
Method Article
DNAを用いた細胞のシンプル、手頃な価格、モジュール式パターニング
要約
ここでは、DNAプログラム付着を用いた単細胞分解能でマイクロパターン細胞に対するプロトコルを提示する。このプロトコルは、ベンチトップフォトリソグラフィプラットフォームを使用して、ガラススライド上にDNAオリゴヌクレオチドのパターンを作成し、市販の相補オリゴヌクレオチドを有する細胞膜にラベルを付けます。オリゴのハイブリダイゼーションは、プログラムされた細胞接着をもたらす。
要約
細胞の相対的な位置付けは、細胞と細胞の相互作用を組織する微小環境の重要な特徴です。同じタイプまたは異なるタイプの細胞間の相互作用を研究するために、マイクロパターン化技術が有用であることが証明された。DNAプログラムによる細胞集合体(DPAC)は、DNAハイブリダイゼーションを用いて細胞の基質や他の細胞への接着を標的とするマイクロパターニング技術です。DPACの最も基本的な操作は、脂質修飾オリゴヌクレオチドで細胞膜を修飾し、相補的なDNA配列でパターン化された基質上でそれらを流すことから始まります。細胞は、相補的なDNA配列を見つける場合にのみ、基質に選択的に付着します。非接着細胞は洗い流され、接着細胞のパターンが明らかになります。追加の操作には、細胞基板または細胞-細胞接着のさらなるラウンド、ならびに長期培養のための埋め込みヒドロゲルにDPACによって形成されたパターンを移すが含まれる。以前は、表面上のオリゴヌクレオチドをパターン化し、DNA配列を持つ細胞を修飾する方法には、それぞれ特殊な機器とカスタムDNA合成が必要でした。我々は、モジュラー形式を使用して展開された安価なベンチトップフォトリソグラフィーセットアップおよび市販のコレステロール修飾オリゴヌクレオチド(CMO)を利用して、プロトコルの更新版を報告する。CMO標識細胞はDNAパターン化基質に対して高効率で接着する。このアプローチは、複数の細胞タイプを高精度に一度にパターン化し、細胞外マトリックス内に埋め込まれたマイクロ組織の配列を作成するために使用することができます。この方法の利点は、その高解像度、マイクロパターンを破壊することなく3次元微小環境に細胞を埋め込む能力、および任意の細胞タイプのパターン化における柔軟性を含む。
概要
組織における細胞の位置合わせは、微小環境1、2、3、4の重要な特徴である。空間制御された配置に生細胞をパターン化するために使用される技術は、分化4、5、6、7、8、細胞運動性9、形態形成10、11、代謝13、および細胞細胞相互作用7、14を研究するための貴重な実験ツールである.細胞のパターニングにはさまざまな方法があり、それぞれに利点と欠点3、4があります。マイクロコンタクト印刷やレーザーカットステンシルなど、細胞外マトリックス(ECM)タンパク質の接着島を作る方法は、シンプルでスケーラブルです。しかし、異なるECM分子に対する異なる細胞型の接着特性が15、16、17に類似していることが多いため、一度に1つまたは2つ以上の細胞型をパターン化することは困難である。より複雑な微小パターンは、光誘導分子吸着(LIMAP)を用いて作成することができ、UV光を用いてPEG被覆領域をアブレーションし、その後のタンパク質吸着を可能にする技術である18、19。このプロセスを繰り返して、複数の細胞タイプの高解像度マイクロパターンを作成できます。しかし、異なるタンパク質パッチへの細胞のクロス結合が起こり得、パターン特異性19に劣るパターンを生じる。マイクロメカニカル再構成可能な培養装置に細胞を播種するなどの物理的方法は、動的制御を伴う構造化された共培養を作成することができるが、マイクロコンタクト印刷またはLIMAP14,8のパターン設計において柔軟性がない。他の技術とは異なり、バイオプリンティングはヒドロゲル20,21内の細胞の3次元配置を作成することができる。しかし、バイオプリント構造は、他のマイクロパターニング技術よりもはるかに低い解像度を有し、数百ミクロン22の平均特徴サイズを有する。理想的な細胞パターニング法は、高解像度、パターン複数の細胞タイプ、簡単にアクセスできる機器や試薬を使用し、3次元(3D)細胞培養のためのヒドロゲルに成功したパターンを埋め込む能力を有するであろう。本稿では、DNAハイブリダイゼーションの柔軟性と速度を利用して細胞接着をターゲットとする細胞マイクロパターニング技術であるCMO-DPACについて紹介します。この方法は、以前のプロトコル23、24から、より手頃な価格、モジュラー、アクセス可能にしました。現在のプロトコルを使用すると、あらゆるラボが、特殊な機器や専門知識を持たない完全に機能するシステムをセットアップできるはずです。
DNAプログラムによる細胞組立(DPAC)は、細胞間の間隔と組織形状を正確に制御して、単細胞解像度で細胞をパターン化する強力な組織工学技術です。DPACでは、細胞膜にハイブリダイズするように設計された2つの脂質修飾オリゴを使用して、細胞膜をDNAオリゴヌクレオチド(オリゴ)で修飾しています。オリゴは疎水性脂質に結合しているため、それらがハイブリダイズする細胞膜25に迅速に分け、非共有結合分子の純疎水性を増加させ、それによって細胞表面26での寿命を増強する。オリゴは、他の細胞またはDNA機能化ガラススライド上の相補オリゴとハイブリダイズして、所定の組成、細胞間間隔、および幾何学23、24を有する定義された2Dまたは3D細胞パターンを作成することができる方法で細胞表面に提示される。パターン化されたマイクロ組織は、表面から酵素的に切断され、長期間にわたる3D培養のためにヒドロゲルに埋め込むことができます。原発細胞または幹細胞と組み合わせて使用すると、細胞の結果として得られるコレクションは、形態形成を受け、オルガノイド23、27、28に形成することができる。DPACは、競合するシグナル6,29に応答して成人の神経幹細胞運命のダイナミクスを調べ、乳腺上皮細胞23,28の自己組織化を研究し、間葉縮合27を通じて「組織折り紙」を生成するために適用された。
DPACは、複数の細胞集団の正確な配置を可能にし、(ミクロンの順序で)22、23の押出ベースのバイオプリンターよりも実質的に優れた解像度を有する。また、マイクロコンタクト印刷などのECMベースのパターン化方法とは異なり、DPACは、ECM被覆面15,23に対して異なる細胞タイプの差動接着を必要としない。組織の組成がその挙動にどのような影響を与えるか、細胞が決定を下す際に複数の細胞と微小環境の手掛かりをどのように統合するか、および細胞のペアがどのように相互作用するかについての質問に答えるのに理想的です。他のマイクロパターン化法よりもこの方法の利点は、単一の画像化平面における3D細胞培養に使用できることであり、組織自己組織化およびオルガノイド形態形成23、27、30のタイムラプス研究を促進する。
これらの利点にもかかわらず、DPACの実装は、カスタムオリゴヌクレオチド試薬の合成とDNAパターニング23、24のための特殊な機器へのアクセスを必要とし、広範な採用を制限しています。例えば、元のプロトコルで使用される最適脂質修飾オリゴ(DMO)は、カスタム合成、リグノセリック酸またはパルミチン酸で修飾、精製26としなければならない。このプロセスでは、DNAシンセサイザーと高速液体クロマトグラフィー装置の使用、ならびに機関および連邦規制の対象となるメチルアミンなどの関連試薬の購入が必要です。代わりに、DMOを一括購入することもできますが、これにはこの技術に対する前もってかなりの投資が必要です。
これらの制限を克服するために、カスタム合成されたMMOの代わりに市販のコレステロール修飾オリゴ(CMO)を使用するDPACの改訂版を開発しました。さらにコストを削減し、プラットフォームの柔軟性を高めるために、モジュラー、3オリゴシステムに変更しました。このプロトコルのユーザーは、各々の細胞集団に対して新しいコレステロール修飾オリゴを注文する代わりに、すべての細胞集団に同じコレステロール修飾オリゴ(「ユニバーサルアンカー」と「ユニバーサル共同アンカー」)を使用し、ユニバーサルアンカーと別のタイプのアダプタストランドの両方でハイブリダイズする安価で変更されていないオリゴ(「アダプタストランド」)を採用することができます。
元のDPACプロトコルのもう一つの制限は、高解像度の液体プリンタ(例えば、ナノeナブラー、バイオフォースナノサイエンス)23、24を用いてDNAパターンスライドを作成することであった。この器械は異常な決断および低い試薬の要求を自慢するが、ほとんどの機関にとって利用できない、比較的低い印刷率(およそ1特徴は毎秒パターン化される)。近年、DNAの特徴を表面にパターン化する2つのフォトリソグラフィ法が開発されました。今回、Viiolaたちの研究グループは、UV光30への曝露時に一本鎖DNAオリゴを共有結合するポリアクリルアミドおよびベンゾフェノンコーティングを使用した。この方法を用いて、細胞の収縮性と自己組織化の結果として、大規模なプログラムされた形状変化を受けた組織足場を作成することができた。シャイデラーらは、陽性フォトレジストのUV曝露を用い、アミン修飾DNAオリゴをアルデヒド機能化スライド29に選択的に露出させる方法を開発した。焼成および還元的アミノ化の後、アミン修飾DNAは表面に共有結合される。この方法は、空間的に自己再生および分化の手掛かりを提示する成人神経幹細胞の応答を調べるのに用いた。この記事では、シャイデラーらのプロトコルを適応させ、CMO標識細胞を捕捉するDNAパターンを作成します。このフォトパターニングプロトコルは、クリーンルームを使用せずに実行できます。それは簡単にベンチトップやヒュームフードに展開されている安価で市販の機器を使用しています。安価なまたはDIY(日曜大工)フォトリソグラフィー装置を使用すると、クリーンルーム設備にアクセスすることなく研究者にアクセスでき、研究者は時間やリソース31,32の大きな投資なしに技術を試すことができます。しかし、クリーンルーム施設で一般的に見られる市販のスピンコーターとマスクアライナーを使用することで、より良い分解能と複数のDNA特徴のアライメントを達成することができます。
ここでは、DNAベースの接着を用いて単細胞分解能で細胞をパターン化する方法について述べる。まず、正のフォトレジストを用いたフォトパターニングを使用して、アルデヒド修飾ガラス基板上にアミン修飾DNAの高解像度パターンを作成します。次に、スライドは非特異的な細胞の結合を減らすために処理され、PDMSフローセルはパターン化された領域の上に細胞を閉じ込めるために作成される。次に、細胞はコレステロールで機能する短いDNAオリゴヌクレオチドで標識され、その結果、細胞膜に挿入されます。その後、細胞はDNAマイクロパターンの上に流されます。ガラス表面上の細胞表面DNAとDNAとのハイブリダイゼーションにより、DNAパターンに対する細胞の特異的接着が生じます。非接着細胞は洗い流され、接着細胞パターンが明らかになります。このプロセスを繰り返して、複数のセルタイプをパターン化したり、多層構造を作成したりできます。必要に応じて、3D細胞培養のために細胞をECMに完全に埋め込むことができます。
プロトコル
1. 設計実験
- 目的の実験を計画し、特徴のサイズ、特徴の間隔、関係するセルの数、および互いに対するセルの配置を考慮します。実験計画のガイドである 補足ファイル 1、およびサンプルのオリゴシーケンスを含む 補足ファイル 2を参照してください。
- コンピュータ支援設計ソフトウェアを使用してフォトマスクを設計します。フォトマスクの例は 、補足ファイル 3にあります。
- 標準的な顕微鏡スライド(25 mm x 75 mm)の寸法の長方形を描きます。
- 幅10mm、長さ10mmの長方形の4つの領域をスライド全体に均等に配置します。
- 各領域内で、実験の目的のサイズ、形状、間隔を表すフィーチャを描画します。セルは、実験でこれらの機能にのみ接着します。
- 複数のセルタイプに整列したフォトマスクを作成するには、すべての機能セットを含むマスター図面を作成し、各セルタイプに対応するバージョンを保存します。
- この CAD 図面から高解像度 (少なくとも 20,000 ドット/インチ) の透明フォトマスクを、1.2.3 透明で描画されたフィーチャと大きい領域を黒で並べてください。
2. アルデヒド機能化スライド上のフォトパターンDNA(シャイデラー等から適応したプロトコル)
- 複数の細胞タイプをパターニングする場合は、DNAパターニングの前に、アルデヒド機能化スライド上に受託マーカーを作製し、特徴のアライメントを容易にする。補助 的なファイル 1で、受託者マーカーを作成するための別の方法が提案されています。
- 金属受託マーカーを作成するには、ステップ2.3から2.11に記載されているようにS1813陽性フォトレジストを適用します。後で簡単に整列できる大きな機能を含むフォトマスクを使用します。DNAパターニングに使用されるフォトマスクの設計にこれらの特徴を組み込みます。
- 電子銃蒸発29を用いて、チタンの薄膜(100オングストローム)をスライドに堆積する。アセトンを用いて余分な金属とフォトレジストを除去し、次にDNAフォトパターニングに進みます。
- DNAバッファーに5'アミン修飾オリゴの20 μM溶液を調製します(50 mMのリン酸ナトリウム水、pH = 8.5)。推奨されるオリゴ シーケンスについては、補足ファイル2 を参照してください。
注:一部のパターンや用途ではアミン修飾オリゴを5μMまで使用できるため、表面DNA濃度を最適化する必要があります。 - ホットプレートを100°Cに予熱する。
- 両面テープまたは真空を使用して、スピンコーターのローターにアルデヒド機能ガラススライドを取り付けます。
注意: スピンコーティング中のスライド剥離は安全上のリスクです。常に、アクリルボックスなどの蓋付きの容器にスピンコートを使用してください。
注: ガラスを傷つけるように、ダイヤモンドの筆記者または類似の実装を使用して、スライドの隅にラベルを付けます。これは、フォトレジストが洗い流された後のスライドの識別および向きに役立つ。 - 使い捨てのピペットを使用して、正のフォトレジストをアルデヒドスライドに落とします。コーティングの場合でも、真ん中に1つの大きな滴ではなく、スライド全体にフォトレジストの小さな滴を追加します(補足図1A)。
- スピンコーターを使用して、スライドを3000 rpmで30s回転させます。
- スライドを100°Cのホットプレートに1.5分間置き(ソフトベーク)してフォトレジストを架橋します。
- ホットプレートからスライドを取り外します。スライドの上にこの実験に必要な機能を持つフォトマスクを置き、ガラス片でフォトマスクの重量を量ります(補足図1B、C)。設定全体を不透明なボックス(補足図1D)で覆います。2分間のUVランプ(365 nm波長、360 mW、スライドからの5インチ、全放射エネルギー密度100 mJ/cm2)で露出します。
注:UV光はフォトマスクの透明な領域の下のフォトレジストのポリマー結合を破壊し、DNAが後で付着できる領域を作り出します。 - 3-5分間の現像液に浸漬してスライドを開発する(補足図1E)。
- 余分な開発液を水で洗い流してください。空気または窒素の流れの下で乾燥します。(補足図1F)。
- 顕微鏡下のスライドを見てフォトリソグラフィが成功したことを確認します。フォトレジストは UV 光に敏感であるため、この手順を迅速に実行し、他のスライドを準備しながら、暗闇の中でスライドを保存します (該当する場合)。
注: 正常にパターン化されたスライドでは、各フィーチャのエッジが鋭く定義され、亀裂が発生しない、エッジにフィーチャの歪みがない必要があります。正しいフォトリソグラフィと誤ったフォトリソグラフィーの例は 、補足図 2Aに示されています。フォトリソグラフィで目的の機能品質が得られていない場合のトラブルシューティングの推奨事項については、表 1 を参照してください。 - 20 μMアミン修飾オリゴ溶液(ステップ2.1)の液滴をスライドのフォトパターン化された各領域に追加します。ピペットチップを使用して、スライドを傷つけないように注意して、領域全体に液滴をそっと広げます。(補足図1G)。
- 65~70°Cのオーブンで、DNA溶液がスライド表面に完全に乾燥するまで(約1時間)焼きます。
- パターン化された焼きスライドを15cmの細胞培養皿に入れ、シェーカーの上にヒュームフードに入れることで還元的なアミノ化を行います。100mgのホウ水素ナトリウムを計量する。ヒュームフードに、40 mLのリン酸緩衝生理食塩水(PBS)を加え、そっと混ぜ、パターン化されたスライドを含む皿に加えます。穏やかな揺れで15分間反応を進めます。
注:オリゴのアミンは、スライド表面にアルデヒドを持つシフベースを最初に形成します。これは、DPACで使用する前に不可逆結合に変換する必要がある可逆的な共有結合です。還元剤(ホウ水素ナトリウム)の添加は、還元的アミノ化によりシッフ塩基を二次アミンに変換する。
注意:水素化水素ナトリウムと水の反応は水素ガスを作り出し、反応が始まった後も何時間も何日も続きます。ヒュームフードで還元的アミノ化ステップを実行し、すべてのホロハイドライド溶液廃棄物を、少なくとも24時間ヒュームフードに開いた容器または緩くキャップされた容器に入れておきます。 - 未反応のDNAを、0.1%のドデシル硫酸ナトリウム(SDS)で2回水に洗浄し、蒸留水で3回洗浄して除去します。スライドを窒素または空気の流れの下で乾燥させます。
- 残りのフォトレジストを除去するためにアセトンでスライドをすすいでください。
注:この時点で、DNAは不可逆的かつ共有的にスライドに結合しており、未反応のアルデヒド官能基はすべてアルコールに変換されています。フォトレジストは不要になりました。 - 複数のオリゴをパターン化する場合は、ステップ 2.4 に戻り、フォトマスクを受託者マークに合わせて、繰り返します。
注: ここで実験を一時停止できます。真空デシケータにスライドを保管します。乾燥した条件下では、スライドは品質を損なうことなく最大3ヶ月間保存することができます。
3. スライド疎水性(オプション)を作る(Todhunterらから適応したプロトコル)
注: スライドの表面の化学を変更して、より不活性で疎水性を高めるのは、有利ですが、必須ではありません。非特異的な細胞の付着は、これらの表面33上で減少し、それによって、スライドの非パターン化領域への細胞の非特異的結合を緩和する。さらに、パターン化された細胞が最終的にヒドロゲル内に埋め込まれ、スライドから移動する場合、表面処理は歪みや引き裂きを伴わずにスライドを横切る細胞を含むヒドロゲルの信頼性の高い動きのために不可欠です。ジメチルクロロシラン(トリデカフルオロ-1,1,2-テトラヒドロホクチル)を用いたシラニン素化は、滑り表面に疎水性フルオロアルキル基の存在をもたらす。
注意:酢酸および塩化メチレンの煙への暴露を防ぐために化学煙フードで3.1以降のすべてのステップを実行します。
- 10%酢酸でスライドし、次に空気流で乾燥させます。
- ガラスコプリンジャーに、60 mL塩化メチレン(ジクロロメタン)、トリエチルアミン0.6 mL、0.6 mL(トリデカフルオロ-1,1,2,2-テトラヒドロコクチル)ジメチルクロロシランの溶液を調製します。金属ヘラでかき混ぜて混ぜます。
注:これらの試薬は水に敏感です。それらは乾燥した条件下で貯え、できるだけ新鮮に使用するべきです。 - シラン溶液を含むコプリン瓶にスライドを追加します。コプリンジャーを軌道シェーカー(60-80 rpmに設定)に置き、シランとスライドの反応を15分間進行させます。
- 金属鉗子を使用して、シラン溶液からスライドを取り外します。塩化メチレンを含むコプリン瓶に1分間浸し、スライドから余分なシランを取り除きます。
- スライドをエタノールを含む50 mL円錐形チューブに浸します。煽る。蒸留水を含む50 mL円錐管にスライドを浸します。煽る。
注:塩化メチレンと水は混和性ではないので、最終的な水のすすぎの前に余分な塩化メチレンを除去するためにエタノールリンスが必要です。 - 水からスライドを取り出し、点検します。スライドは、90°以上の接触角を有する水滴で、かなり乾燥している必要があります。スライドを完全に乾燥させ、使用するまで真空デシケーターに保管します。
注: ここで実験を一時停止できます。乾燥した状態でスライドを保管してください。
4. 実験用のPDMSフローセルとスライドを準備する
メモ: 長方形の PDMS フロー セルは、スライドのパターン化された領域にセルを集中させるために使用されます。3Dで培養した実験の場合、フローセルはヒドロゲルの金型を形成する。
- PDMS フローセルの金型として使用する SU-8 マスターを作成します。
- ホットプレートを95°Cに予熱する。
- シリコンウエハにSU-8 2075の5 mLを加えます。
- 10sの場合は500 rpmでウエハースのSU-8をスピンコートし、30年代には1,000rpmをスピンコートします。これにより、高さ34で最大240 μmの機能を作成する必要があります。
- ホットプレートのウエハーを少なくとも45分間柔らかく焼きます。
- ホットプレートからウエハーを取り外します。フォトマスク( 補足ファイル4を参照)(エマルジョン側を下に)ウエハーの上に置き、ガラスディスクで重み付けして、フォトマスクとスライドの接触を確認します。
- 350 mJ/cm2の放射エネルギー密度のための UV ライト(365 nm)で公開します。
- ホットプレートでウエハーを12~15分間焼きます。
- ウエハーを広いガラス容器に入れます。SU-8開発者ソリューションでウェハーをカバーします。シェーカーの上に置き、少なくとも15分間攪拌しながら開発します。
- 鉗子を使用して、開発液からウエハを取り除きます。噴出ボトルからより多くの開発者溶液を噴霧することにより、5 sのためにリンス。イソプロピルアルコールをスプレーしてすすいでください。白色の沈殿物が現れる場合は、ウエハを現液に戻し、より長く開発します。
- 空気または窒素の流れの下で乾燥したウエハー。
- スライドを5分間焼きます。
注: マスターウェーハを作成したら、フィーチャーがそのまま残っている限り、無期限に再利用できます。
- PDMS を準備します。
- 重量を量るボートで、ポリジメチルシロキサンエラストマーおよび架橋剤を10:1の比率(質量)で加える。激しくかき混ぜて、混ぜ合わせさえ確実にします。
- PDMSを真空デシケータで15〜30分間脱ガスし、気泡が見えないまで下剤を使用します。
- マスターウエハーを15cmのティッシュ培養皿に入れます。ウエハーの上にPDMSを注ぎます。気泡が現れた場合は、真空デシケータで数分間脱気します。
- 60°Cのオーブンで3時間焼きます。
注:ベイク処理後、PDMSフローセルはベンチトップに無期限に保存できます。
- 実験用に PDMS フロー セルを準備します。
- CMO-DPAC実験を開始する少し前に、マスターウエハから必要な数のPDMSフローセルを切り取ります。プラズマは、表面親水性をレンダリングするために90 sのための10 cc / min部屋の空気で酸化します。
- 各側に1~2mmのPDMSが残るように個々のフローセルを切り取り、フローセルの上下を切り開いて入口と出口を作ります。
- 手順 2 と 3 で作成したパターン化されたスライドを取得します。フォトマスクの上に揃えます。
- フォトマスクを参照として使用して、各パターン化された領域の位置に、スライド上に PDMS フローセルを配置します。
- リン酸緩衝生理食塩水(PBS)+1%ウシ血清アルブミン(BSA)を各フローセルの入口に加える( 補足図1H参照)。フローセルがPBS+1%BSAによって完全に充填され、大きな気泡がないことを確認します。直ちに手順 5 と 6 に進みます。
注: BSA によるブロッキングにより、スライド表面への非特定セル接着が最小限に抑えられます。
5. コレステロール修飾DNAを持つ細胞を持ち上げ、標識する
- コレステロール修飾DNA溶液を調製します。
- 実験の各セルセットについて、コレステロール修飾ユニバーサルアンカーストランドの3μLの3μLを、アダプタストランドの100 μMストック溶液の3μLと混ぜ合わせます。1分間インキュベートします。オリゴを事前にハイブリダイズします。69 μL のリン酸緩衝生理食塩水 (PBS) を加え、4 μM ユニバーサル アンカー + アダプター ソリューションを作成します。
- 実験中の細胞のセットごとに、100 μM ユニバーサルコレステロール修飾コアンカーストランドストック溶液の3 μLを12 μLのPBSに加え、20 μMの溶液を作成します。
- 単細胞懸濁液を準備します。
- 接着性細胞の場合、トリプシンまたは他の解離剤を使用して、培養フラスコから細胞を除去する。トリプシンと遠心分離機を中和する培養培地を細胞にペレット化する。非接着細胞の場合、細胞懸濁液および遠心分離機を採取して細胞をペレット化する。
- 細胞ペレットを1mLの氷冷PBSまたは無血清培地で再懸濁する。1.5 mLマイクロ遠心チューブに100万~300万個の細胞を移します。遠心分離機 160 x g で 4 分間
注:使用されている細胞タイプが凝集/凝集しやすい場合は、すべての洗浄工程にカルシウムイオンとマグネシウムイオンを含まないPBSを使用して、不要な細胞凝集を減らしてください。使用される細胞タイプに対して生存率が特に懸念される場合は、PBSの代わりに無血清培地を使用してください。ウシ胎児血清を含む培地は、脂質修飾オリゴの組み込みを妨げる可能性がありますので、細胞標識には推奨されません。35
- コレステロール修飾オリゴで細胞にラベルを付けます。
- 75 μLの氷冷PBSまたは無血清培地で細胞ペレットを再懸濁します。細胞の生存率を最大化し、細胞表面からのコレステロール修飾オリゴの損失を最小限に抑えるために、ラベリングおよび洗浄プロセス全体を通して細胞をアイスバケツに保管してください。
注:DNAを追加する前に細胞を再懸濁すると、DNAの分布が細胞集団全体で均一になります。 - ステップ5.1.1で作成した4μMユニバーサルアンカー+アダプタソリューションの75μLを、セルサスペンションを含むマイクロ遠心分離管に追加します。ピペットで十分に混ぜます。氷の上で5分間インキュベートします。
- マイクロ遠心チューブにユニバーサル共同アンカーソリューションの15 μLを追加します。ピペットで十分に混ぜます。氷の上で5分間インキュベートします。
- 細胞懸濁液から余分なオリゴを取り除きます。マイクロ遠心分離チューブに1mLの氷冷PBSまたは無血清培地を加えます。P1000ピペットと混ぜます。遠心分離機 160 x g 4 °Cで 4 分間 上清を捨てます。さらに 2 回繰り返します。
注:細胞が凝集しやすい場合は、最終洗浄の前に40μmのフィルターを通して細胞懸濁液を通過させます。細胞がマイクロ遠心チューブの側面に吸着しやすい場合は、カゼインでチューブを事前遮断することを検討してください。
- 75 μLの氷冷PBSまたは無血清培地で細胞ペレットを再懸濁します。細胞の生存率を最大化し、細胞表面からのコレステロール修飾オリゴの損失を最小限に抑えるために、ラベリングおよび洗浄プロセス全体を通して細胞をアイスバケツに保管してください。
6. DNA標識細胞のパターン
- 氷冷PBSまたは無血清培地中の細胞を再懸濁して、少なくとも2500万個の細胞/mLの細胞密度の高い溶液を作成します。
注:ステップ4で説明した10mm x 15mm x 200 μm PDMSフローセルの4つを使用した1つのスライドでは、この緻密なセル懸濁液の約100 μLが必要です。これらの細胞のほとんどはパターンに付着せず、最終的には廃棄されますが、パターン上に非常に濃縮された細胞溶液を持つことは、細胞パターニングの効率を劇的に向上させます。 - スライドをピックアップし、それを少し傾けます。パターン化されたスライドの各フローセルの入口に25μLのセル懸濁液を加えます。出口からPBS+1%BSA溶液を取り出し、細胞懸濁液がPDMSフローセルを充填することを可能にする。氷の上で、または室温で30sのインキュベート。
注:この時点で、顕微鏡下のフローセルを見ると、細胞間に隙間がほとんどまたはまったく見えない密に詰まった細胞が表示されます。 補足図 2B を参照してください。 - スライドの出口から5μLの細胞懸濁液を吸引し、それを入口に戻します。フローセルごとに 10 回繰り返します。
注:CMO標識細胞のDNAパターンスライドへの接着はほぼ瞬時に行われます。パターン上を複数回にわたって細胞を流すと、細胞が所定のDNAスポット上を流れ、捕捉される確率が高くなります。 - ピペットPBSまたは無血清培地を各フローセルの入口に静かに入れ、余分な細胞を洗い流す。出口からの細胞懸濁液を集める。2~4回繰り返すか、顕微鏡下でスライドを目視検査して余分な細胞が残っていないことが確認されるまで繰り返します。
注:最初の洗浄から余分な細胞を保存することが有利です。パターニング効率が不十分な場合、過剰な細胞は遠心分離され、より低い量のPBSで再懸濁され、より細胞密度の高い溶液を作成し、ステップ6.2からプロセスを繰り返すことができます。 - パターン内のセルのセットごとに手順 6.1 ~ 6.4 を繰り返します。複数のセルタイプがサーフェステンプレートによって直接パターン化されるパターンの場合は、パターンの最小のセルタイプから始め、最も豊富なセルタイプで終了します。
注: 細胞が直交 DNA 配列でラベル付けされている条件であっても、セルをプールする代わりに、セルラー アセンブリの各ラウンドを順番に行うことをお勧めします。細胞をプールすると、各細胞集団が効果的に希釈され、パターニング効率が低下します。 - セルアセンブリの最終ラウンドが完了すると、次の手順は特定の実験に基づいて異なります。セルがガラスの上に残ることを意図している場合は、スライドを含むペトリ皿にメディアを追加し、フォースを使用してスライドからPDMSフローセルを微調整します。細胞がヒドロゲルに埋め込まれ、3Dで培養される場合は、ステップ7に進みます。
7.3D培養用ヒドロゲルへの移入(オプション)
- 2%DNaseを含むヒドロゲル前駆体溶液を調製する。
注: ソリューションの構成は、実験用の設定によって異なります。マトリゲルとマトリゲルとコラーゲンの混合物は、私はこのプロトコルでよく働くが、他のヒドロゲルも可能である。 - 各フローセルの入口に、2%のDNaseを含むヒドロゲル溶液50μLを加えます。流出口から余分な流体を吸引し、ヒドロゲル溶液をフローセル内に駆動させる。粘性ヒドロゲル前駆体の場合、流量セルにヒドロゲルが流れるのを助けるためにスライドをわずかに傾ける必要がある。
- 37°Cで30〜45分間(ヒドロゲルゲル化キネティクスに依存)でスライドをインキュベートし、ヒドロゲルを設定し、細胞と表面の間のDNAベースの接着を切断させます。
- スライドから各フローセルを取り出し、ヒドロゲル前駆体溶液の上に置きます。
- 2ウェルチャンバースライドまたは6ウェルプレートのウェルに50 μLのヒドロゲル前駆体を加えます。
- 各フローセルの両側にPBSのピペット10μL。
- カミソリの刃または細かいピンセットを使用してフローセルの全長に沿ってPBSを分配し、PBSがヒドロゲルの下に急ぐようにフローセルの側面をそっと持ち上げます。
注:これは、歪みや引き裂くことなく転送できるように、スライド全体にハイドロゲルを「浮かぶ」でしょう。 - カミソリの刃を使用して、フローセルをガラススライドの端にそっと動かします。
- スライドを反転します。カミソリの刃で、スライドからフローセルを動かして、カミソリの刃の上に着陸させます。
- 湾曲した鉗子を使用して、カミソリの刃からフローセルを取り出します。細胞が底部にあるようにフローセルを反転し、ヒドロゲル前駆体溶液の液滴の上に置きます。
- 各フローセルに対してステップ 7.4.1 ~ 7.4.6 を繰り返します。
- パターン化された細胞を含むヒドロゲルがヒドロゲル下敷に結合できるように少なくとも30分間インキュベートし、パターン化された細胞の完全な埋め込みをもたらす。
- PDMS フロー セルを削除します。
- PDMS フローセルを浸すのに十分なメディアを追加します。
注:メディアの流入は、ヒドロゲルとPDMSフローセルとの間の接着を緩めます。 - フローセルの長い軸に沿って向き合った曲線鉗子を使用して、フローセルが飛び出してメディアに浮かぶまで流量セルを穏やかに微調整します。鉗子とフローセルを収集し、破棄します。
注: 最適な結果を得るには、湾曲した鉗子を広げ、PDMS フローセルの壁に穏やかな圧力をかけます。流れセルの長軸方向に力を加えます。
- PDMS フローセルを浸すのに十分なメディアを追加します。
8. CMO でセルのラベル付けが成功したことを確認する(オプション、トラブルシューティング用)
- 実験で使用されているアダプター・ストランドの表面接着配列と相補的な蛍光修飾(FAMまたはAF647)オリゴヌクレオチドをオーダーします。
- CMO DNAで細胞にラベルを付け、ステップ5で説明したように過剰なDNAを洗い流す。200 μLの氷冷PBSで再中断します。
- PBSで蛍光標識された相補オリゴヌクレオチドの4μM溶液を構成する。この溶液の200μLを細胞懸濁液に加えます。氷の上で5分間インキュベートします。
- 氷冷PBSを1mL加えます。細胞を混ぜてペレットにします。上清を除去する。このプロセスをさらに2回繰り返して、ハイブリダイズしていないDNAを洗い流します。
- 細胞表面にDNAが存在する量を定量するために分析的なフローサイトメトリーを行う。
- フローサイトメーターで、DNAで標識されていないコントロール細胞を解析します。この人口に基づいてゲートを設定します。
- 蛍光標識された相補オリゴヌクレオチドで処理されたCMO標識細胞を解析します。
- 平均蛍光強度を計算します。
結果
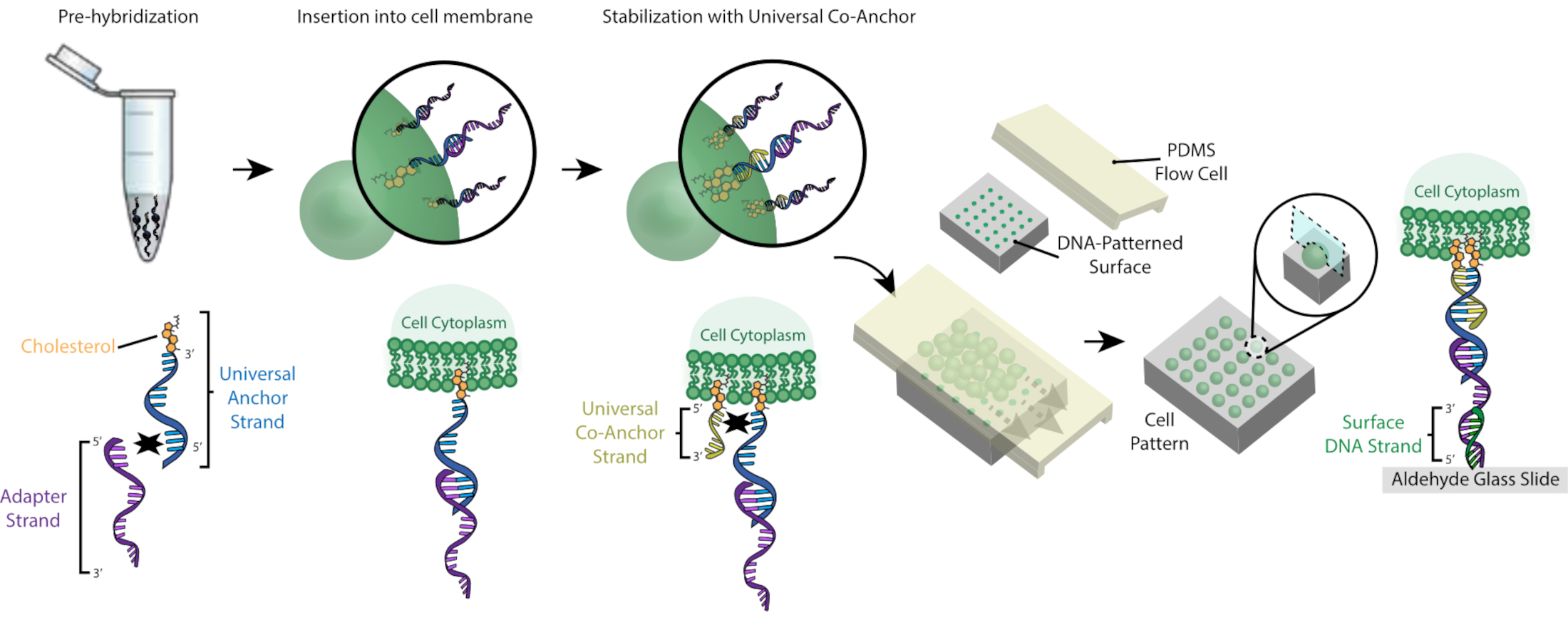
このプロトコルは、高精度で、カスタム試薬や高価なクリーンルーム機器を使用せずに、2Dと3Dのパターンセルを可能にします。 図 1 は、プロトコルの概要を示しています。まず、DNA機能化スライドはフォトリソグラフィを用いたものです。次に、セルに CMO のラベルが付けられます。その後、細胞はスライド上に流れ、そこでスライドのDNA機能領域にのみ接続します。余分な細胞が洗い流された後、細胞の所望のパターンが明らかにされる。これらの細胞は、スライド上で培養するか、DNaseを含むヒドロゲルに埋め込んで、3D細胞培養のためにスライドから転送することができる。
CMOを使用した細胞のラベリングにより、DNAパターンスライドへの付着が可能になります(図2)。まず、コレステロール修飾ユニバーサルアンカーストランドは、アダプタストランドと事前ハイブリダイズされます。次に、ユニバーサルアンカー+アダプタソリューションをセルサスペンションと1:1混合します。ユニバーサルアンカー+アダプタ複合体のコレステロールは、細胞膜に挿入します。ユニバーサルアンカーストランドとハイブリダイズするコレステロール修飾ユニバーサルコアンカーストランドの添加は、複合体26のネット疎水性を高めることによって細胞膜中のCMO複合体の安定性を向上させる。細胞懸濁液から余分なDNAを洗浄した後、細胞はスライド上に流される。アダプター・ストランドと表面DNAストランドのハイブリダイゼーションにより、スライドのDNAパターン領域に細胞が付着します。
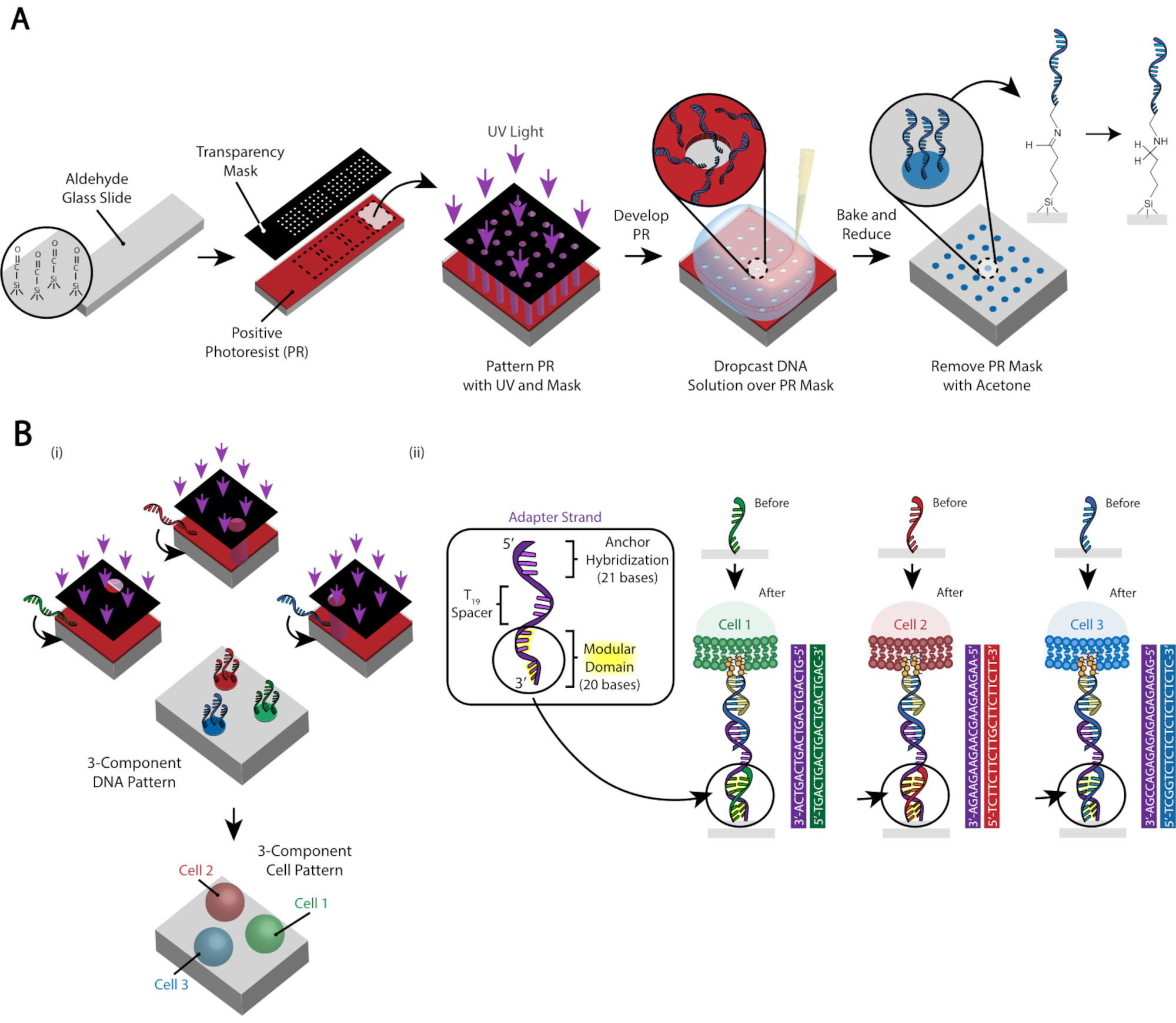
細胞のパターンは、アルデヒド修飾ガラススライド29 の特定領域へのアミン修飾DNAオリゴの付着を制限するフォトリソグラフィ法を用いて作成される(図3A)。正のフォトレジストは、アルデヒド機能化スライド上にスピンコートされる。透明フォトマスクがスライドの上に配置され、スライドが UV ライトに露出します。開発後、UV光にさらされたスライドの領域は、もはやフォトレジストでコーティングされず、アルデヒド基を露出している。アミン修飾DNAオリゴの20 μM溶液をスライドに落とし、パターン化された領域をカバーするために広げます。ベーキングに続いて還元的アミノ化は、アミン修飾DNAとスライド間の共有結合をもたらす。驚くべきことに、このプロセスは、以前にパターン化されたオリゴの機能性を損なうことなく、複数のオリゴをパターン化するために繰り返すことができます(図3B)。しかし、パターンの重複を避けるために注意が必要です, それは減少濃度で両方のオリゴの存在になります (補足図 3).複数のセルの集団は、モジュールドメイン(3'の端に最も近い20のベース)で異なるアダプタストランドを使用して、順番にパターン化することができます。
このフォトパターニングプロトコルは、クリーンルームの文脈でシャイドラーらによって開発されましたが、化学発煙フード内に簡単に収まる安価な「自家製」フォトリソグラフィーセットアップで同様の結果を達成することが可能であることを実証しました。セットアップには、DCモーター、デジタルコントローラ、CDケーキボックスで作られた400ドルのスピンコーターと、個々のコンポーネントから組み立てられ、転用されたシャープコンテナに収納されたUVランプが含まれています(補足図1)。自家醸造フォトリソグラフィーのセットアップの主な利点は、単一セルサイズの機能を作成しながら、非常に手頃な価格(すべての機器に対して< $1000)であることです。しかし、安価な機器の使用には限界があります - 例えば、マスクアライナーを使用せずに複数のDNAオリゴをパターン化するために受託者マーカーを正確に整列させることはより困難です。クリーンルームに便利にアクセスできないラボや、大きな投資をせずにこの方法を試してみたいラボでは、この安価なフォトリソグラフィーのセットアップをお勧めします。
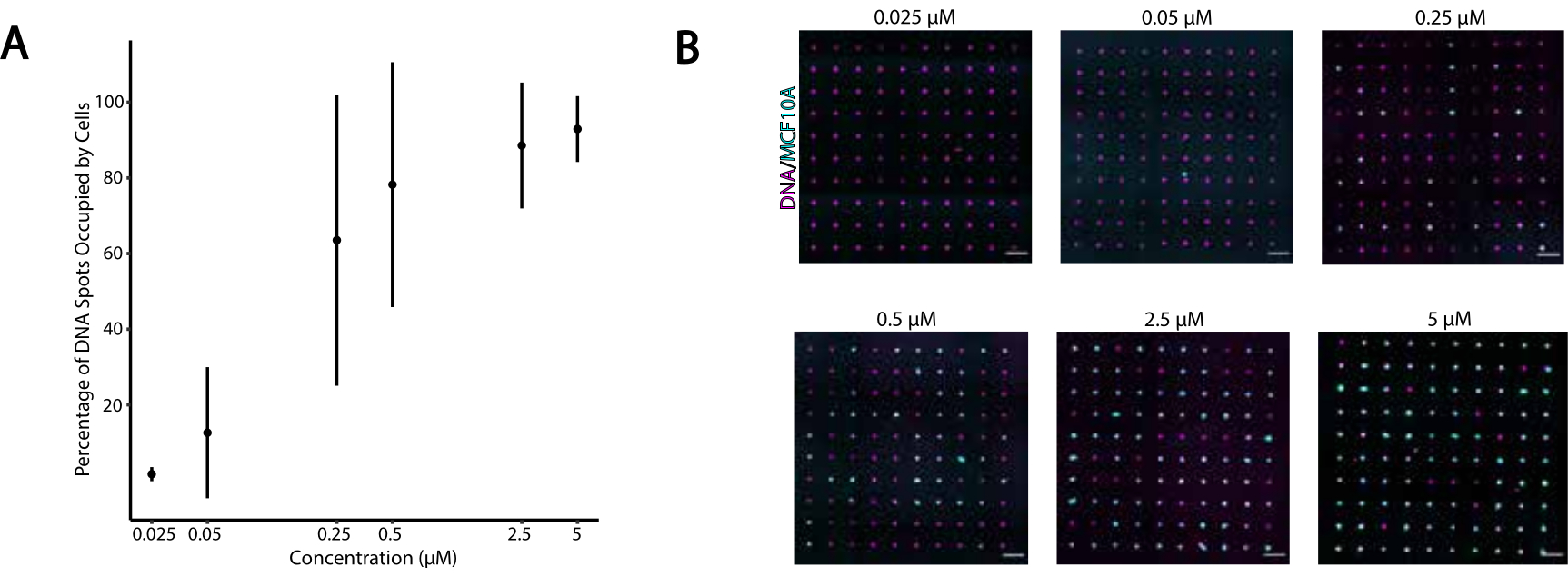
DNAプログラムによる細胞接着の最適条件を特定するために、細胞表面上のDNA鎖の濃度を系統的に変化させ、DNA修飾ガラス表面への細胞接着効率を測定した。標識液におけるユニバーサルアンカー+アダプタストランドおよびユニバーサル共同アンカーの濃度は、数桁(図4A,B)にまたがって変化し、1細胞当たり104~106個のDNA複合体が得られた(補足図4)。細胞接着は用量依存性であり、細胞が0.05μM以下の濃度でCMOで標識されたときのDNAパターンへの接着は最小限で、濃度は2.5μM以上で高い占有率であった。そのため、ユニバーサルアンカー+アダプタストランドの2μMソリューションとユニバーサル共同アンカーの2μMソリューションを使用しました。ガラス表面に使用されるDNAの量が29減少した場合、またはアダプタストランドと表面ストランドの間の不一致が増加した場合、細胞接着も減少すると予想されます。アダプタ ストランド シーケンス設計の詳細については、「補足ファイル 2」を参照してください。CpG反復を伴わないアダプタストランドを用いたCMO標識は、マウスTLR9を発現するHEK細胞においてTLR9を刺激しなかった(補足図5)。
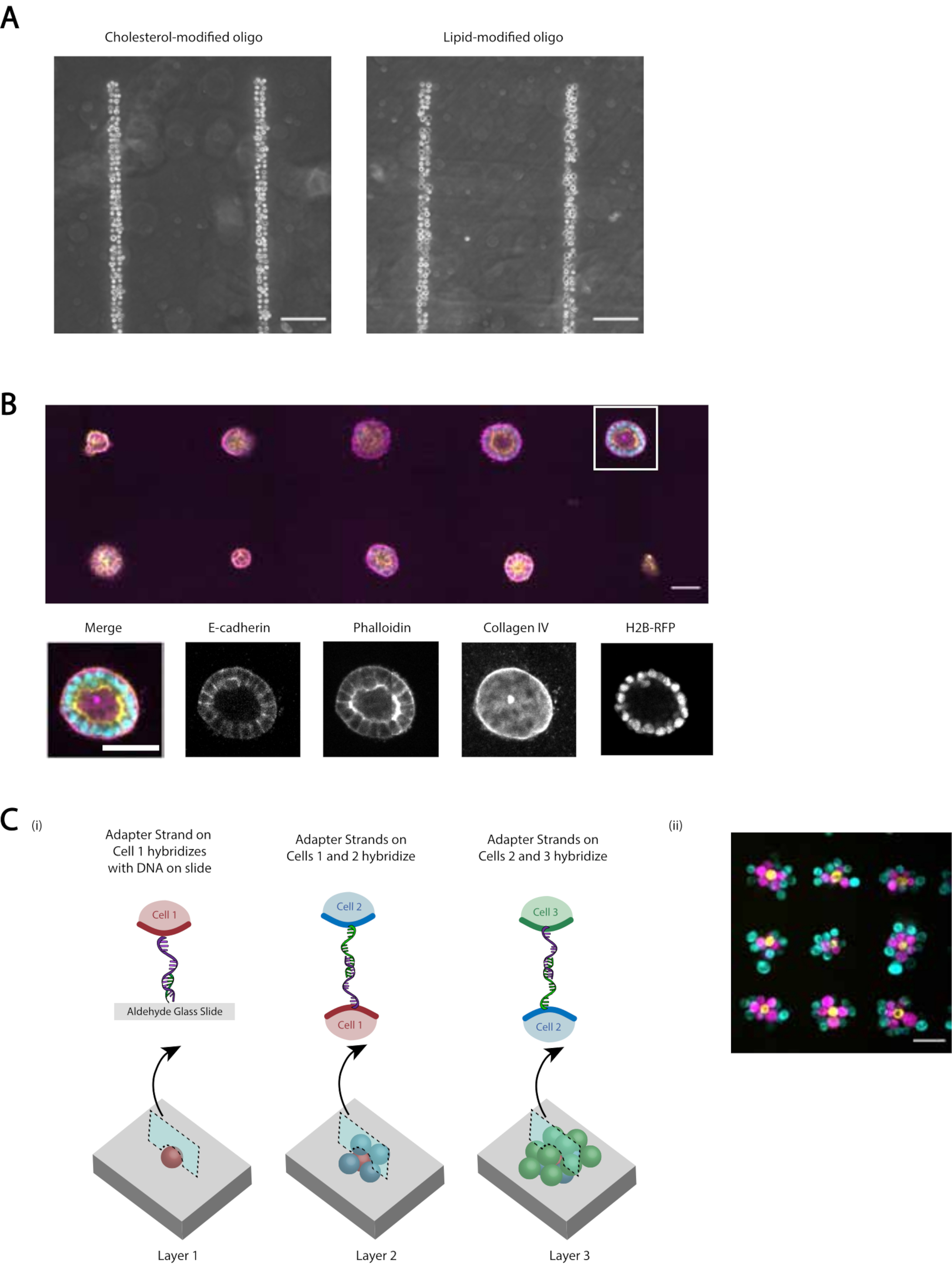
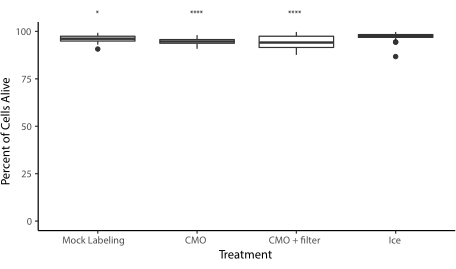
改訂されたプロトコルが再現可能で効率的なDNAプログラム細胞接着を提供するいくつかのデモンストレーションを行います。例えば、MMOで標識されたヒト臍静脈内皮細胞(HUVEC)は、DNAパターンに高効率で付着した。CMO ラベル付きの HUVEC と LMO ラベル付き HUVEC が付着しています (図 5A)。 CMO-DPACを使用してパターン化された細胞は、その生存率と機能性を保持した。CMOで標識された細胞は、生存率を評価するためにカルセインAMおよびエチジウムホモジマーによって染色された(補足図6)。非標識対照細胞と比較して生存率の差は小さかった(94%対97%)。CMO-DPACを介してパターン化され、Matrigelに移された単一のMDCKは、培養5日後に増殖し、正しく偏光することができました(図5B)。DPAC はまた、セルのパターンを第 3 次元に詳細化する手段も提供します (図 5C)。たとえば、多層的な多細胞集合体は、相補的なCMOで標識された細胞の層を交互に作成することができる(図5C)。これらの実験は、プロトコルが再現可能であり、細胞の生存率や機能性に悪影響を及ぼさず、3D ECMの単一のイメージングプレーン内で正常に培養できる細胞パターンを生み出す。
直交DNA配列を提供して細胞接着を指示することで、DPACは単一の表面上で複数の細胞タイプをパターニングする手段を提供します。DPACのこの特徴を実現するには、フォトリソグラフィによって生成されたDNAパターンを互いに整列させる必要があります。スライド上に堆積した金属受託マーカーは、複数のフォトマスクの整列を可能にし、したがって、複数の細胞タイプのパターニングを一度に可能にする。異なるユニークな染料で染色されたMCF10Asは、直交CMOでラベル付けされ、UCバークレーとUCSFのロゴの視覚化を作成するためにパターン化されました(図6)。この実験は、複数のユニークな細胞集団を高精度で、クロスコンタミネーションなしでパターン化できることを実証した。
CMO-DPACを用いた細胞のパターニングに成功するには、高品質のフォトリソグラフィ、細胞表面上のオリゴの十分な濃度、パターン上の細胞の高密度、および十分な洗浄が必要です。これらのいずれかのステップの失敗は、最終結果に影響を与えます。 補足図2 は、正しいフォトリソグラフィーと誤ったフォトリソグラフィーの画像例(補足的な図2A)、完全に占めるパターンを作成するためのパターン上の望ましい細胞密度(補足図2B)、DPACの後続のステップ中に過度に活発なピペット形成によるパターン化細胞の喪失(補足図2C)、および細胞の望ましくない束(補足図2D)を含む。 表 1 に、一般的な障害ポイントと推奨されるトラブルシューティングの一覧を示します。蛍光相補オリゴの使用は、スライド上のパターン化されたDNAの存在とフローサイトメトリーによる細胞表面上のCMOの存在を確認するためのトラブルシューティング用のツールとして推奨されます(プロトコルのステップ8を参照)。

図1: CMO-DPAC プロトコルの概要 まず、アルデヒド機能化ガラススライドに正のフォトレジストを塗布し、透明マスクで所望のパターンで覆い、UV光に曝すことで、DNAパターン化されたスライドを作成します。UV露光フォトレジストは現像者と一緒に洗い流され、アルデヒドスライドの露出領域を残し、アミン機能化されたDNAの表面への結合を可能にする。その後、セルにCMOのラベルが付き、サーフェス上を流れます。細胞膜上のDNAは表面のDNAにハイブリダイズし、接着を生じる。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:ステップワイズプロセスでセルにCMOのラベルが付いています。 まず、コレステロール修飾ユニバーサルアンカーストランドは、アダプタストランドと事前ハイブリダイズされます。次に、ユニバーサルアンカー+アダプタソリューションをセルサスペンションと混合します。ユニバーサルアンカー+アダプタ複合体のコレステロールは、細胞膜に挿入します。インキュベーション後、コレステロール修飾ユニバーサル共同アンカーストランドを細胞懸濁液に加え、ユニバーサルアンカーストランドとハイブリダイズし、細胞膜に挿入します。第2コレステロール分子の添加は、DNA複合体のネット疎水性を増大させ、膜26内で安定化させる。過剰なDNAを洗浄した後、細胞を濃縮し、パターン化された表面の上にPDMSフローセルに添加する。アダプター・ストランドの3'末端は、ガラススライド上の表面DNAストランドとハイブリダイズし、相補DNAで機能する領域でスライドに特異的に接着します。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:細胞の配置を最終的に指示するDNAパターンスライドを作成するためにフォトリソグラフィを使用する。 (A) フォトリソグラフィプロセスの概要アルデヒド機能化スライドは、正のフォトレジストでスピンコートされています。UV光は、透明な透明なフォトマスクを通してスライドに光を当てます。スライドが開発された後、以前にUV光にさらされていた領域は、現在、アルデヒド基を露出しています。アミン機能化されたDNAオリゴの20 μM溶液をスライドに落とし、パターン化された領域に広げます。スライドは、次いで、アミンとアルデヒド基との間にシフ結合(C=N)の形成を誘導するために焼成され、可逆的な共有結合29である。その後、PBS中の0.25%のホウ水素ナトリウムを用いた還元的アミノ化は、還元的アミノ化によってシッフ塩基を二次アミンに変換し、DNAとスライド間の不可逆的結合をもたらす。残りのフォトレジストは、アセトンでリンスすることにより除去することができる。(B)このプロセスを繰り返して、複数成分DNAパターンを作成し、複数の細胞集団を用いて実験を行うことができる。(i)最初のオリゴがパターン化された後、スライドは再びフォトレジストでコーティングされ、プロトコルは以前のように進行する。複数のDNA鎖をパターニングするには、受託者マーカーを用いたフォトマスクのアライメントが必要である。(ii) パターン化される各セルタイプは、アダプタストランドの20ベースモジュラードメインで異なります。補体オリゴの直交セットを使用することにより、複数の細胞タイプを交差接着なしでパターン化することができます。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:標識時のCMO濃度の関数として、CMO標識細胞のDNAパターンへの接着性が高まる。この実験では、ユニバーサルアンカー+アダプタストランド(プレハイブリダイズ)とユニバーサル共同アンカーを等濃度で使用しました。濃度は、細胞のCMO標識中の細胞懸濁液中のCMOの濃度をいう。(A)CMO標識MCF10A細胞が細胞標識中にCMO濃度の関数として占めていた直径15μmのDNAスポットの割合を定量化する。3つの実験から標準偏差±平均として表されるデータ。(B)CMOの異なる濃度でDNAパターン(マゼンタ)およびMCF10As(シアン)を接着した代表的な画像。スケールバー= 100 μm.この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図5:CMO-DPACを使用して、培養用の3次元ヒドロゲルに埋め込んだり、層構造を作成したりできる2次元細胞パターンを作成できます。(A)CMO標識ヒト臍帯静脈内皮細胞(HUVEC)とLMO標識HUVECとの直接比較が、線形DNAパターンに付着した。細胞標識の両方の方法は、DNAパターンのほぼ100%の占有率をもたらす。(B)H2B-RFPを発現する単一のマダン・ダービー・カイン・イングネル細胞(MDCK)を、直径15μmのスポットに200μm離してパターン化し、その後マトリゲルに埋め込んだ。120時間の培養後、得られた上皮嚢胞を固定し、Eカドヘリン、アクチン、およびコラーゲンIVについて染色した。白いボックスの回転楕円体を詳細に示します。スケールバー= 50 μm(C) 多層セル構造は、相補的なアダプタストランドを使用して個別のセル集団にラベルを付け、セルの新しい追加がそれ以前のセル層に付着するようにシーケンシャルにパターニングすることによって作成できます。(i) 多層構造を作成するための細胞集団のシーケンシャルなパターン化の概略。(ii)MCF10Asの3層細胞集合体(色素を用いて可視化)を、このプロセスを用いて作成した。スケールバー= 50 μm.この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図6:複数の細胞タイプは、交差汚染や接着性の喪失なしにパターン化することができます。 複数のアミン修飾DNAオリゴをアルデヒドスライドに順番にパターン化し、金属受託マーカーを使用して整列させた。MCF10A(シアン、マゼンタ、イエロー)の3つの集団は、相補的なCMOでラベル付けされたユニークな染料で染色され、スライドにパターン化され、UCバークレーとUCSFのロゴの画像が得られました。スケールバー 1 mm. この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
補足図1:ベンチトップフォトリソグラフィの設定例(A)スピンコーティングの前に、正のフォトレジストで覆われたスピンコーターのスライド。(B) 透明フォトマスクの画像。(C)露光時にフォトマスクはフォトレジストコーティングスライドとガラスディスクの間に挟まれ、UVランプ用ハウジングは、再利用されたシャープ容器から作られた。(E) 開発者向けソリューションに浸漬されたスライド。(F)スライドを開発。(G)アミン修飾DNA溶液がスライドのパターン化された領域に広がる。(H) スライドのパターン化された領域の上に配置された PDMS フロー セル。 このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
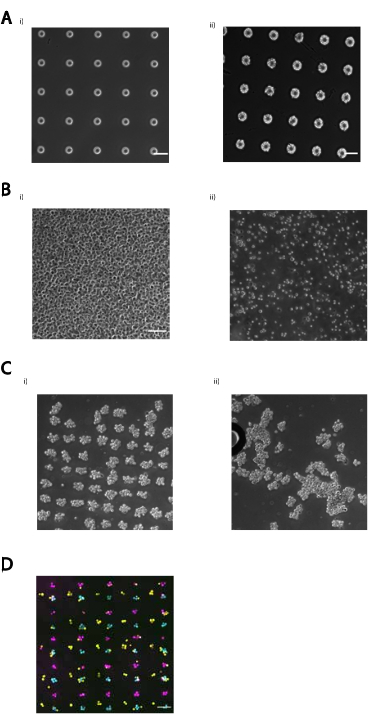
補足図 2: このプロトコルの一般的な障害の例。 (A) (i) 露光後の UV 露光または過剰開発機能の前に焼き小さい焼き上げは、エッジがギザギザで、サイズが不規則なフィーチャになる可能性があります。(ii) フィーチャの周囲にエッジがクリーンで、均一なフィーチャ サイズ、パターンに明らかな亀裂がない、正しいフォトパターンスライドの例。スケールバー= 50 μm(B)細胞密度はパターニング効率にとって重要です。顕微鏡下でパターンの上の細胞を観察する場合、左側の例の画像で証明されるように、細胞間に隙間はほとんど存在しないはずです。スケールバー = 50 μm(C) パターン化された細胞は、過度に激しいピペット処理から生じる流体力に敏感であり、パターン化された細胞を損傷して取り除くことができます。多層セル集合体は、最下部の 1 つのセルが複数のセルの構造をサポートしているので、特に脆弱です。(i) マトリゲルに正常に埋め込まれたセル集合体の配列。(ii) 粘性マトリゲルを激しくピペット化した結果として外れた細胞凝集体のグリッド。(D) 細胞の凝集は、特に上皮細胞で起こり得る。これらの塊は通常、均質であるが、細胞が特に粘着性である場合、異質であり得る(既に異なるタイプのパターン化された細胞に付着した細胞)。画像は、MCF10Aの3つの異なる集団を、3つの異なる単細胞サイズのDNAスポット(15μm)で構成されるアレイにパターン化したことを示しています。ほとんどのDNAスポットには2〜4個の細胞が付着しています。凝集は、EDTA処理によって、またはパターン化する前に束をフィルタリングすることによって解決することができる。スケールバー= 100 μm このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
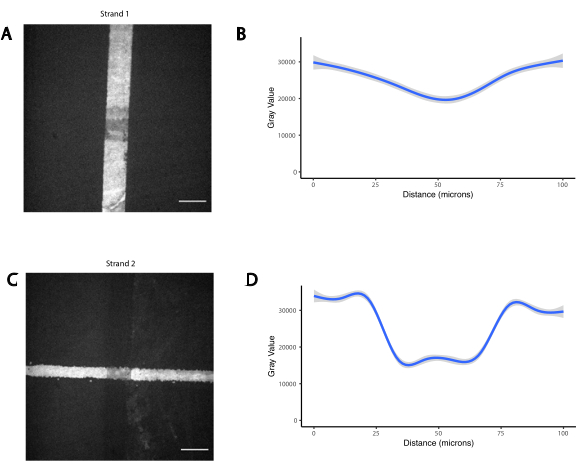
補足図3:重なり合うフォトパターンは、濃度が低下した場合に両方のオリゴの存在を生じる。 2つの直交アミン修飾オリゴを連続してフォトパターン化し、最初に垂直線(ストランド1)、それに重なる水平線(ストランド2)を続けた。オリゴを蛍光相補オリゴとのハイブリダイゼーションにより可視化した。(A)ストランド1の蛍光画像。(B)重なり合う100μm垂直線上のストランド1の蛍光プロファイルの定量化。(C)ストランド2の蛍光画像。(D)重なり合う100μm水平線上のストランド2の蛍光プロファイルの定量化。スケールバー= 50 μm このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
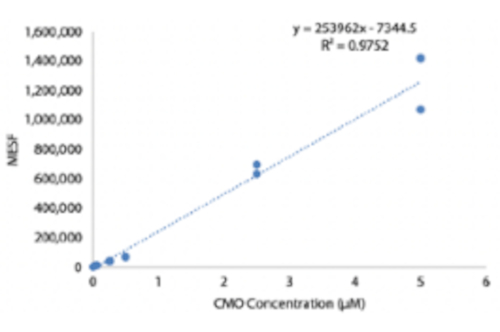
補足図4:CMO標識濃度の関数としての細胞表面上のDNA複合体の定量化。 ハベックスは、異なる濃度のCMO溶液で標識し、洗浄し、次いで蛍光相補鎖でインキュベートした。MESF(等価可溶性フルオロクロムの分子)ミクロスフェアキットを使用して、定量的フローサイトメトリーを行い、標識中のCMO濃度の関数として細胞表面上のDNA複合体の数を推定しました。 このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
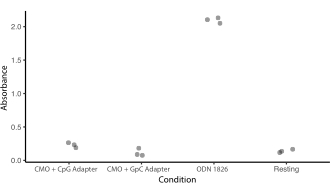
補足図5:CMO標識はTLR9応答を刺激しない。 CMO標識がTLR9のDNA検出メカニズムを引き起こすかどうか、そしてこれがアダプタストランド配列のCpGの影響を受けるかどうかを調べる実験を行った。マウスTLR9を発現するHEK細胞は、ODN 1826(CpG含有TLR9アゴニスト)、CMOユニバーサルアンカー+ユニバーサルコアンカー+アダプタストランド(ODN 1826(CMO-CpG)、またはCMOユニバーサルアンカー+ユニバーサルアンカー+ユニバーサルコアンカー+アダプタストランドの0.2 μMで一晩インキュベートしました。TLR9刺激はSEAP(分泌された胚性アルカリホスファターゼ)の産生をもたらすであろう。SEAP分泌は、色測定アッセイ(吸光度)によって定量した。治療条件は、PBSのみで処理した安静細胞と比較した。CMO-GPCによるインキュベーションはTLR9発現を刺激しなかった。CMO-CpGによるインキュベーションは、静止細胞よりもわずかに高いが、ODN-1826よりもはるかに低かった。 このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
補足図6:CMO標識処理後の細胞の生存率プロトコルが生存率にどのような影響を与えるかを評価するために、HUVECは4つの集団に分かれました:1つは氷の上に1時間残り、1つはPBSで模擬標識されましたが、それ以外のすべての遠心分離機と洗浄ステップを通して撮影され、1つはCMOでラベル付けされ、1つはCMOでラベル付けされ、40 μmのフィルターを通して塊を除去しました。次に、カルセインAMおよびエチジウムホモジマーで細胞を染色し、生細胞および死細胞の数を評価した。すべての治療は氷の制御(Tukeyポストホック分析を用いた一方のANOVA)よりも有意に生存率を低下させたが、CMO標識(フィルタリングの有無にかかわらず)の生存率の中央値は約94%であった。3つの独立した実験から収集されたデータ。* = p < 0.05.= p < 0.0001 このファイルをダウンロードするには、ここをクリックしてください。
{kind=link}
| 結果 | 考えられる原因 | 推奨される修正 |
| フォトリソグラフィー - 特徴がひび割れている | 一貫性のない、または不十分なソフトベーク | ソフトベークの時間を3分まで増やします。ホットプレートの実際の温度を確認し、必要に応じて温度を上昇させる |
| フォトリソグラフィ - 特徴は鋭くないか、それらの中に残っているフォトレジストを持っています | 開発不足 | スライドが開発者ソリューションに費やす時間を増やします。穏やかな攪拌を組み込む |
| フォトリソグラフィー - スライド全体で一貫性のない機能 | UVライトが中心になっていないか、適切に焦点を合わせていない可能性があります | UVライトの設定を調整して、均一な強度のコリメート光を確保 |
| 細胞は高効率のパターン化された斑点に付着しない | 表面に十分なDNAがない | 蛍光相補オリゴでスライドをハイブリダイズし、顕微鏡でイメージングすることにより、DNAが表面に存在することを確認します。 |
| 細胞はCMOで不十分に標識される | 細胞懸濁液に蛍光相補オリゴを加え、フローサイトメトリーを介して蛍光を確認する | |
| パターンの上に十分なセルがありません | PDMSフローセル、遠心分離機から洗浄して細胞を回収し、より低い体積で再中断して細胞を濃縮する | |
| 細胞懸濁液に残り過ぎるCMO、スライド上のDNAとのハイブリダイズ | 別の洗浄ステップを追加します。洗い物ごとにできるだけ多くの上清を取り除くようにしてください。 | |
| 時間と温度によるCMOの内部化が多すぎる | CMO でセルにラベルを付けた後、すばやく作業します。細胞を保持し、氷の上にスライドし、氷冷試薬を使用する | |
| 細胞の束 | トリプシン化中に細胞が十分に分離されなかった | 細胞のすすがみで PBS + 0.04% EDTA を使用します。最終洗浄前に35 μmフィルターを通過するセル懸濁液 |
| 細胞は非特異的に付着する | 特定の領域に - スライドの傷、PDMSフローセルのずれ、またはパターン領域外のDNAの流出が原因である可能性がある場合 | 傷を避け、PDMSフローセルをパターン領域に合わせるのに注意してください |
| 細胞がどこにでも付着している場合 - 不十分なブロッキングまたは洗浄 | 細胞をパターン化した後、より多くのするしを追加します。洗浄中に、より精力的にピペット;細胞のパターニングを開始する前に、より長い1%BSAでブロックします。シラナイズスライド(オプションのステップ3)または水滴の接触角を測定することによって、シラナイゼーションが成功したことを確認 | |
| フローセル内の泡フォーム | ピペット化誤差、プラズマ酸化中に生み出される不均一な親水性表面 | 気泡が小さい場合は、フローセルの入口にPBSを加え、洗浄してもよい。気泡が大きい場合は、PDMSフローセルに穏やかな圧力をかけ、入口または出口に向かって気泡を引き寄せます。 |
| 細胞は最初はパターンに付着するが、他の細胞タイプのパターニング、またはヒドロゲル前駆体の追加中に除去される | あまりにも激しくピペットからのせん断力は、細胞が表面から剥離する可能性があります | その後の打ち上げ、細胞のパターニングのラウンド、またはヒドロゲル前駆体の追加中に、より穏やかにピペット。ヒドロゲル前駆体は粘性があるため、パターンが外れる可能性が高いため、特に注意してください。多層構造は、トップヘビーになる傾向があり、外れやすくなります。 |
| 3D転送中の組織変形 | ハイドロゲルはスライドに固執する | 接触角度測定を用いたスライドの疎水性の確認 |
| かみそりブレードを使用してPDMSを両エッジに完全に持ち上げ、PBSが組織の下に浮かべるようにする | ||
| これは純粋なコラーゲンヒドロゲルで起こり得る - ヒドロゲルのタンパク質濃度または組成を調整することを検討してください | ||
| 細胞はヒドロゲルと一緒に移動しないし、スライドに残る | ターボDNAse濃度を増加させるか、インキュベーション時間を増加させる | |
| ヒドロゲルは十分に固い | 問題のヒドロゲルのインキュベーション時間および/またはゲル化機構を増加させる(例えば、コラーゲンの場合、pHが正しいことを確認してください) | |
| PDMSを取り除く時のヒドロゲルの涙 | 実験を開始する前に、実験を開始する前に、PDMSフローセルを親水性にして、メディアを添加する際に容易に取り外されるようにします。PDMS を取り外すには、フォースを非常に穏やかに使用してください。 |
表 1: このプロトコルから発生する可能性のある障害を特定し、解決するためのトラブルシューティング ガイド。 特に、パターンに対する細胞の接着性が悪いと、多くの根本原因が考えられる可能性があり、このガイドは、これらの問題の特定と解決に役立つはずです。
補足ファイル 1.このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル 2. このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル 3.このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル 4.このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
本稿では、インビトロ細胞培養実験用の2Dおよび3Dにおける細胞の高解像度パターン化のための詳細なプロトコルを紹介する。この方法の以前に公開されたバージョンとは異なり、ここで紹介するプロトコルは使いやすさを重視しています:それは高度に特殊な装置を必要とせず、すべての試薬は、カスタム合成を必要とするのではなく、ベンダーから購入することができます。他の細胞顕微鏡法とは異なり、この方法は、細胞型の迅速かつ無数である:細胞外マトリックスタンパク質15に特異的な接着を必要としない。CMO-DPACによってパターン化された細胞は、マトリゲルまたはコラーゲンのような細胞外マトリックス内に埋め込むことができるので、その結果、押出印刷ベースの方法22で現在可能であるよりもはるかに高い空間分解能を有する3D培養が得られる。CMO-DPAC を使用して、スライドごとに数百から数千の顕微鏡機能を作成できるため、同時に多数の複製を実行できます。
このプロトコルの成功における最も重要なパラメータの1つは、パターン化されたスライドの上のフローセルに追加される細胞の密度です。理想的には、密度は少なくとも2500万個の細胞/mLでなければなりません。フローセルにロードすると、この細胞密度は、パターンの上にほぼ密接に詰まった単層の細胞をもたらす(補足図2B)。これらの高い細胞密度は、細胞がDNAスポットの上に直接落ち着き、接着する確率を最大化します。細胞密度を下げると、全体的なパターニング効率が低下します。このプロトコルのもう一つの重要なステップは、CMO溶液を追加する前にPBSまたは無血清培地中の細胞を完全に再懸濁させることです。CMOは細胞膜に非常に急速に分割し、CMO溶液を細胞ペレットに直接加えることは細胞の異種標識をもたらす。CMO溶液を細胞懸濁液に添加した後、細胞がCMOで均一に標識されるようにピペッティングして十分に混合することが重要です。インキュベーション後、過剰なCMOを複数の遠心分離と洗浄工程で徹底的に洗い流す必要があります。細胞懸濁液中に存在する過剰な遊離CMOは、ガラススライド上のパターン化されたアミン修飾DNAに結合し、懸濁液中のCMO修飾細胞のハイブリダイゼーションおよび接着を遮断する。時間もこのプロトコルの重要な考慮事項です。CMOを使用する場合は、できるだけ迅速に作業し、CMOの内部化を最小限に抑え、細胞の生存率を最大化するために、細胞を氷上に保つことが重要です。フローサイトメトリー実験では、CMOはMMOのように細胞表面上に長く持続せず、氷上での2時間にわたるCMO複合体の25%の損失が示されている36。さらに、細胞の処理時間が長くなるにつれて細胞の生存率が低下する。生存率は、迅速に働き、細胞を氷上に保ち、氷冷試薬を使用し、無血清培地を使用して栄養素を提供することで最大化することができます。
CMO-DPACは、細胞を高精度にパターニングすることで細胞生物学を研究する強力な方法ですが、限界があります。CMO-DPAC 実験は、特に複数の細胞タイプ、層、または3D細胞培養(補足ファイル1)で実験の複雑さが加えられるため、困難な場合があります。このプロトコルを起動する際に、実験的な障害が一般的になる可能性があります(表 1に示すように)。そのため、品質管理チェック(スライドにDNAが存在することを確認し、細胞にDNAが十分に標識されていることを確認する(ステップ8)、余分な細胞が完全に洗い流されていることを確認する)ことをお勧めします。この原稿に記載されている情報とその補足ファイルが、必要なトラブルシューティングを容易にすることを期待しています。
コレステロールは、細胞代謝、遺伝子発現、膜流動性37,38に影響を及ぼす生理活性分子である。これまでの研究では、単一細胞RNAシーケンシングを用いてCMO標識細胞およびLMO標識細胞の遺伝子発現に及ぼす影響を比較した。CMO標識HEK細胞は、非標識およびLMO標識細胞36と比較して遺伝子発現を変化させた。CMOを有する標識細胞は、コレステロールおよびスフィンゴ脂質輸送(GeneCards)に関連したAP2B1およびMALAT1、コレステロール蓄積39を調節する長い非コードRNAを含む、標識されていないコントロールに対する8つの遺伝子の差動発現(>1.5倍)をもたらした。軽微ながら、問題の実験が細胞内の代謝、膜動態、または他のコレステロール関連経路を研究している場合、これらの転写応答はそれにもかかわらず懸念される可能性があります。
このプロトコルは柔軟性があり、各実験のニーズに合わせて調整することができます。CMOは、特定の受容体を使用する代わりに脂質膜に挿入するので、この方法は細胞型に依存しない(HUVEC、MCF10A、HEKs、およびMCKがここで実証されている)。コレステロールは以前に発表されたLmoとは異なる疎水性アンカーですが、これまでのところ、同様に振る舞うことがわかりました。したがって、CMOは、神経幹細胞、線維芽細胞、末梢血単核細胞、腫瘍細胞、原発性乳腺上皮細胞6、23、27、29、36を含むがこれらに限定されない、これまでMMOで発表した多種多様な細胞タイプと連携することを期待する。.CMO標識はTLR9を刺激せず、プロトコルが免疫細胞と互換性があることを示唆している。CMOの膜取り込みは、細胞グリコカリックス35における全細胞サイズと負電荷の程度の関数である。このように、迅速な最適化に適した膜の組み込みの程度をテストするためのプロトコル(ステップ8)を含んでいます。各細胞パターンの特徴は、実験計画によって必然的に異なります(詳細については、補足ファイル1を参照)。DNAのパターン化のために上記のフォトパターニングプロトコルが推奨されますが、高分解能の液滴プリンタの使用など、アミン-DNA溶液の液滴を空間的に閉じ込める方法は何でも機能するはずです。パターンの解像度と最小フィーチャ間隔は、使用する方法によって異なります。また、このプロトコルのDNAフォトパターニングセクションを、膜発現亜鉛指40にハイブリダイズしたDNA、NHS共役DNA41を用いたDNA、および細胞表面のアジドシアル酸残基をホスフィン結合DNA42と反応させるなど、DNAで細胞を標識するために使用された他の方法と組み合わせることも理論的には可能である。.CMO-DPACは、細胞のペア間の相互作用の研究、「送信側」細胞から「受信機」細胞へのシグナルの伝達を調べた共培養実験、幹細胞分化に対する近くの細胞外手がかりの影響の研究など、細胞間の相互作用を厳しく制御する必要がある様々な実験に適用できます。.この方法は、細胞の移動を3次元で研究するために使用できるマイクロ組織、組織23、27への細胞の自己組織化、および細胞とECM27との動的相互作用を作成するためにも使用することができる。このプロトコルが、研究者自身の研究室で高解像度DNAベースの細胞パターニングの新しいアプリケーションを探求するためのアクセス可能なプラットフォームを研究者に提供することを期待しています。
開示事項
Z.J.G.は、プロヴァンスバイオサイエンスのアドバイザー兼エクイティホルダーです。
謝辞
著者らは、このプロトコルをテストしたジェレミー・ガルシアと、UCSFバイオメディカルマイクロとナノテクノロジーコアで機器に関するトレーニングを提供してくれたブーシャン・ハルビカールに感謝したいと考えています。この研究は、国防総省乳がん研究プログラム(W81XWH-10-1-1023およびW81XWH-13-1-0221)、NIH(U01CA199315、乳癌研究プログラム)からの助成金によって部分的にサポートされました。 DP2 HD080351-01、1R01CA190843-01、1R21EB019181-01A、および1R21CA182375-01A1、NSF(MCB1330864)、およびUCSFセルラー構築センター(DBI-154829)、NSF技術センターO.J.Sは、NSF大学院研究フェローシップ、シーベル奨学金、P.E.O.奨学金によって資金提供されました。Z.J.GとA.R.A.はチャン・ザッカーバーグ・バイオハブの調査官です。
資料
| Name | Company | Catalog Number | Comments |
| 2-well Chambered Coverglass w/ non-removable wells | Thermo Fisher Scientific | 155379 | |
| Acetic Acid | Sigma-Aldrich | A6283 | |
| Adapter with External SM1 Threads and Internal SM3 Thread | ThorLabs | SM3A1 | |
| Aldehyde Functionalized Slides | Schott | Nexterion Slide AL | Store under dry conditions after opening. |
| All Plastic Syringes, 1 mL | Fisher Scientific | 14-817-25 | |
| Amine-Modified DNA Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Aspheric Condenser Lens | ThorLabs | ACL7560 | |
| Borosilicate Disc, 6in Diameter X 1/2in Thick | Chemglass | CG-1906-23 | |
| Cell Culture Dishes 60x15 mm style | Corning | 353002 | |
| Cholesterol-Modified Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Diamond Scribe | Excelta | 475B | |
| DNA Oligonucleotide | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190250 | |
| Isopropyl Alcohol | Sigma-Aldrich | 278475 | |
| Matrigel Matrix, Growth Factor Reduced | Corning | 354230 | |
| Methylene Chloride (Stabilized/Certified ACS) | Fisher Scientific | D37-4 | |
| MF-321 Developer | Kayaku Advanced Materials | n/a | |
| Microposit S1813 Positive Photoresist | Kayaku Advanced Materials | n/a | |
| Ø3" Adjustable Lens Tube, 0.81" Travel | ThorLabs | SM3V10 | |
| Oven | Thermo Scientific | 51-028-112H | |
| PE-50 Compact Benchtop Plasma Cleaning System | Plasma Etch | PE-50 | |
| Photomask (custom) | CAD/Art Services | n/a | Minimum feature size guaranteed by CAD/Art Services is 10 microns. |
| Razor Blades | Fisher Scientific | 12-640 | |
| RCT Basic Hot Plate | IKA | 3810001 | |
| Silicon Wafer (100 mm) | University Wafer | 590 | |
| Sodium Borohydride, 98%, granules | Acros Organics | 419471000 | |
| Spin Coater Kit | Instras | SCK-200 | This is a low cost option, but any spin coater that can maintain a speed of 3000 rpm will suffice. |
| SU-8 2075 | Microchem | Y111074 0500L1GL | |
| SU-8 Developer | Microchem | Y020100 4000L1PE | |
| Sylgard 184 Silicone Elastomer Kit | Dow | 2646340 | |
| Syringe Needles | Sigma-Aldrich | Z192341 | |
| T-Cube LED Driver, 1200 mA Max Drive Current | ThorLabs | LEDD1B | |
| Tridecafluoro-1,1,2,2-tetrahydrooctyl dimethylchlorosilane | Gelest | SIT8170.0 | |
| Triethylamine | Sigma-Aldrich | 90335 | |
| Turbo DNase | Thermo Fisher Scientific | AM2238 | |
| Tweezers Style N7 | VWR | 100488-324 | The curved shape of these tweezers is essential for delicately picking up the PDMS flow cells containing patterned tissues. |
| UV LED (365 nm, 190 mW (Min) Mounted LED, 700 mA) | ThorLabs | M365L2 | |
| Wafer Tweezers | Agar Scientific | T5063 | |
| WHEATON Dry-Seal vacuum desiccator | Millipore Sigma | W365885 |
参考文献
- Kreeger, P. K., Strong, L. E., Masters, K. S. Engineering approaches to study cellular decision-making. Annual Review of Biomedical Engineering. , 49-72 (2018).
- Goubko, C. a., Cao, X. Patterning multiple cell types in co-cultures: A review. Materials Science and Engineering C. 29 (6), 1855 (2009).
- Sun, W., et al. The bioprinting roadmap. Biofabrication. 12 (2), 022002 (2020).
- Liu, W. F., Chen, C. S. Cellular and multicellular form and function. Advanced Drug Delivery Reviews. 59 (13), 1319-1328 (2007).
- Duffy, R. M., Sun, Y., Feinberg, A. W. Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle. Annals of Biomedical Engineering. 44 (6), 2076-2089 (2016).
- Chen, S., et al. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nature Communications. 7, 10309 (2016).
- Shaya, O., et al. Cell-cell contact area affects notch signaling and notch-dependent patterning. Developmental Cell. 40 (5), 505-511 (2017).
- Rao, N., et al. A co-culture device with a tunable stiffness to understand combinatorial cell-cell and cell-matrix interactions. Integrative Biology. 5 (11), 1344 (2013).
- Sriraghavan, V., Desai, R. A., Kwon, Y., Mrksich, M., Chen, C. S. Micropatterned dynamically adhesive substrates for cell migration. Langmuir. 26 (22), 17733-17738 (2010).
- Wong, L., Pegan, J. D., Gabela-Zuniga, B., Khine, M., McCloskey, K. E. Leaf-inspired microcontact printing vascular patterns. Biofabrication. 9 (2), 021001 (2017).
- Chen, T. H., et al. Directing tissue morphogenesis via self-assembly of vascular mesenchymal cells. Biomaterials. 33 (35), 9019-9026 (2012).
- Laurent, J., et al. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nature Biomedical Engineering. 1 (12), 939-956 (2017).
- Lin, C., Khetani, S. R. Micropatterned co-cultures of human hepatocytes and stromal cells for the assessment of drug clearance and drug-drug interactions. Current Protocols in Toxicology. 2017, 1-23 (2017).
- Hui, E. E., Bhatia, S. N. Micromechanical control of cell-cell interactions. Proceedings of the National Academy of Sciences of the United States of America. 104 (14), 5722-5726 (2007).
- D'Arcangelo, E., McGuigan, A. P. Micropatterning strategies to engineer controlled cell and tissue architecture in vitro. BioTechniques. 58 (1), 13-23 (2015).
- Martinez-Rivas, A., González-Quijano, G. K., Proa-Coronado, S., Séverac, C., Dague, E. Methods of micropatterning and manipulation of cells for biomedical applications. Micromachines. 8 (12), (2017).
- Lee, S., et al. Simple lithography-free single cell micropatterning using laser-cut stencils. Journal of Visualized Experiments. (158), e60888 (2020).
- Strale, P. O., et al. Multiprotein printing by light-induced molecular adsorption. Advanced Materials. 28 (10), 2024-2029 (2016).
- Melero, C., et al. Light-induced molecular adsorption of proteins using the primo system for micro-patterning to study cell responses to extracellular matrix proteins. Journal of Visualized Experiments. (152), e60092 (2019).
- Reid, J. A., Mollica, P. M., Bruno, R. D., Sachs, P. C. Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Research. , 1-13 (2018).
- Wang, Z., Lee, S. J., Cheng, H. -. J., Yoo, J. J., Atala, A. 3D bioprinted functional and contractile cardiac tissue constructs. Acta Biomaterialia. 70, 48-56 (2018).
- Miri, A. K., et al. Effective bioprinting resolution in tissue model fabrication. Lab on a Chip. 19 (11), 2019-2037 (2019).
- Todhunter, M. E., et al. Programmed synthesis of three-dimensional tissues. Nature Methods. 12 (10), 975-981 (2015).
- Todhunter, M. E., Weber, R. J., Farlow, J., Jee, N. Y., Gartner, Z. J. Fabrication of 3D microtissue arrays by DNA programmed assembly of cells. Current Protocols in Chemical Biology. 8 (3), 147-178 (2016).
- Csizmar, C. M., Petersburg, J. R., Wagner, C. R. Programming cell-cell interactions through non-genetic membrane engineering. Cell Chemical Biology. 25 (8), 931-940 (2018).
- Weber, R. J., Liang, S. I., Selden, N. S., Desai, T. A., Gartner, Z. J. Efficient targeting of fatty-acid modified oligonucleotides to live cell membranes through stepwise assembly. Biomacromolecules. 15 (12), 4621-4626 (2014).
- Hughes, A. J., et al. Engineered tissue folding by mechanical compaction of the mesenchyme. Developmental Cell. 44 (2), 165-178 (2018).
- Weber, R. J., et al. Rapid organoid reconstitution by chemical micromolding. ACS Biomaterials Science & Engineering. 2 (11), 1851-1855 (2016).
- Scheideler, O. J., et al. Recapitulating complex biological signaling environments using a multiplexed, DNA-patterning approach. Science Advances. 6 (12), (2020).
- Viola, J. M., et al. Guiding cell network assembly using shape-morphing hydrogels. Advanced materials (Deerfield Beach, Fla.). , 2002195 (2020).
- Mohammad, A., Davis, M., Aprelev, A., Ferrone, F. A. Note: Professional grade microfluidics fabricated simply. Review of Scientific Instruments. 87 (10), 1-4 (2016).
- Lee, O. J., Chuah, H. S., Umar, R., Chen, S. K., Yusra, A. F. I. Construction of cost effective homebuilt spin coater for coating amylose-amylopectin thin films. Journal of Fundamental and Applied Sciences. 9 (2), 279 (2018).
- Webb, K., Hlady, V., Tresco, P. A. Relative importance of surface wettability and charged functional groups on NIH 3T3 fibroblast attachment, spreading, and cytoskeletal organization. Journal of Biomedical Materials Research. 41 (3), 422-430 (1998).
- Processing Guidelines for: SU-8 2025, SU-8 2035, SU-8 2050, SU-8 2075. Microchem SU-8 2000 Permanent Expoxy Negative Photoresist Available from: https://kayakuam.com/wp-content/uploads/2019/09/SU-82000DataSheet2025thru2075Ver4.pdf (2019)
- Palte, M. J., Raines, R. T. Interaction of nucleic acids with the glycocalyx. Journal of the American Chemical Society. 134 (14), 6218-6223 (2012).
- McGinnis, C. S., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods. 16 (7), 619-626 (2019).
- Maxfield, F. R., van Meer, G. Cholesterol, the central lipid of mammalian cells. Current Opinion in Cell Biology. 22 (4), 422-429 (2010).
- Luo, J., Yang, H., Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology. 21 (4), 225-245 (2020).
- Liu, L., Tan, L., Yao, J., Yang, L. Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Molecular Medicine Reports. 21 (4), 1761-1770 (2020).
- Mali, P., Aach, J., Lee, J. H., Levner, D., Nip, L., Church, G. M. Barcoding cells using cell-surface programmable DNA-binding domains. Nature Methods. 10 (5), 403-406 (2013).
- Hsiao, S. C., et al. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir. 25 (12), 6985-6991 (2009).
- Gartner, Z. J., Bertozzi, C. R. Programmed assembly of 3-dimensional microtissues with defined cellular conductivity. Proceedings of the National Academy of Sciences. (17), 1-5 (2009).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved