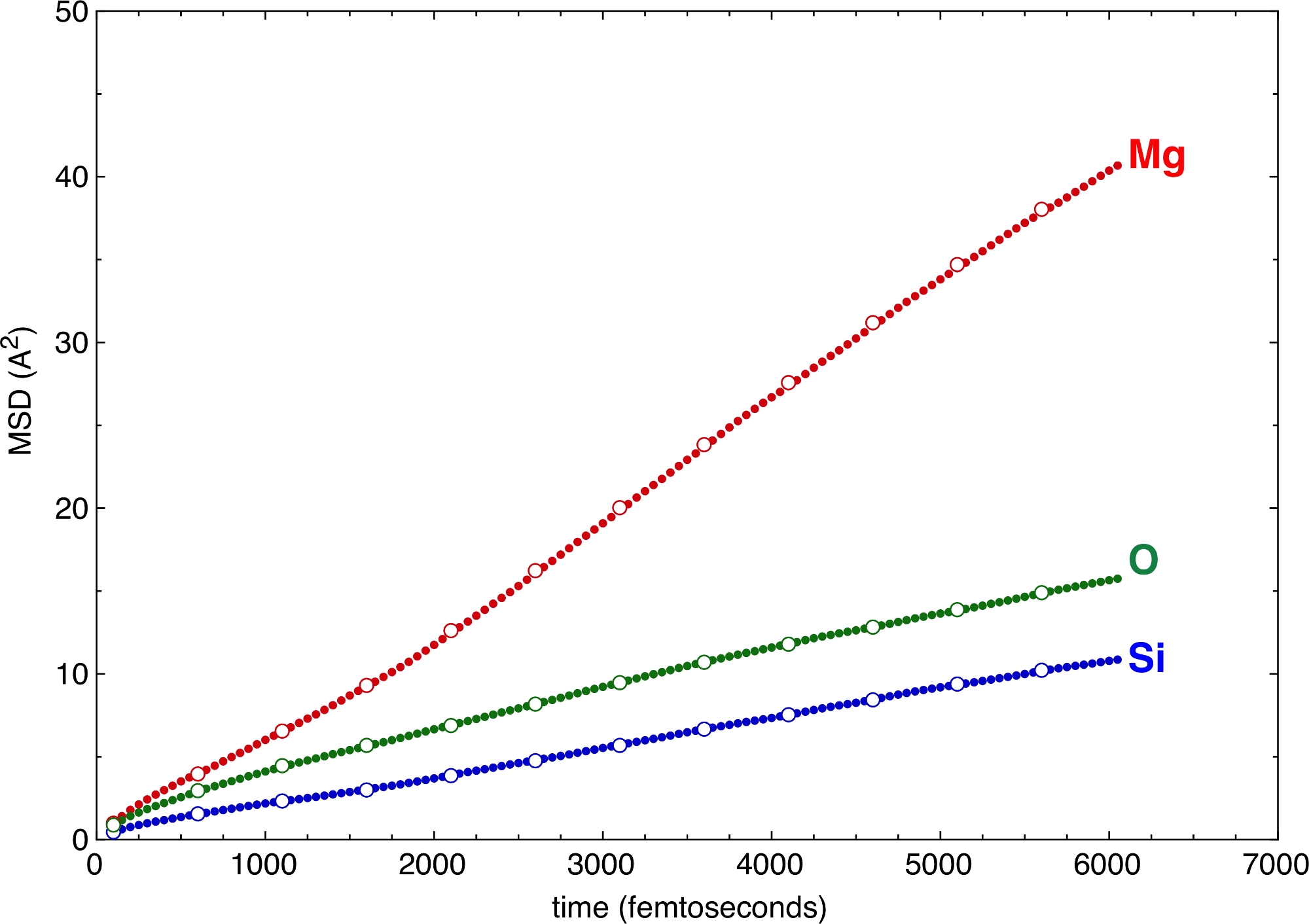{kind=link}
Finalmente, as funções atômicas de VAC produzem o espectro vibracional do derretimento. A Figura 7 mostra o espectro nas mesmas condições de pressão e temperatura acima. Representamos as contribuições dos átomos de Mg, Si e O, bem como o valor total. Em frequência zero há um valor finito do espectro, que corresponde ao caráter de difusão do derretimento. A extração das propriedades termodinâmicas do espectro vibracional precisa remover esse caráter difusivo semelhante ao gás de zero, mas também levar em conta adequadamente sua decadência em frequências mais altas.

Figura 7: O espectro vibracional do pirolito derrete.
A parte real da transformação fourier da função de auto-correlação velocidade atômica produz o espectro vibracional. Aqui o espectro é computado para um derretimento de silicato multicompontado. Os fluidos têm um caráter difusivo não-zero em termos de gás em zero frequência. Clique aqui para ver uma versão maior desta figura.
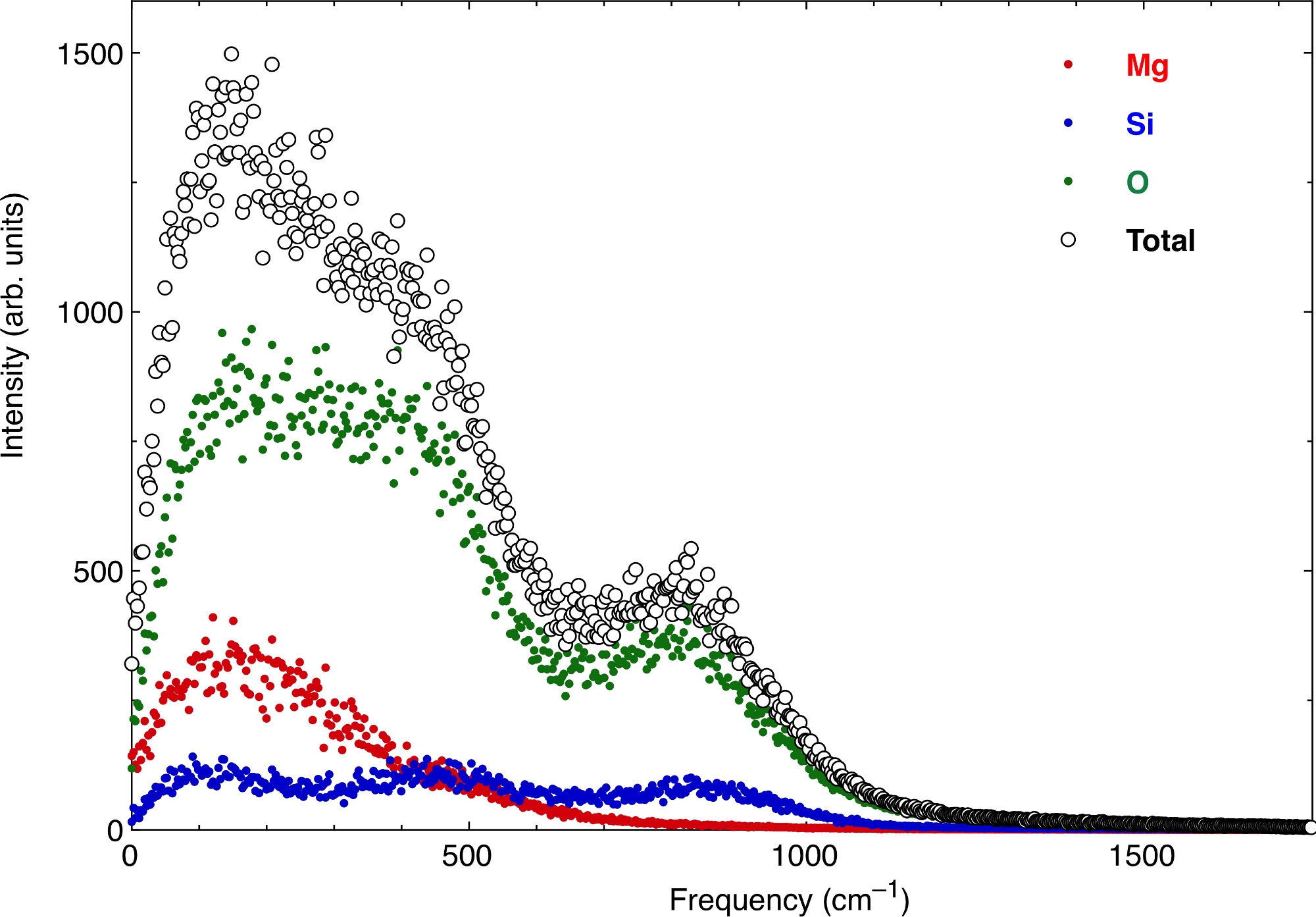{kind=link}
Discussão
O pacote UMD foi projetado para funcionar melhor com simulações ab initio, onde o número de instantâneos é tipicamente limitado a dezenas a centenas de milhares de instantâneos, com algumas centenas de átomos por célula de unidade. Simulações maiores também são tratáveis desde que a máquina na qual a pós-processamento seja executada tenha recursos de memória ativos suficientes. O código distingue-se pela variedade de propriedades que pode calcular e por sua licença de código aberto.
Os arquivos umd.dat são apropriados para os conjuntos que preservam o número de partículas inalteradas ao longo da simulação. O pacote UMD pode ler arquivos decorrentes de cálculos onde a forma e o volume da caixa de simulação variam. Estes cobrem os cálculos mais comuns, como NVT e NPT, onde o número de partículas, N, temperatura T, volume, V e/ou pressão, P, são mantidos constantes.
Para o tempo começam a função de distribuição de pares, bem como todos os scripts que precisam estimar as distâncias interatômicas, como os scripts de especiação, funcionam apenas para células unitárias ortogonais, ou seja, para células cúbicas, tetragonais e ortotómicas, onde os ângulos entre os eixos são de 90°.
As principais linhas de desenvolvimento para a versão 2.0 são a remoção da restrição de ortogonalidade para distâncias e a adição de mais recursos para os scripts de especiação: analisar ligações químicas individuais, analisar os ângulos interatômicos e implementar a segunda esfera de coordenação. Com a ajuda da colaboração externa, estamos trabalhando na portabilidade do código em uma GPU para análise mais rápida em sistemas maiores.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho foi apoiado pelo Conselho Europeu de Pesquisa (ERC) no âmbito do programa de pesquisa e inovação da União Europeia Horizon 2020 (número de subvenção 681818 IMPACT ao RC), pela Diretoria de Física Extrema e Química do Observatório de Carbono Profundo, e pelo Conselho de Pesquisa da Noruega através de seu programa de financiamento centros de excelência, número do projeto 223272. Reconhecemos o acesso aos supercomputadores GENCI através da série stl2816 de bolsas de computação eDARI, ao supercomputador Irene AMD através do projeto PRACE RA4947, e ao supercomputador Fram através do UNINETT Sigma2 NN9697K. A FS foi apoiada por um projeto marie skłodowska-curie (contrato de concessão ABISSE No.750901).
Materiais
| Name | Company | Catalog Number | Comments |
| getopt library | open-source | ||
| glob library | open-source | ||
| matplotlib library | open-source | ||
| numpy library | open-source | ||
| os library | open-source | ||
| Python software | The Python Software Foundation | Version 2 and 3 | open-source |
| random library | open-source | ||
| re library | open-source | ||
| scipy library | open-source | ||
| subprocess library | open-source | ||
| sys library | open-source |
Referências
- Frenkel, D., Smit, B. . Understanding Molecular Simulation. From Algorithms to Applications. , (2001).
- Allen, M. P., Tildesley, D. J., Allen, T. . Computer Simulation of Liquids. , (1989).
- Zepeda-Ruiz, L. A., Stukowski, A., Oppelstrup, T., Bulatov, V. V. Probing the limits of metal plasticity with molecular-dynamics simulations. Nature Publishing Group. 550 (7677), 492-495 (2017).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modeling. 14 (1), 33-38 (1996).
- Momma, K., Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 44 (6), 1272-1276 (2011).
- Brehm, M., Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. Journal of Chemical Information and Modeling. 51 (8), 2007-2023 (2011).
- Stixrude, L. Visualization-based analysis of structural and dynamical properties of simulated hydrous silicate melt. Physics and Chemistry of Minerals. 37 (2), 103-117 (2009).
- Kresse, G., Hafner, J. Ab initio Molecular-Dynamics for Liquid-Metals. Physical Review B. 47 (1), 558-561 (1993).
- Gygi, F. Architecture of Qbox: A scalable first-principles molecular dynamics code. IBM Journal of Research and Development. 52 (1-2), 137-144 (2008).
- Harvey, J. P., Asimow, P. D. Current limitations of molecular dynamic simulations as probes of thermo-physical behavior of silicate melts. American Mineralogist. 100 (8-9), 1866-1882 (2015).
- Caracas, R., Hirose, K., Nomura, R., Ballmer, M. D. Melt-crystal density crossover in a deep magma ocean. Earth and Planetary Science Letters. 516, 202-211 (2019).
- Green, M. S. Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena. II. Irreversible Processes in Fluids. The Journal of Chemical Physics. 22 (3), 398-413 (1954).
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. Journal of the Physical Society of Japan. 12 (6), 570-586 (1957).
- Lin, S. T., Blanco, M., Goddard, W. A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. The Journal of Chemical Physics. 119 (22), 11792-11805 (2003).
- Meyer, E. R., Kress, J. D., Collins, L. A., Ticknor, C. Effect of correlation on viscosity and diffusion in molecular-dynamics simulations. Physical Review E. 90 (4), 1198-1212 (2014).
- Soubiran, F., Militzer, B., Driver, K. P., Zhang, S. Properties of hydrogen, helium, and silicon dioxide mixtures in giant planet interiors. Physics of Plasmas. 24 (4), 041401-041407 (2017).
- Flyvbjerg, H., Petersen, H. G. Error estimates on averages of correlated data. The Journal of Chemical Physics. 91 (1), 461-466 (1989).
- Tuckerman, M. E. . Statistical mechanics: theory and molecular simulation. , (2010).
- McDonough, W. F., Sun, S. S. The composition of the Earth. Chemical Geology. 120, 223-253 (1995).
- Elkins-Taton, L. T. Magma oceans in the inner solar system. Annual Review of Earth and Planetary Sciences. 40, 113-139 (2012).
- Lock, S. J., et al. The origin of the Moon within a terrestrial synestia. J. Geophysical Research: Planets. 123, 910-951 (2018).
- Solomatova, N. V., Caracas, R. Pressure-induced coordination changes in a pyrolitic silicate melt from ab initio molecular dynamics simulations. Journal of Geophysical Research: Solid Earth. 124, 11232-11250 (2019).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados