
Method Article
Einfache, erschwingliche und modulare Musterung von Zellen mit DNA
In diesem Artikel
Zusammenfassung
Hier stellen wir ein Protokoll für Mikrostrukturzellen mit Einzelzellauflösung unter Verwendung von DNA-programmierter Adhäsion vor. Dieses Protokoll verwendet eine Benchtop-Photolithographie-Plattform, um Muster von DNA-Oligonukleotiden auf einem Glasobjektträger zu erzeugen und dann Zellmembranen mit kommerziell erhältlichen komplementären Oligonukleotiden zu markieren. Die Hybridisierung der Oligos führt zu einer programmierten Zelladhäsion.
Zusammenfassung
Die relative Positionierung von Zellen ist ein Schlüsselmerkmal der Mikroumgebung, die Zell-Zell-Interaktionen organisiert. Um die Wechselwirkungen zwischen Zellen des gleichen oder eines anderen Typs zu untersuchen, haben sich Mikrostrukturierungstechniken als nützlich erwiesen. DNA Programmed Assembly of Cells (DPAC) ist eine Mikrostrukturierungstechnik, die auf die Adhäsion von Zellen an einem Substrat oder anderen Zellen mittels DNA-Hybridisierung abzielt. Die grundlegendsten Operationen in DPAC beginnen mit der Dekoration von Zellmembranen mit lipidmodifizierten Oligonukleotiden und fließen sie dann über ein Substrat, das mit komplementären DNA-Sequenzen strukturiert wurde. Zellen haften nur dort selektiv am Substrat, wo sie eine komplementäre DNA-Sequenz finden. Nicht adhärente Zellen werden weggewaschen, wodurch ein Muster von adhärenten Zellen aufgedeckt wird. Weitere Operationen umfassen weitere Runden der Zellsubstrat- oder Zell-Zell-Adhäsion sowie die Übertragung der von DPAC gebildeten Muster in ein Einbettungshydrogel für die Langzeitkultur. Bisher erforderten Methoden zur Strukturierung von Oligonukleotiden auf Oberflächen und zur Dekoration von Zellen mit DNA-Sequenzen spezielle Geräte bzw. eine benutzerdefinierte DNA-Synthese. Wir berichten über eine aktualisierte Version des Protokolls, die ein kostengünstiges Tisch-Photolithographie-Setup und kommerziell erhältliche cholesterinmodifizierte Oligonukleotide (CMOs) verwendet, die in einem modularen Format eingesetzt werden. CMO-markierte Zellen haften mit hoher Effizienz an DNA-gemusterten Substraten. Dieser Ansatz kann verwendet werden, um mehrere Zelltypen gleichzeitig mit hoher Präzision zu strukturieren und Arrays von Mikrogeweben zu erzeugen, die in eine extrazelluläre Matrix eingebettet sind. Zu den Vorteilen dieser Methode gehören ihre hohe Auflösung, die Fähigkeit, Zellen in eine dreidimensionale Mikroumgebung einzubetten, ohne das Mikromuster zu stören, und die Flexibilität bei der Strukturierung jedes Zelltyps.
Einleitung
Die Positionierung von Zellen zueinander in einem Gewebe ist ein wichtiges Merkmal der Mikroumgebung1,2,3,4. Techniken, die verwendet werden, um lebende Zellen in räumlich kontrollierte Anordnungen zustrukturieren,sind wertvolle experimentelle Werkzeuge zur Untersuchung der Differenzierung4,5,6,7,8, Zellmotilität9, Morphogenese10,11,12, Metabolismus13und Zell-Zell-Interaktionen7,14 . Es gibt eine Vielzahl von Methoden zur Strukturierung von Zellen, jede mit ihren eigenen Vor- und Nachteilen3,4. Methoden, die Klebstoffinseln aus extrazellulären Matrixproteinen (ECM) erzeugen, wie Mikrokontaktdruck und lasergeschnittene Schablonen, sind einfach und skalierbar. Es ist jedoch schwierig, mehr als einen oder zwei Zelltypen gleichzeitig zu strukturieren, da die Adhäsionseigenschaften verschiedener Zelltypen zu verschiedenen ECM-Molekülen oft ähnlich sind15,16,17. Komplexere Mikromuster können mit lichtinduzierter molekularer Adsorption (LIMAP) erzeugt werden, einer Technik, die UV-Licht verwendet, um PEG-beschichtete Regionen abzutragen und eine nachfolgende Proteinadsorption zu ermöglichen18,19. Dieser Vorgang kann wiederholt werden, um hochauflösende Mikromuster mit mehreren Zelltypen zu erstellen. Es kann jedoch zu einer Kreuzbindung von Zellen an die verschiedenen Proteinpflaster kommen, was zu einer schlechten Musterspezifität führt19. Physikalische Methoden wie das Seeding von Zellen auf mikromechanische rekonfigurierbare Kulturgeräte können strukturierte Co-Kulturen mit dynamischer Kontrolle erzeugen, jedoch ohne die Flexibilität im Musterdesign von Mikrokontaktdruck oder LIMAP14,8. Im Gegensatz zu den anderen Techniken kann bioprinting dreidimensionale Anordnungen von Zellen innerhalb der Hydrogele20,21 erzeugen. Biogedruckte Konstrukte haben jedoch eine viel geringere Auflösung als andere Mikrostrukturierungstechniken, mit einer durchschnittlichen Merkmalsgröße in der Größenordnung von Hunderten von Mikrometern22. Eine ideale Zellmusterungsmethode hätte eine hohe Auflösung, mustert mehrere Zelltypen, verwendet Geräte und Reagenzien, die leicht zugänglich sind, und hätte die Fähigkeit, erfolgreiche Muster in ein Hydrogel für die dreidimensionale (3D) Zellkultur einzubetten. In diesem Artikel stellen wir CMO-DPAC vor, eine Zellmikrostrukturierungstechnik, die die Flexibilität und Geschwindigkeit der DNA-Hybridisierung nutzt, um die Zelladhäsion an einem Substrat zu erreichen. Diese Methode wurde von unseren vorherigen Protokollen23,24 angepasst, um sie erschwinglicher, modularer und zugänglicher zu machen. Mit dem aktuellen Protokoll sollte jedes Labor in der Lage sein, ein voll funktionsfähiges System ohne spezielle Ausrüstung oder Fachwissen einzurichten.
DNA Programmed Assembly of Cells (DPAC) ist eine leistungsstarke Tissue-Engineering-Technik, die Zellen mit Einzelzellauflösung mit präziser Kontrolle über den Zellabstand und die Gewebegeometrie mustert. In DPAC werden Zellmembranen mit DNA-Oligonukleotiden (Oligos) dekoriert, wobei zwei lipidmodifizierte Oligos verwendet werden, die auf der Zellmembran hybridisiert werden sollen. Da die Oligos an hydrophobe Lipide konjugiert sind, teilen sie sich schnell in die Zellmembran25 auf, wo sie hybridisieren, wodurch die Nettohydrophobie der nicht kovalent gebundenen Moleküle erhöht und dadurch ihre Lebensdauer an der Zelloberflächeverlängert wird 26. Die Oligos werden auf der Zelloberfläche so dargestellt, dass sie mit komplementären Oligos auf anderen Zellen oder DNA-funktionalisierten Objektträgern hybridisieren können, um definierte 2D- oder 3D-Zellmuster mit vorgeschriebener Zusammensetzung, Zell-Zell-Abstand und Geometrie zu erzeugen23,24. Die gemusterten Mikrogewebe können enzymatisch von der Oberfläche abgespalten und für eine längere 3D-Kultur in ein Hydrogel eingebettet werden. In Kombination mit Primärzellen oder Stammzellen können die resultierenden Zellansammlungen einer Morphogenese unterzogen werden und sich zu organoiden23,27,28 bilden. DPAC wurde angewendet, um die Dynamik des Schicksals adulter neuraler Stammzellen als Reaktion auf konkurrierende Signale zu untersuchen6,29, um die Selbstorganisation von Brustepithelzellen23,28zu untersuchen und "Gewebeorigami" durch mesenchymale Kondensation27zu erzeugen.
DPAC ermöglicht die präzise Platzierung mehrerer Zellpopulationen und hat eine wesentlich bessere Auflösung als extrusionsbasierte Bioprinter (in der Größenordnung von Mikrometern)22,23. Darüber hinaus erfordert DPAC im Gegensatz zu ECM-basierten Strukturierungsverfahren wie dem Mikrokontaktdruck keine differentielle Haftung der verschiedenen Zelltypen auf einer ECM-beschichteten Oberfläche15,23. Es ist ideal für die Beantwortung von Fragen darüber, wie die Zusammensetzung eines Gewebes sein Verhalten beeinflusst, wie Zellen mehrere zelluläre und mikroumweltliche Hinweise integrieren, wenn sie Entscheidungentreffen 6,29, und wie Zellpaare miteinander interagieren. Ein Vorteil dieser Methode gegenüber anderen Mikrostrukturierungsmethoden besteht darin, dass sie für die 3D-Zellkultur in einer einzigen Bildgebungsebene verwendet werden kann, was Zeitrafferstudien der Gewebeselbstorganisation und der Organoidmorphogeneseermöglicht 23,27,30.
Trotz dieser Vorteile erforderte die erfolgreiche Implementierung von DPAC die Synthese von kundenspezifischen Oligonukleotidreagenzien und den Zugang zu spezialisierten Geräten für die DNA-Musterung23,24, was die breite Akzeptanz einschränkte. Zum Beispiel müssen die optimalen lipidmodifizierten Oligos (LMOs), die im ursprünglichen Protokoll verwendet werden, kundenspezifisch synthetisiert, mit Lignocerinsäure oder Palmitinsäure modifiziert und gereinigtwerden 26. Dieser Prozess erfordert die Verwendung eines DNA-Synthesizers und eines Hochleistungsflüssigkeitschromatographiegeräts sowie den Kauf der zugehörigen Reagenzien wie Methylamin, einer kontrollierten Substanz, die sowohl institutionellen als auch bundesstaatlichen Vorschriften unterliegt. Alternativ können LMOs in großen Mengen individuell erworben werden, was jedoch eine erhebliche Vorabinvestition in die Technologie erfordert.
Um diese Einschränkungen zu überwinden, haben wir eine überarbeitete Version von DPAC entwickelt, die kommerziell erhältliche cholesterinmodifizierte Oligos (CMOs) anstelle der kundenspezifisch synthetisierten LMOs verwendet. Um die Kosten weiter zu senken und die Flexibilität der Plattform zu erhöhen, haben wir auf ein modulares Drei-Oligo-System umgestellt. Anstatt für jede einzelne Zellpopulation ein neues cholesterinmodifiziertes Oligo zu bestellen, kann ein Benutzer dieses Protokolls stattdessen für jede Zellpopulation die gleichen cholesterinmodifizierten Oligos ("Universal Anchor" und "Universal Co-Anchor") verwenden und dann ein kostengünstiges, unverändertes Oligo ("Adapter Strand") verwenden, das sowohl mit dem Universal Anchor als auch entweder mit der aminfunktionalisierten DNA auf der Oberfläche oder dem Adapterstrang eines anderen Zelltyps hybridisiert.
Eine weitere Einschränkung des ursprünglichen DPAC-Protokolls bestand darin, dass es die DNA-gemusterten Dias mit einem hochauflösenden Flüssigkeitsdrucker (z. B. Nano eNabler, BioForce Nanosciences)erstellte 23,24. Obwohl dieses Instrument eine außergewöhnliche Auflösung und geringe Reagenzienanforderungen aufweist, ist es für die meisten Institutionen nicht verfügbar und hat eine relativ niedrige Druckrate (ca. 1 Merkmalsmuster pro Sekunde). Vor kurzem wurden zwei photolithographische Methoden entwickelt, um DNA-Merkmale auf Oberflächen zu strukturieren. Viola und Kollegen verwendeten eine Polyacrylamid- und Benzophenonbeschichtung, die einzelsträngige DNA-Oligos bei Exposition gegenüber UV-Licht kovalent bindet30. Mit dieser Methode konnten sie Gewebegerüste herstellen, die durch Zellkontraktilität und Selbstorganisation großflächige, programmierte Formveränderungen erfuhren. Scheideler et al. entwickelten eine Methode, die die UV-Belichtung eines positiven Fotolacks nutzt, um aminmodifizierte DNA-Oligos selektiv einem Aldehyd-funktionalisierten Objektträger29auszusetzen. Nach dem Backen und der reduktiven Aminierung wird die aminmodifizierte DNA kovalent an die Oberfläche gebunden. Diese Methode wurde verwendet, um die Reaktion adulter neuraler Stammzellen auf räumlich dargestellte Selbsterneuerungs- und Differenzierungshinweise zu untersuchen. Dieser Artikel passt das Protokoll von Scheideler et al. an, um die DNA-Muster zu erstellen, die CMO-markierte Zellen erfassen. Dieses Fotomusterungsprotokoll kann ohne Verwendung eines Reinraums durchgeführt werden. Es verwendet kostengünstige und handelsübliche Geräte, die leicht auf einem Tisch oder Abzug eingesetzt werden können. Die Verwendung von kostengünstigen oder DIY-Photolithographiegeräten (Do-it-yourself) erhöht die Zugänglichkeit für Forscher ohne Zugang zu Reinraumeinrichtungen und ermöglicht es den Forschern, die Technik ohne großen Zeit- oder Ressourcenaufwand auszuprobieren31,32. Eine bessere Auflösung und die Ausrichtung mehrerer DNA-Merkmale kann jedoch durch die Verwendung des kommerziellen Spin coaters und Mask Aligners erreicht werden, die üblicherweise in Reinraumeinrichtungen zu finden sind.
Hier beschreiben wir eine Methode, um Zellen mit Einzelzellauflösung mittels DNA-basierter Adhäsion zu mustern. Zunächst wird die Fotostrukturierung mit einem positiven Fotolack verwendet, um hochauflösende Muster von aminmodifizierter DNA auf einem aldehydmodifizierten Glassubstrat zu erzeugen. Als Nächstes wird die Folie behandelt, um die unspezifische Zellanheftung zu reduzieren, und PDMS-Flusszellen werden erstellt, um Zellen über strukturierte Bereiche einzugrenzen. Zellen werden dann mit kurzen DNA-Oligonukleotiden markiert, die mit Cholesterin funktionalisiert werden und dadurch in die Zellmembran eingebracht werden. Die Zellen werden dann über die DNA-Mikromuster geflossen. Die Hybridisierung zwischen der Zelloberflächen-DNA und der DNA auf der Glasoberfläche führt zu einer spezifischen Adhäsion der Zellen an das DNA-Muster. Nicht adhärente Zellen werden weggewaschen, wodurch das adhärente Zellmuster enthüllt wird. Dieser Vorgang kann wiederholt werden, um mehrere Zelltypen zu strukturieren oder mehrschichtige Strukturen zu erzeugen. Auf Wunsch können die Zellen vollständig in ein ECM für die 3D-Zellkultur eingebettet werden.
Protokoll
1. Design-Experiment
- Planen Sie das gewünschte Experiment unter Berücksichtigung der Feature-Größe, des Feature-Abstands, der Anzahl der beteiligten Zelltypen und der Anordnung der Zellen zueinander. Siehe Supplemental File 1, ein Handbuch für experimentelles Design, und Supplemental File 2, die Beispiel-Oligosequenzen enthält.
- Entwerfen Sie Fotomasken mit computergestützter Designsoftware. Eine Beispiel-Fotomaske finden Sie in Supplemental File 3.
- Zeichnen Sie ein Rechteck mit den Abmessungen eines Standard-Objektträgers (25 mm x 75 mm).
- Zeichnen Sie vier rechteckige Bereiche mit einer Breite von 10 mm und einer Länge von 10 mm, die gleichmäßig über den Schlitten verteilt sind.
- Zeichnen Sie innerhalb jedes Bereichs Features, die die gewünschte Größe, Form und den gewünschten Abstand für das Experiment aufweisen. Zellen werden sich nur an diese Merkmale im Experiment halten.
- Um ausgerichtete Fotomasken für mehrere Zellentypen zu erstellen, erstellen Sie eine Masterzeichnung mit allen Feature-Sätzen, und speichern Sie dann Versionen, die den einzelnen Zelltypen entsprechen.
- Bestellen Sie aus dieser CAD-Zeichnung eine hochauflösende (mindestens 20.000 Punkte pro Zoll) Transparenz-Fotomaske mit den in 1.2.3 transparent gezeichneten Merkmalen und den größeren Bereichen schwarz.
2. Photostruktur-DNA auf aldehydfunktionalisierten Objektträgern (Protokoll angepasst an Scheideler et al.29 )
- Wenn Sie mehrere Zelltypen strukturieren, stellen Sie vor jeder DNA-Musterung fiduciale Marker auf dem Aldehyd-funktionalisierten Objektträger her, um die Ausrichtung der Merkmale zu erleichtern. Alternative Methoden zum Erstellen von Filialmarkern werden in Ergänzungsdatei 1vorgeschlagen.
- Um Metalltreuhandmarkierungen zu erstellen, wenden Sie den positiven S1813-Fotolack an, wie in den Schritten 2.3 - 2.11 beschrieben. Verwenden Sie eine Fotomaske, die große Features enthält, die später leicht auszurichten sind. Integrieren Sie diese Merkmale in das Design der Fotomasken, die für die DNA-Musterung verwendet werden.
- Legen Sie einen dünnen Film (100 Angström) aus Titan mit Elektronenkanonenverdampfung29auf dem Objektträger ab. Entfernen Sie überschüssiges Metall und Fotolack mit Aceton und fahren Sie dann mit der DNA-Fotostrukturierung fort.
- Herstellen einer 20 μM Lösung eines 5'-aminmodifizierten Oligos im DNA-Puffer (50 mM Natriumphosphat in Wasser, pH = 8,5). Siehe Ergänzende Datei 2 für vorgeschlagene Oligosequenzen.
HINWEIS: Es ist möglich, für einige Muster und Anwendungen nur 5 μM aminmodifiziertes Oligo zu verwenden, so dass die Oberflächen-DNA-Konzentration möglicherweise optimiert werden muss. - Eine Heizplatte auf 100 °C vorheizen.
- Verwenden Sie doppelseitiges Klebeband oder ein Vakuum, um einen aldehydfunktionalisierten Glasschieber am Rotor eines Spin Coaters zu befestigen.
ACHTUNG: Das Ablösen des Schiebers während der Schleuderbeschichtung ist ein Sicherheitsrisiko. Verwenden Sie den Spin Coater immer in einem geschlossenen Behälter mit Deckel, z. B. einer Acrylbox.
HINWEIS: Beschriften Sie eine Ecke des Objektträgers mit einem Diamantritzer oder einem ähnlichen Gerät, um das Glas zu zerkratzen. Dies hilft bei der Identifizierung und Ausrichtung des Objektträgers, nachdem der Fotolack weggespült wurde. - Verwenden Sie eine Einwegpipette, um den positiven Fotolack auf den Aldehydträger fallen zu lassen. Für gleichmäßige Beschichtungen fügen Sie kleine Tropfen des Fotolacks über das Objektträger hinzu, anstatt einen großen Tropfen in der Mitte (Ergänzende Abbildung 1A).
- Drehen Sie den Schlitten mit dem Spin Coater 30 s lang mit 3000 U / min.
- Legen Sie den Objektträger für 1,5 min auf die 100 °C Warme Platte (Soft Bake), um Fotolack zu vernetzen.
- Entfernen Sie den Schieber von der Kochplatte. Legen Sie eine Fotomaske mit den für dieses Experiment gewünschten Merkmalen auf das Objektträger und wiegen Sie die Fotomaske mit einem Stück Glas ab (Ergänzende Abbildung 1B, C). Decken Sie das gesamte Setup in einer undurchsichtigen Box ab (Ergänzende Abbildung 1D). Belichten Sie mit einer UV-Lampe (365 nm Wellenlänge, 360 mW, 5 Zoll vom Objektträger, Gesamtstrahlungsenergiedichte 100 mJ/cm2)für 2 min.
HINWEIS: UV-Licht bricht die Polymerbindungen im Fotolack unter transparenten Bereichen der Fotomaske und schafft Bereiche, in denen dna später haften kann. - Entwickeln Sie die Folie, indem Sie 3-5 Minuten in die Entwicklerlösung eintauchen (ergänzende Abbildung 1E).
- Spülen Sie überschüssige Entwicklerlösung mit Wasser ab. Trocknen Sie unter einem Luft- oder Stickstoffstrom. (Ergänzende Abbildung 1F).
- Bestätigen Sie, dass die Photolithographie erfolgreich war, indem Sie den Objektträger unter dem Mikroskop betrachten. Da der Fotolack UV-lichtempfindlich ist, führen Sie diesen Schritt schnell aus und bewahren Sie das Dia dann im Dunkeln auf, während Sie andere Dias vorbereiten (falls zutreffend).
HINWEIS: Ein erfolgreich gemustertes Schlitten sollte scharf definierte Kanten für jedes KE, keine Risse und keine KE-Verzerrungen an den Kanten aufweisen. Beispiele für korrekte und falsche Photolithographie finden Sie in ergänzender Abbildung 2A. In Tabelle 1 finden Sie Vorschläge zur Fehlerbehebung, wenn die Fotolithografie nicht die gewünschte Funktionsqualität bietet. - Fügen Sie einen Tröpfchen der 20 μM aminmodifizierten Oligolösung (Schritt 2.1) auf jeden photostrukturierten Bereich des Objektträgers hinzu. Verwenden Sie eine Pipettenspitze, um den Tröpfchen vorsichtig über die gesamte Region zu verteilen, wobei Sie darauf achten, dass der Schlitten nicht zerkratzt wird. (Ergänzende Abbildung 1G).
- Den Objektträger in einem 65-70 °C heißen Ofen backen, bis die DNA-Lösung vollständig auf der Objektträgeroberfläche getrocknet ist (ca. 1 h).
- Führen Sie eine reduktive Aminierung durch, indem Sie die gemusterten, gebackenen Objektträger in eine 15 cm große Zellkulturschale legen und in einen Abzug auf einen Shaker legen. 100 mg Natriumborhydrid abwiegen. In einem Abzug 40 ml phosphatgepufferte Kochsalzlösung (PBS) hinzufügen, vorsichtig mischen und in die Schüssel geben, die die gemusterten Objektträger enthält. Lassen Sie die Reaktion 15 Minuten unter leichtem Schütteln ablaufen.
HINWEIS: Das Amin auf dem Oligo bildet zunächst mit den Aldehyden auf der Objektträgeroberfläche eine Schiff-Base. Dies ist eine reversible kovalente Bindung, die vor der Verwendung in DPAC in eine irreversible Bindung umgewandelt werden muss. Die Zugabe eines Reduktionsmittels (Natriumborhydrid) wandelt die Schiff-Base durch reduktive Aminierung in ein sekundäres Amin um.
VORSICHT: Die Reaktion von Natriumborhydrid mit Wasser erzeugt Wasserstoffgas und wird dies stunden- oder tagelang nach Beginn der Reaktion tun. Führen Sie den reduktiven Aminierungsschritt in einem Abzug durch und bewahren Sie alle Natriumborhydrid-Lösungsabfälle mindestens 24 Stunden lang in einem offenen oder lose verschlossenen Behälter im Abzug auf. - Entfernen Sie nicht umgesetzte DNA, indem Sie sie zweimal mit 0,1% Natriumdodecylsulfat (SDS) in Wasser und dann dreimal mit destilliertem Wasser waschen. Trocknen Sie den Objektträger unter einem Stickstoff- oder Luftstrom.
- Spülen Sie das Dia mit Aceton ab, um den restlichen Fotolack zu entfernen.
HINWEIS: Zu diesem Zeitpunkt wurde die DNA irreversibel und kovalent an den Objektträger gebunden und alle nicht umgesetzten funktionellen Aldehydgruppen wurden in Alkohole umgewandelt. Der Fotolack wird nicht mehr benötigt. - Wenn mehrere Oligos gemustert sind, kehren Sie zu Schritt 2.4 zurück, richten Sie die Fotomaske mit Fiducialmarken aus und wiederholen Sie den Vorgang.
HINWEIS: Das Experiment kann hier angehalten werden. Bewahren Sie die Objektträger in einem Vakuum-Exsikkator auf. Unter trockenen Bedingungen können die Objektträger bis zu 3 Monate ohne Qualitätsverlust gelagert werden.
3. Objektträger hydrophob machen (optional) (Protokoll angepasst von Todhunter et al.24)
HINWEIS: Es ist vorteilhaft, aber nicht erforderlich, die Oberflächenchemie des Objektträgers zu modifizieren, um ihn inerterter und hydrophober zu machen. Die unspezifische Zellbindung wird auf diesen Oberflächen reduziert33, wodurch die unspezifische Bindung von Zellen an ungemusterte Bereiche des Objektträgers verringert wird. Wenn die gemusterten Zellen letztendlich in ein Hydrogel eingebettet und vom Objektträger übertragen werden, ist die Oberflächenbehandlung für eine zuverlässige Bewegung des zellbeladenen Hydrogels über den Objektträger ohne Verzerrung oder Reißen unerlässlich. Die Silanisierung mit (Tridecafluor-1,1,2,2-tetrahydrooctyl)dimethylchlorsilan führt zur Anwesenheit von hydrophoben Fluoralkylgruppen auf der Objektträgeroberfläche.
VORSICHT: Führen Sie alle Schritte ab 3.1 in einem chemischen Abzug aus, um eine Exposition gegenüber Essigsäure- und Methylenchloriddämpfen zu vermeiden.
- Spülen Sie den Objektträger mit 10% Essigsäure ab und trocknen Sie ihn dann unter einem Luftstrom ab.
- In einem Coplinglasglas eine Lösung aus 60 ml Methylenchlorid (Dichlormethan), 0,6 ml Triethylamin und 0,6 ml (Tridecafluor-1,1,2,2-tetrahydrooctyl) dimethylchlorsilan herstellen. Mit einem Metallspatel zum Mischen umrühren.
HINWEIS: Diese Reagenzien sind empfindlich gegenüber Wasser. Sie sollten unter trockenen Bedingungen gelagert und so frisch wie möglich verwendet werden. - Geben Sie den Objektträger in das Coplin-Glas mit der Silanlösung. Legen Sie das Coplin-Glas auf einen Orbitalschüttler (eingestellt auf 60-80 U / min) und lassen Sie die Reaktion des Silans und des Schlittens 15 Minuten lang fortschreiten.
- Verwenden Sie eine Metallzange, um den Objektträger von der Silanlösung zu entfernen. Tauchen Sie den Objektträger 1 Minute lang in ein Coplin-Glas mit Methylenchlorid ein, um überschüssiges Silan aus dem Objektträger zu entfernen.
- Tauchen Sie den Objektträger in ein 50 ml großes konisches Röhrchen mit Ethanol. Agitieren. Tauchen Sie den Objektträger in ein 50 ml langes konisches Röhrchen mit destilliertem Wasser. Agitieren.
HINWEIS: Methylenchlorid und Wasser sind nicht mischbar, daher ist eine Ethanolspülung erforderlich, um überschüssiges Methylenchlorid vor dem endgültigen Wasserspülen zu entfernen. - Entfernen Sie die Rutsche aus dem Wasser und inspizieren Sie sie. Der Objektträger sollte ziemlich trocken sein, wobei alle Wassertröpfchen einen Kontaktwinkel von mehr als 90° aufweisen sollten. Lassen Sie die Objektträger vollständig trocknen und lagern Sie sie bis zum Gebrauch im Vakuum-Exsikkator.
HINWEIS: Das Experiment kann hier angehalten werden. Lagern Sie die Rutsche unter trockenen Bedingungen.
4. PDMS-Durchflusszellen und Objektträger für das Experiment vorbereiten
HINWEIS: Rechteckige PDMS-Flusszellen werden verwendet, um die Zellen über die gemusterten Bereiche des Objektträgers zu konzentrieren. Für in 3D kultivierte Experimente bilden die Flusszellen eine Form für das Hydrogel.
- Stellen Sie SU-8 Master her, um es als Form für PDMS-Durchflusszellen zu verwenden.
- Heizplatte auf 95 °C vorheizen.
- Fügen Sie 5 mL SU-8 2075 zu einem Siliziumwafer hinzu.
- Drehen Sie den SU-8 auf dem Wafer mit 500 U / min für 10s, gefolgt von 1.000 U / min für 30s. Dadurch sollen Merkmale bis zu einer Höhe von 240 μmerzeugt werden 34.
- Die Waffel auf der Kochplatte mindestens 45 min weich backen.
- Nehmen Sie den Wafer von der Kochplatte. Legen Sie die Fotomaske (siehe Ergänzungsdatei 4) (Emulsionsseite nach unten) auf den Wafer und beschweren Sie sie mit einer Glasscheibe, um den Kontakt zwischen der Fotomaske und dem Objektträger sicherzustellen.
- Mit UV-Licht (365 nm) belichten, um eine Strahlungsenergiedichte von 350 mJ/cm2 zu erhalten.
- Waffel auf der Kochplatte für 12-15 min backen.
- Legen Sie den Wafer in einen breiten Glasbehälter. Cover Wafer mit SU-8 Entwicklerlösung. Auf einen Shaker geben und unter Rühren mindestens 15 min weiterentwickeln.
- Verwenden Sie eine Pinzette, um den Wafer aus der Entwicklerlösung zu entfernen. Spülen Sie für 5 s, indem Sie mehr Entwicklerlösung aus einer Spritzflasche sprühen. Mit Isopropylalkohol zum Ausspülen besprühen. Wenn ein weißer Niederschlag auftritt, geben Sie den Wafer an die Entwicklerlösung zurück und entwickeln Sie länger.
- Trockener Wafer unter einem Luft- oder Stickstoffstrom.
- Rutsche für 5 Min. backen.
HINWEIS: Sobald der Master-Wafer erstellt wurde, kann er unbegrenzt wiederverwendet werden, solange die Eigenschaften intakt bleiben.
- Bereiten Sie PDMS vor.
- In einem Wiegeboot Polydimethylsiloxan-Elastomer und Vernetzer im Verhältnis 10:1 (nach Masse) hinzufügen. Rühren Sie kräftig um, um eine gleichmäßige Vermischung zu gewährleisten.
- Entgasen Sie das PDMS in einem Vakuum-Exsikkator für 15-30 min, bis keine Blasen mehr sichtbar sind.
- Legen Sie die Masterwaffel in eine 15 cm große Gewebekulturschale. PDMS über den Wafer gießen. Wenn Blasen auftreten, entgasen Sie einige Minuten in einem Vakuum-Exsikkator.
- Im 60 °C Ofen 3 h backen.
HINWEIS: Nach dem Backen können PDMS-Durchflusszellen unbegrenzt auf der Tischplatte gelagert werden.
- Bereiten Sie PDMS-Flusszellen für das Experiment vor.
- Schneiden Sie kurz vor Beginn eines CMO-DPAC-Experiments die erforderliche Anzahl von PDMS-Flusszellen aus dem Master-Wafer aus. Plasma oxidiert mit 10 cc/min Raumluft für 90 s, um die Oberfläche hydrophil zu machen.
- Schneiden Sie jede einzelne Durchflusszelle aus, so dass auf jeder Seite 1-2 mm PDMS verbleiben, und schneiden Sie dann die Ober- und Unterseite der Durchflusszelle auf, um einen Ein- und Auslass zu erzeugen.
- Rufen Sie die in den Schritten 2 und 3 erstellte gemusterte Folie ab. Auf der Fotomaske ausrichten.
- Verwenden Sie die Fotomaske als Referenz und platzieren Sie die PDMS-Flusszellen auf dem Objektträger an der Position jedes gemusterten Bereichs.
- 50 μL phosphatgepufferte Kochsalzlösung (PBS) + 1% Rinderserumalbumin (BSA) in den Einlass jeder Durchflusszelle geben, wie in ergänzender Abbildung 1Hdargestellt. Vergewissern Sie sich, dass die Durchflusszelle vollständig mit dem PBS + 1% BSA gefüllt ist und dass keine großen Blasen vorhanden sind. Fahren Sie sofort mit den Schritten 5 und 6 fort.
HINWEIS: Die Blockierung mit BSA minimiert die unspezifische Zellhaftung an der Objektträgeroberfläche.
5. Heben und markieren Sie Zellen mit cholesterinmodifizierter DNA
- Bereiten Sie die cholesterinmodifizierten DNA-Lösungen vor.
- Mischen Sie für jeden Zellsatz im Experiment 3 μL einer 100 μM-Stammlösung des cholesterinmodifizierten Universal Anchor Strangs mit 3 μL einer 100 μM-Stammlösung eines Adapterstrangs. Inkubieren Sie für 1 Minute. Dies wird die Oligos vorhybridisieren. Fügen Sie 69 μL phosphatgepufferte Kochsalzlösung (PBS) hinzu, um eine 4 μM Universal Anchor + Adapter-Lösung zu erstellen.
- Fügen Sie für jeden Zellsatz im Experiment 3 μL einer 100 μM Universal-Cholesterin-modifizierten Co-Anchor-Strang-Stammlösung zu 12 μL PBS hinzu, wodurch eine 20 μM-Lösung entsteht.
- Bereiten Sie die Einzelzellsuspension(en) vor.
- Verwenden Sie für adhärente Zellen Trypsin oder ein anderes Dissoziationsmittel, um die Zellen aus dem Kulturkolben zu entfernen. Fügen Sie Kulturmedien hinzu, um das Trypsin zu neutralisieren, und zentrifugieren Sie, um die Zellen zu pelletieren. Sammeln Sie bei nicht adhärenten Zellen die Zellsuspension und zentrifugieren Sie sie, um die Zellen zu pelletieren.
- Resuspendieren Sie das Zellpellet in 1 ml eiskaltem PBS oder serumfreien Medien. Übertragen Sie 1-3 Millionen Zellen in ein 1,5 ml Mikrozentrifugenröhrchen. Zentrifuge bei 160 x g für 4 min.
HINWEIS: Wenn der verwendete Zelltyp anfällig für Verklumpung / Aggregation ist, verwenden Sie PBS ohne Calcium- und Magnesiumionen für alle Waschschritte, um unerwünschte Zellaggregationen zu reduzieren. Wenn die Lebensfähigkeit für den verwendeten Zelltyp ein besonderes Anliegen ist, verwenden Sie serumfreie Medien anstelle von PBS. Medien, die fötales Rinderserum enthalten, werden nicht für die Zellmarkierung empfohlen, da sie den Einbau von lipidmodifizierten Oligos behindern können. 35 Einwohner
- Markieren Sie die Zellen mit cholesterinmodifizierten Oligos.
- Resuspendieren Sie das Zellpellet in 75 μL eiskaltem PBS oder serumfreien Medien. Bewahren Sie die Zellen während des gesamten Markierungs- und Waschprozesses in einem Eiskübel auf, um die Lebensfähigkeit der Zellen zu maximieren und den Verlust der cholesterinmodifizierten Oligos von der Zelloberfläche zu minimieren.
HINWEIS: Die Wiederverwendung der Zellen vor dem Hinzufügen der DNA stellt sicher, dass die Verteilung der DNA über die Zellpopulation gleichmäßig ist. - Die 75 μL der in Schritt 5.1.1 erstellten 4 μM Universal Anchor + Adapter-Lösung in das Mikrozentrifugenröhrchen mit der Zellsuspension geben. Durch Pipettieren gründlich mischen. Inkubieren Sie für 5 min auf dem Eis.
- Geben Sie 15 μL der Universal Co-Anchor Solution in das Mikrozentrifugenröhrchen. Durch Pipettieren gründlich mischen. Inkubieren Sie für 5 min auf dem Eis.
- Entfernen Sie überschüssige Oligos aus der Zellsuspension. Geben Sie 1 ml eiskaltes PBS oder serumfreie Medien in das Mikrozentrifugenröhrchen. Mit einer P1000 Pipette mischen. Zentrifuge bei 160 x g für 4 min bei 4 °C. Verwerfen Sie den Überstand. Wiederholen Sie dies noch zweimal.
HINWEIS: Wenn Zellen anfällig für Verklumpungen sind, führen Sie die Zellsuspension vor dem endgültigen Waschen durch einen 40 μm-Filter. Wenn Zellen zur Adsorption an der Seite des Mikrozentrifugenröhrchens neigen, sollten Sie erwägen, das Röhrchen mit Casein vorzublocken.
- Resuspendieren Sie das Zellpellet in 75 μL eiskaltem PBS oder serumfreien Medien. Bewahren Sie die Zellen während des gesamten Markierungs- und Waschprozesses in einem Eiskübel auf, um die Lebensfähigkeit der Zellen zu maximieren und den Verlust der cholesterinmodifizierten Oligos von der Zelloberfläche zu minimieren.
6. Mustern Sie die DNA-markierten Zellen
- Resuspendieren Sie die Zellen in eiskalten PBS- oder serumfreien Medien, um eine zelldichte Lösung von mindestens 25 Millionen Zellen / ml zu erzeugen.
HINWEIS: Für einen Objektträger mit vier der in Schritt 4 beschriebenen 10 mm x 15 mm x 200 μm PDMS-Durchflusszellen werden etwa 100 μL dieser dichten Zellsuspension benötigt. Obwohl sich die meisten dieser Zellen nicht an das Muster halten und letztendlich verworfen werden, verbessert eine extrem konzentrierte Lösung von Zellen über dem Muster die Effizienz der Zellstrukturierung dramatisch. - Heben Sie die Folie auf und neigen Sie sie leicht. Geben Sie 25 μL Zellsuspension in den Einlass jeder Durchflusszelle auf dem gemusterten Objektträger. Entfernen Sie die PBS + 1% BSA-Lösung aus dem Auslass, damit die Zellsuspension die PDMS-Durchflusszelle füllen kann. Auf Eis oder bei Raumtemperatur 30 s inkubieren.
HINWEIS: Zu diesem Zeitpunkt sollte die Betrachtung der Flusszelle unter einem Mikroskop dicht gepackte Zellen mit wenig bis gar keinen Lücken zwischen den Zellen zeigen. Siehe ergänzende Abbildung 2B. - 5 μL Zellsuspension aus dem Auslass des Objektträgers absaugen und wieder in den Einlass geben. Wiederholen Sie dies 10 Mal pro Durchflusszelle.
HINWEIS: Die Adhäsion von CMO-markierten Zellen an dem DNA-gemusterten Objektträger erfolgt nahezu augenblicklich. Wenn die Zellen mehrmals über das Muster fließen, erhöht sich die Wahrscheinlichkeit, dass eine Zelle über einen bestimmten DNA-Punkt fließt und erfasst wird. - Pipettieren Sie vorsichtig PBS oder serumfreie Medien in den Einlass jeder Durchflusszelle, um überschüssige Zellen auszuwaschen. Sammeln Sie die Zellsuspension aus dem Auslass. Wiederholen Sie dies 2-4 Mal oder bis eine visuelle Inspektion des Objektträgers unter dem Mikroskop bestätigt, dass keine überschüssigen Zellen mehr vorhanden sind.
HINWEIS: Es kann vorteilhaft sein, die überschüssigen Zellen ab der ersten Wäsche zu speichern. Wenn die Strukturierungseffizienz unbefriedigend ist, können die überschüssigen Zellen zentrifugiert und in einem geringeren PBS-Volumen resuspendiert werden, um eine zelldichtere Lösung zu erzeugen, und dann kann der Prozess ab Schritt 6.2 wiederholt werden. - Wiederholen Sie die Schritte 6.1-6.4 für jeden Satz von Zellen im Muster. Bei Mustern, bei denen mehrere Zelltypen direkt von der Flächenvorlage gemustert werden, beginnen Sie mit dem am wenigsten häufig vorkommenden Zelltyp des Musters und enden Sie mit dem am häufigsten vorkommenden Zelltyp.
HINWEIS: Es ist ratsam, jede Runde der zellulären Assemblierung nacheinander durchzuführen, anstatt die Zellen zu poolen, selbst unter Bedingungen, unter denen die Zellen alle mit orthogonalen DNA-Sequenzen markiert sind. Die Bündelung der Zellen verdünnt effektiv jede Zellpopulation und reduziert die Mustereffizienz. - Nachdem die letzte Runde der Zellmontage abgeschlossen ist, variieren die nächsten Schritte basierend auf dem spezifischen Experiment. Wenn die Zellen auf dem Glas verbleiben sollen, fügen Sie einer Petrischale, die den Objektträger enthält, Medien hinzu und verwenden Sie dann vorsichtig eine Pinzette, um die PDMS-Flusszellen vom Objektträger zu stoßen. Wenn die Zellen in ein Hydrogel eingebettet und in 3D kultiviert werden, fahren Sie mit Schritt 7 fort.
7. Transfer in Hydrogel für 3D-Kultur (optional)
- Bereiten Sie eine Hydrogel-Vorläuferlösung vor, die 2% DNase enthält.
HINWEIS: Die Zusammensetzung der Lösung variiert je nach Versuchsaufbau. Matrigel und Mischungen aus Matrigel und Kollagen Ich arbeite gut in diesem Protokoll, aber auch andere Hydrogele sind möglich. - 50 μL Hydrogellösung mit 2% DNase in den Einlass jeder Durchflusszelle geben. Saugen Sie die überschüssige Flüssigkeit aus dem Auslass ab und treiben Sie die Hydrogellösung in die Durchflusszelle. Bei viskosen Hydrogelvorläufern kann ein leichtes Kippen des Objektträgers erforderlich sein, um das Hydrogel in die Durchflusszelle fließen zu lassen.
- Inkubieren Sie den Objektträger bei 37 °C für 30-45 min (abhängig von der Hydrogel-Gelierungskinetik), damit sich das Hydrogel festsetzen und die DNA-basierte Adhäsion zwischen den Zellen und der Oberfläche spalten kann.
- Entfernen Sie jede Durchflusszelle aus dem Objektträger und legen Sie sie auf die Hydrogelvorläuferlösung.
- Fügen Sie 50 μL Hydrogelvorläufer zu einer Vertiefung eines 2-Well-Kammerobjektträgers oder einer 6-Well-Platte hinzu.
- Pipettieren Sie 10 μL PBS auf beiden Seiten jeder Durchflusszelle.
- Verwenden Sie eine Rasierklinge oder eine feinpunktige Pinzette, um das PBS über die gesamte Länge der Durchflusszelle zu verteilen, und heben Sie dann vorsichtig die Seiten der Durchflusszelle an, so dass das PBS unter das Hydrogel stürzt.
HINWEIS: Dies "schwebt" das Hydrogel über den Schlitten und ermöglicht eine Übertragung ohne Verzerrung oder Reißen. - Bewegen Sie die Durchflusszelle mit einer Rasierklinge vorsichtig an den Rand des Glasobjektträgers.
- Drehen Sie die Folie um. Stoßen Sie mit der Rasierklinge die Durchflusszelle vom Objektträger ab, so dass sie auf der Rasierklinge landet.
- Heben Sie die Durchflusszelle mit einer gekrümmten Pinzette von der Rasierklinge auf. Invertieren Sie die Flusszelle, so dass sich die Zellen auf der Unterseite befinden, und legen Sie sie dann auf den Tröpfchen der Hydrogel-Vorläuferlösung.
- Wiederholen Sie die Schritte 7.4.1 - 7.4.6 für jede Durchflusszelle.
- Inkubieren Sie für mindestens 30 min, so dass das Hydrogel, das die gemusterten Zellen enthält, an die Hydrogelunterlage binden kann, was zur vollständigen Einbettung der gemusterten Zellen führt.
- Entfernen Sie die PDMS-Durchflusszelle.
- Fügen Sie genügend Medien hinzu, um in die PDMS-Durchflusszelle einzutauchen.
HINWEIS: Der Einstrom von Medien lockert die Haftung zwischen dem Hydrogel und der PDMS-Durchflusszelle. - Verwenden Sie eine gekrümmte Pinzette, die entlang der langen Achse der Flusszelle ausgerichtet ist, um die Durchflusszelle vorsichtig anzustupsen, bis sie abspringt und in das Medium schwebt. Sammeln Sie die Durchflusszelle mit einer Pinzette und entsorgen Sie sie.
HINWEIS: Für optimale Ergebnisse verteilen Sie die gekrümmte Pinzette und üben Sie sanften Druck auf die Wände der PDMS-Durchflusszelle aus. Wenden Sie Kraft in Richtung der Längsachse der Flusszelle an.
- Fügen Sie genügend Medien hinzu, um in die PDMS-Durchflusszelle einzutauchen.
8. Bestätigen Sie die erfolgreiche Kennzeichnung der Zellen mit CMO (optional, zur Fehlerbehebung)
- Bestellen Sie ein fluoreszierend modifiziertes (FAM oder AF647) Oligonukleotid, das die Oberflächenadhäsionssequenz des im Experiment verwendeten Adapterstrangs ergänzt.
- Markieren Sie Zellen mit CMO-DNA und waschen Sie überschüssige DNA wie in Schritt 5 beschrieben aus. Resuspend in 200 μL eiskaltem PBS.
- Bilden Sie eine 4 μM-Lösung des fluoreszenzmarkierten komplementären Oligonukleotids in PBS. 200 μL dieser Lösung in die Zellsuspension geben. 5 Min. auf Eis inkubieren.
- Fügen Sie 1 ml eiskaltes PBS hinzu. Mischen. Zentrifugieren Sie die Zellen, um sie zu pelletieren. Überstand entfernen. Wiederholen Sie diesen Vorgang noch zwei weitere Male, um jede DNA auszuwaschen, die nicht hybridisiert wurde.
- Führen Sie eine analytische Durchflusszytometrie durch, um das Vorhandensein von DNA auf der Zelloberfläche zu quantifizieren.
- Analysieren Sie auf einem Durchflusszytometer Kontrollzellen, die nicht mit DNA markiert wurden. Richten Sie Tore basierend auf dieser Population ein.
- Analysieren Sie CMO-markierte Zellen, die mit einem fluoreszierend markierten komplementären Oligonukleotid behandelt wurden.
- Berechnen Sie die mittlere Fluoreszenzintensität.
Ergebnisse
Dieses Protokoll ermöglicht es, Zellen in 2D und 3D mit hoher Präzision und ohne den Einsatz von kundenspezifischen Reagenzien oder teuren Reinraumgeräten zu strukturieren. Abbildung 1 zeigt eine Übersicht über das Protokoll. Zunächst werden DNA-funktionalisierte Objektträger durch Photolithographie erstellt. Als Nächstes werden die Zellen mit CMOs gekennzeichnet. Die Zellen werden dann über den Objektträger geflossen, wo sie sich nur an die DNA-funktionalisierten Regionen des Objektträgers anheften. Nachdem überschüssige Zellen weggespült wurden, wird das gewünschte Muster der Zellen aufgedeckt. Diese Zellen können auf dem Objektträger kultiviert oder in ein DNase-haltiges Hydrogel eingebettet und für die 3D-Zellkultur vom Objektträger übertragen werden.
Die Markierung von Zellen mit CMOs ermöglicht ihre Anheftung an den DNA-musterten Objektträger (Abbildung 2). Zunächst wird der cholesterinmodifizierte Universal Anchor Strand mit dem AdapterStrang vorhybridisiert. Als nächstes wird die Universal Anchor + Adapter-Lösung 1:1 mit der Zellsuspension gemischt. Das Cholesterin auf dem Universal Anchor + Adapter Komplex wird in die Zellmembran eingesetzt. Die Zugabe des cholesterinmodifizierten Universal Co-Anchor Strangs, der mit dem Universal Anchor Strand hybridisiert, verbessert die Stabilität des CMO-Komplexes in der Zellmembran, indem die Nettohydrophobie des Komplexeserhöht wird 26. Nach dem Auswaschen der überschüssigen DNA aus der Zellsuspension werden die Zellen über den Objektträger geflossen. Die Hybridisierung zwischen dem Adapterstrang und dem Oberflächen-DNA-Strang führt zur Anheftung von Zellen an die DNA-gemusterten Regionen des Objektträgers.
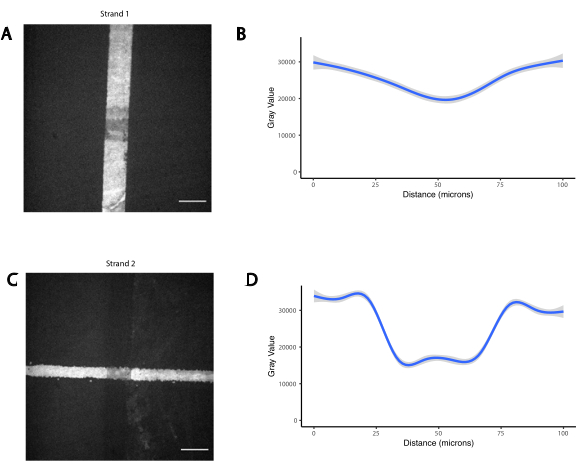
Das Muster der Zellen wird durch Photolithographie erzeugt, um die Bindung von aminmodifizierten DNA-Oligos an bestimmte Regionen eines aldehydmodifizierten Glasobjektträgers29 einzuschränken (Abbildung 3A). Positiver Fotolack wird auf einem aldehydfunktionalisierten Objektträger gedreht. Eine transparente Fotomaske wird dann auf das Dia gelegt und das Dia wird UV-Licht ausgesetzt. Nach der Entwicklung sind die Bereiche des Objektträgers, die UV-Licht ausgesetzt wurden, nicht mehr mit Fotolack beschichtet und weisen somit Aldehydgruppen auf. Eine 20-μM-Lösung aus aminmodifizierten DNA-Oligos wird dann auf den Objektträger fallen gelassen und verteilt, um die gemusterten Regionen abzudecken. Backen mit anschließender reduktiver Amination führt zu einer kovalenten Bindung zwischen der aminmodifizierten DNA und dem Objektträger. Bemerkenswerterweise kann dieser Prozess wiederholt werden, um mehrere Oligos zu mustern, ohne dass die Funktionalität der zuvor gemusterten Oligos verloren geht (Abbildung 3B). Es sollte jedoch darauf geachtet werden, überlappende Muster zu vermeiden, was dazu führt, dass beide Oligos in einer reduzierten Konzentration vorhanden sind (ergänzende Abbildung 3). Mehrere Zellpopulationen können sequenziell strukturiert werden, indem Adapterstränge verwendet werden, die sich in ihrer modularen Domäne unterscheiden (die 20 Basen, die dem 3'-Ende am nächsten sind).
Obwohl dieses Photostrukturierungsprotokoll von Scheideler et al. im Rahmen eines Reinraums entwickelt wurde, haben wir gezeigt, dass es möglich ist, ähnliche Ergebnisse mit einem kostengünstigen, "hausgemachten" Photolithographie-Setup zu erzielen, das leicht in einen chemischen Abzug passt. Das Setup umfasst einen 400-Dollar-Spincoater aus einem Gleichstrommotor, einem digitalen Controller und einer CD-Kuchenbox sowie eine UV-Lampe, die aus einzelnen Komponenten zusammengesetzt und in einem wiederverwendeten Behälter für scharfe Gegenstände untergebracht wurde (ergänzende Abbildung 1). Der Hauptvorteil des hausgemachten Fotolithographie-Setups besteht darin, dass es sehr erschwinglich ist (< $ 1000 für die gesamte Ausrüstung), während es immer noch in der Lage ist, Einzelzellen-Funktionen zu erstellen. Die Verwendung kostengünstiger Geräte hat jedoch ihre Grenzen - zum Beispiel ist es schwieriger, Fiducialmarker präzise auszurichten, um mehrere DNA-Oligos ohne Verwendung eines Mask Aligners zu mustern. Wir empfehlen dieses kostengünstige Fotolithographie-Setup für Labore, die keinen bequemen Zugang zu einem Reinraum haben oder diese Methode ohne große Investitionen ausprobieren möchten.
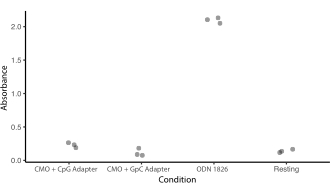
Um optimale Bedingungen für die DNA-programmierte Zelladhäsion zu identifizieren, variierten wir systematisch die Konzentrationen von DNA-Strängen auf Zelloberflächen und maßen die Effizienz der Zelladhäsion an DNA-modifizierten Glasoberflächen. Die Konzentration von Universal Anchor + Adapter Strang und Universal Co-Anchor in Markierungslösungen wurde über mehrere Größenordnungen variiert (Abbildung 4A,B), was zu 104 - 106 DNA-Komplexen pro Zelle führte (Ergänzende Abbildung 4). Die Zelladhäsion war dosisabhängig, mit minimaler Zelladhäsion am DNA-Muster, wenn Zellen mit CMOs in einer Konzentration von 0,05 μM oder weniger markiert wurden, und hoher Belegung bei einer Konzentration von 2,5 μM und höher. Wir haben daher in den meisten Experimenten eine 2 μM-Lösung von Universal Anchor + Adapter Strand und eine 2 μM-Lösung von Universal Co-Anchor verwendet. Es wäre auch zu erwarten, dass die Zelladhäsion abnimmt, wenn die Menge an DNA, die auf der Glasoberfläche verwendet wird, um29 abnimmt oder wenn die Fehlanpassungen zwischen dem Adapterstrang und dem Oberflächenstrang zunehmen. Weitere Informationen zum Design der Adapterstrangsequenz finden Sie in Supplemental File 2. Die CMO-Markierung unter Verwendung von Adaptersträngen ohne CpG-Wiederholungen stimulierte TLR9 in HEK-Zellen, die Maus-TLR9 exprimieren ( ergänzende Abbildung5).
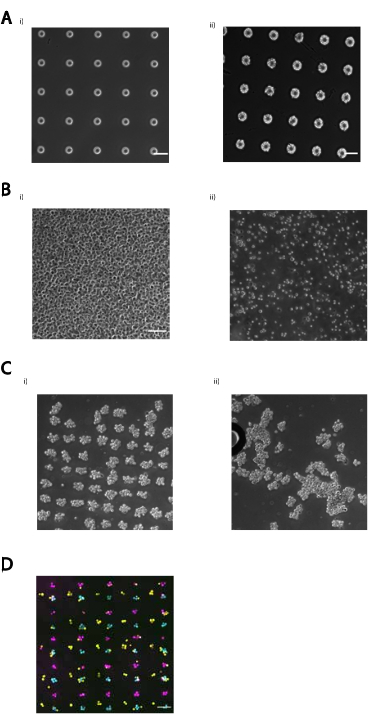
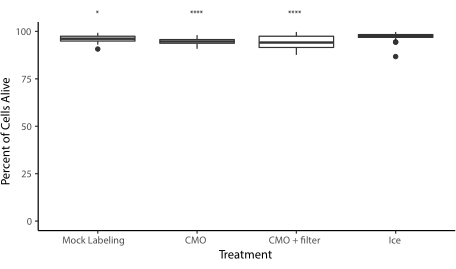
Wir bieten mehrere Demonstrationen, dass das überarbeitete Protokoll eine reproduzierbare und effiziente DNA-programmierte Zelladhäsion bietet. Zum Beispiel hafteten menschliche Nabelschnurvenen-Endothelzellen (HUVECs), die mit CMOs markiert waren, mit hoher Effizienz an DNA-Mustern. CMO-markierte HUVECs sowie LMO-markierte HUVECs (Abbildung 5A). Zellen, die mit CMO-DPAC strukturiert wurden, behielten ihre Lebensfähigkeit und Funktionalität. Zellen, die mit CMOs markiert waren, wurden mit Calcein AM und Ethidium Homodimer gefärbt, um die Lebensfähigkeit zu beurteilen (Ergänzende Abbildung 6). Die Unterschiede in der Lebensfähigkeit im Vergleich zu nicht markierten Kontrollzellen waren gering (94% vs. 97%). Einzelne MDCKs, die über CMO-DPAC strukturiert und in Matrigel übertragen wurden, konnten sich nach 5 Tagen Kultur korrekt vermehren und polarisieren (Abbildung 5B). DPAC bietet auch eine Möglichkeit, Muster von Zellen in die dritte Dimension zu erarbeiten (Abbildung 5C). Beispielsweise können mehrschichtige, mehrzellige Aggregate durch abwechselnde Zellschichten erzeugt werden, die mit komplementären CMOs gekennzeichnet sind (Abbildung 5C). Diese Experimente zeigen, dass das Protokoll reproduzierbar ist, die Lebensfähigkeit oder Funktionalität der Zellen nicht negativ beeinflusst und zelluläre Muster liefert, die innerhalb einer einzigen Bildgebungsebene in einem 3D-ECM erfolgreich kultiviert werden können.
Durch die Bereitstellung orthogonaler DNA-Sequenzen zur direkten Zelladhäsion bietet DPAC die Möglichkeit, mehrere Zelltypen auf einer einzigen Oberfläche zu strukturieren. Um dieses Merkmal von DPAC zu implementieren, müssen DNA-Muster, die durch Photolithographie erzeugt werden, in Bezug aufeinander ausgerichtet werden. Auf dem Objektträger abgeschiedene Metalltreumarker ermöglichten die Ausrichtung mehrerer Fotomasken und damit die Musterung mehrerer Zelltypen gleichzeitig. MCF10As, die mit verschiedenen einzigartigen Farbstoffen gebeizt wurden, wurden mit orthogonalen CMOs markiert und gemustert, um eine Visualisierung der UC Berkeley- und UCSF-Logos zu erstellen (Abbildung 6). Dieses Experiment zeigt, dass mehrere einzigartige Zellpopulationen mit hoher Präzision und ohne Kreuzkontamination zusammen strukturiert werden können.
Eine erfolgreiche Strukturierung von Zellen mit CMO-DPAC erfordert eine qualitativ hochwertige Photolithographie, eine ausreichende Konzentration von Oligo auf der Zelloberfläche, eine hohe Dichte von Zellen über dem Muster und ausreichend Waschen. Das Scheitern eines dieser Schritte wirkt sich auf das Endergebnis aus. Ergänzende Abbildung 2 enthält Beispielbilder der korrekten und falschen Photolithographie (Supplemental Figure 2A), die gewünschte Zelldichte über dem Muster, um vollständig besetzte Muster zu erzeugen (Supplemental Figure 2B), den Verlust von gemusterten Zellen durch übermäßig starkes Pipettieren während nachfolgender Schritte von DPAC (Supplemental Figure 2C) und unerwünschte Verklumpung von Zellen (Supplemental Figure 2D). Tabelle 1 enthält eine Liste der häufigsten Fehlerpunkte und die vorgeschlagene Problembehandlung. Die Verwendung von fluoreszierenden komplementären Oligos wird als Werkzeug zur Fehlerbehebung empfohlen, um das Vorhandensein von gemusterter DNA auf dem Objektträger und das Vorhandensein von CMOs auf der Zelloberfläche durch Durchflusszytometrie zu bestätigen (siehe Schritt 8 des Protokolls).

Abbildung 1: Überblick über das CMO-DPAC-Protokoll. Zunächst wird ein Objektträger mit DNA-Muster erstellt, indem ein aldehydfunktionalisierter Glasobjektträger mit einem positiven Fotolack beschichtet, mit einer Transparenzmaske im gewünschten Muster bedeckt und UV-Licht ausgesetzt wird. Der UV-belichte Fotolack wird mit dem Entwickler weggespült, wodurch exponierte Bereiche des Aldehydobjektträgers verbleiben und die Bindung von aminfunktionalisierter DNA an die Oberfläche ermöglicht wird. Die Zellen werden dann mit CMOs gekennzeichnet und über die Oberfläche geflossen. Die DNA auf der Zellmembran hybridisiert mit der DNA auf der Oberfläche, was zu einer Adhäsion führt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Zellen werden in einem schrittweisen Prozess mit CMOs gekennzeichnet. Zunächst wird der cholesterinmodifizierte Universal Anchor Strand mit dem AdapterStrang vorhybridisiert. Als nächstes wird die Universal Anchor + Adapter-Lösung mit der Zellsuspension gemischt. Das Cholesterin auf dem Universal Anchor + Adapter Komplex wird in die Zellmembran eingesetzt. Nach der Inkubation wird der cholesterinmodifizierte Universal Co-Anchor Strang der Zellsuspension zugesetzt, wo er mit dem Universal Anchor Strang hybridisiert und in die Zellmembran einführt. Die Zugabe des zweiten Cholesterinmoleküls erhöht die Nettohydrophobie des DNA-Komplexes und stabilisiert ihn innerhalb derMembran 26. Nach dem Auswaschen der überschüssigen DNA werden die Zellen konzentriert und zu einer PDMS-Flusszelle auf der gemusterten Oberfläche hinzugefügt. Das 3'-Ende des Adapterstrangs hybridisiert mit dem Surface DNA-Strang auf dem Objektträger, was zu einer Haftung am Objektträger führt, insbesondere in Regionen, die mit komplementärer DNA funktionalisiert sind. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
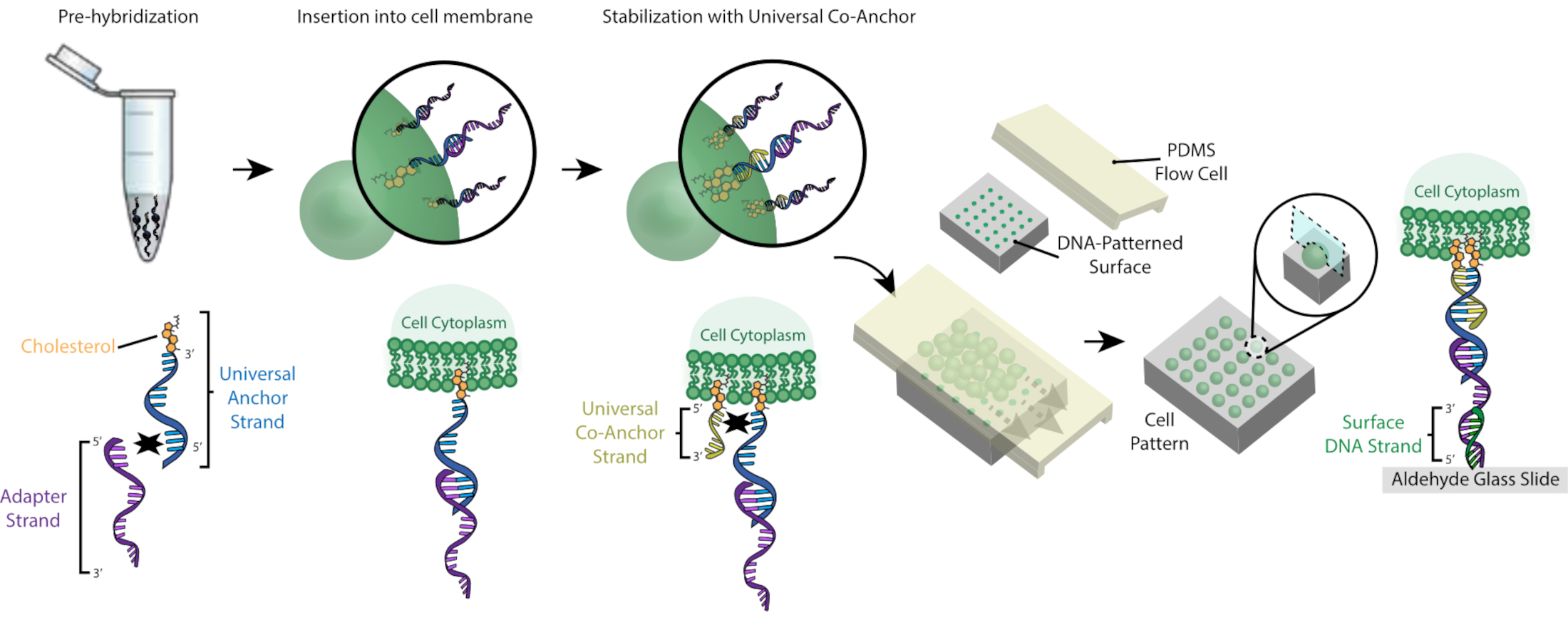{kind=link}

Abbildung 3:Die Photolithographie wird verwendet, um die DNA-gemusterten Objektträger zu erstellen, die letztendlich die Platzierung der Zellen bestimmen. (A) Überblick über den Photolithographieprozess. Ein Aldehyd-funktionalisierter Objektträger ist mit einem positiven Fotolack gedreht. UV-Licht scheint durch eine transparente transparente Fotomaske auf das Objektträger, wo die Zellhaftung gewünscht ist. Nachdem der Objektträger entwickelt wurde, haben die Regionen, die zuvor UV-Licht ausgesetzt waren, nun Aldehydgruppen freigelegt. Eine 20 μM-Lösung eines aminfunktionalisierten DNA-Oligos wird dann auf den Objektträger fallen gelassen und über die gemusterten Regionen verteilt. Der Objektträger wird dann gebacken, um die Bildung von Schiff-Bindungen (C = N) zwischen der Amin- und Aldehydgruppe zu induzieren, eine reversible kovalente Bindung29. Die anschließende reduktive Aminierung mit 0,25% Natriumborhydrid in PBS wandelt die Schiff-Base durch reduktive Aminierung in ein sekundäres Amin um, was zu einer irreversiblen Bindung zwischen der DNA und dem Objektträger führt. Der restliche Fotolack kann dann durch Spülen mit Aceton entfernt werden. (B) Dieser Prozess kann wiederholt werden, um Mehrkomponenten-DNA-Muster zu erzeugen und somit Experimente mit mehreren Zellpopulationen durchzuführen. (i) Nachdem das erste Oligo gemustert wurde, wird das Dia wieder mit Fotolack beschichtet und das Protokoll verläuft wie zuvor. Die Ausrichtung der Fotomasken mit treuhänderischen Markern ist notwendig, um mehrere DNA-Stränge zu strukturieren. (ii) Jeder Zelltyp, der gemustert wird, unterscheidet sich in der modularen 20-Basis-Domäne des Adapterstrangs. Durch die Verwendung orthogonaler Sätze komplementärer Oligos können mehrere Zelltypen ohne Kreuzadhäsion strukturiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
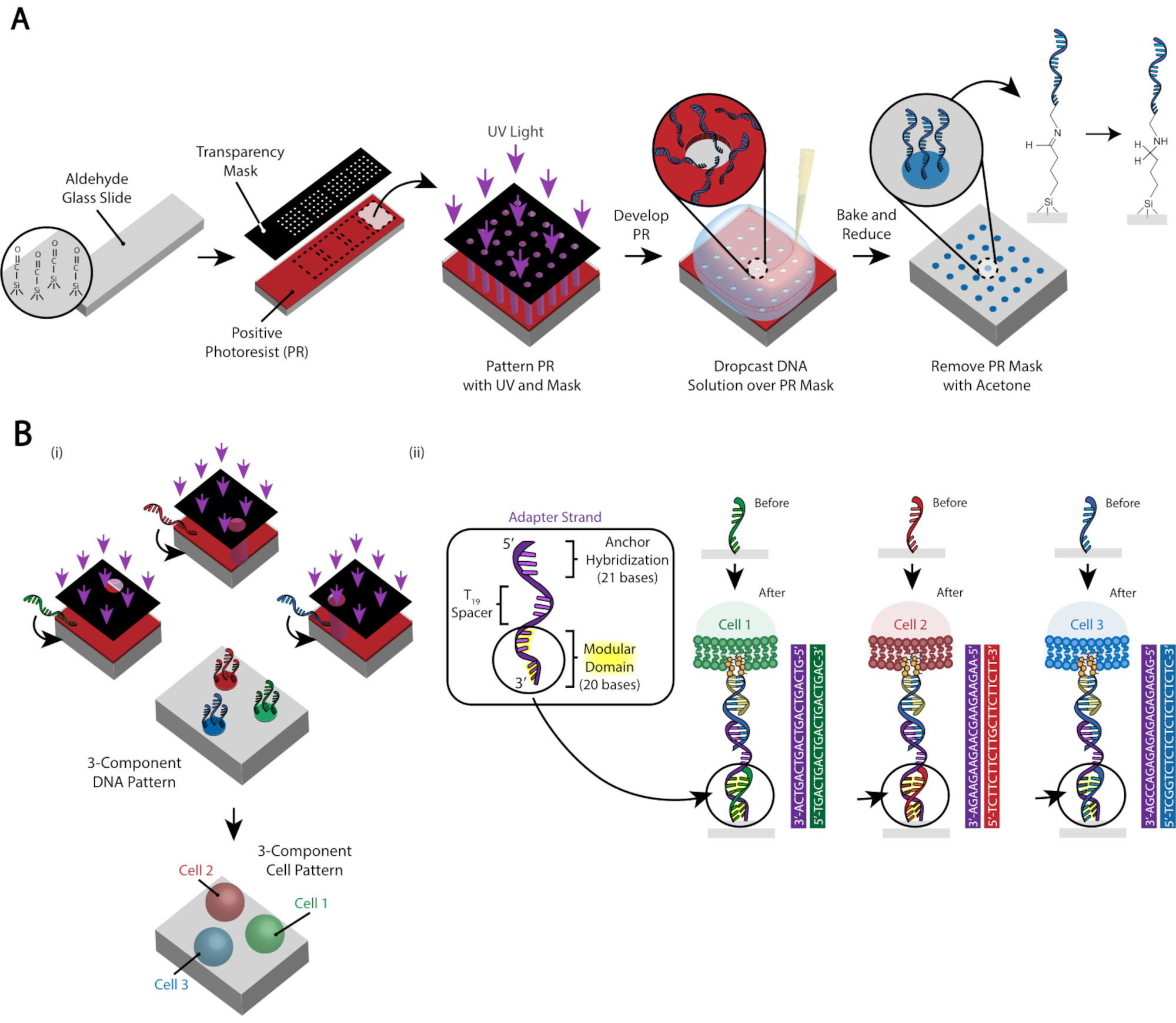{kind=link}

Abbildung 4: Die Adhäsion von CMO-markierten Zellen an DNA-Mustern nimmt in Abhängigkeit von der CMO-Konzentration während der Markierung zu. In diesem Experiment wurden der Universal Anchor + Adapter Strang (vorhybridisiert) und der Universal Co-Anchor in gleichen Konzentrationen verwendet. Die Konzentration bezieht sich auf die Konzentration von CMO in der Zellsuspension während der CMO-Markierung von Zellen. (A) Quantifizierung des Prozentsatzes der DNA-Spots mit einem Durchmesser von 15 μm, die von CMO-markierten MCF10A-Zellen als Funktion der CMO-Konzentration während der Zellmarkierung besetzt waren. Die Daten werden als Mittelwert ± Standardabweichung von drei Experimenten dargestellt. (B) Repräsentative Abbildungen der DNA-Muster (Magenta) und adhärenten MCF10As (Cyan) bei unterschiedlichen CMO-Konzentrationen. Maßstabsbalken = 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
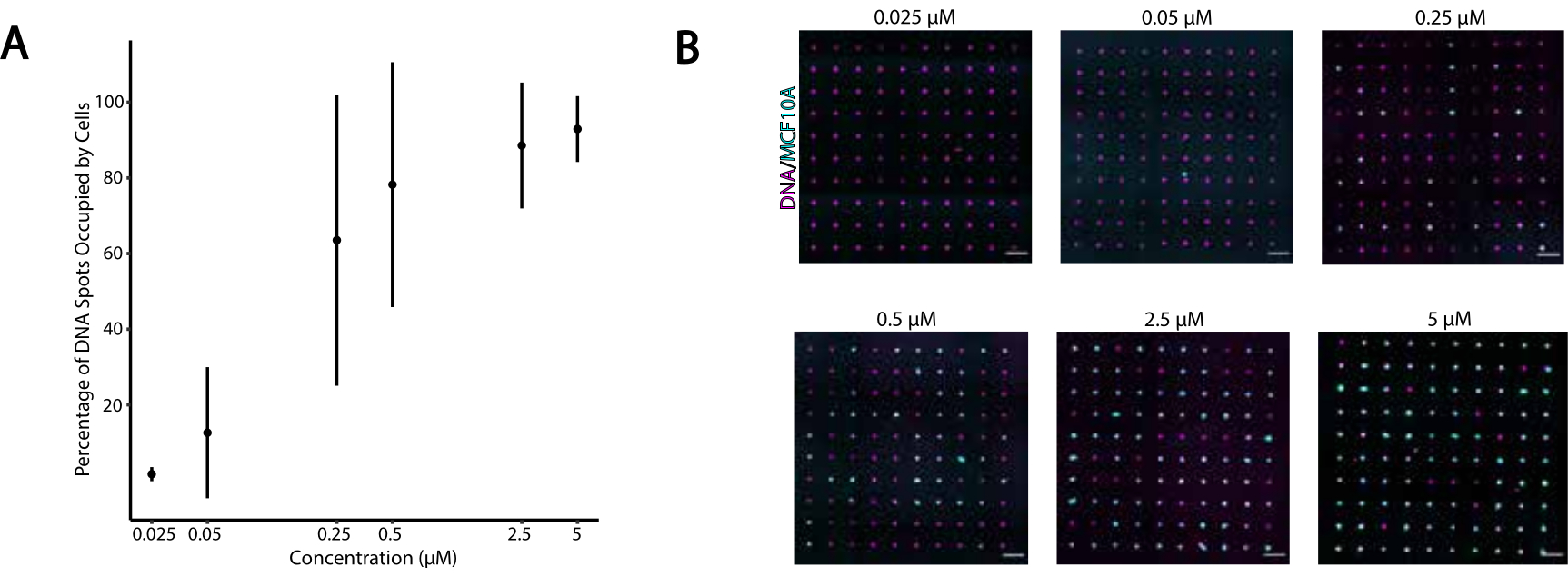{kind=link}

Abbildung 5:CMO-DPAC kann verwendet werden, um zweidimensionale Zellmuster zu erzeugen, die anschließend in ein dreidimensionales Hydrogel zur Kultur eingebettet und/oder geschichtet werden können, um mehrschichtige Strukturen zu erzeugen. (A) Der direkte Vergleich zwischen CMO-markierten menschlichen Nabelschnurvenen-Endothelzellen (HUVECs) und LMO-markierten HUVECs klebte an einem linearen DNA-Muster. Beide Methoden der Zellmarkierung führen zu einer fast 100%igen Belegung des DNA-Musters. (B) Einzelne Madin-Darby Canine Kidney Cells (MDCKs), die H2B-RFP exprimieren, wurden auf Flecken mit einem Durchmesser von 15 μm im Abstand von 200 μm strukturiert und anschließend in Matrigel eingebettet. Nach 120 h Kultur wurden die resultierenden Epithelzysten fixiert und für E-Cadherin, Aktin und Kollagen IV gefärbt. Sphäroid in weißer Box wird im Detail gezeigt. Maßstabsbalken = 50 μm. (C) Mehrschichtige zelluläre Strukturen können geschaffen werden, indem separate Zellpopulationen mit komplementären Adaptersträngen markiert und sequenziell strukturiert werden, so dass jede neue Zugabe von Zellen an der Zellschicht davor haftet. (i) Ein Schema der sequentiellen Strukturierung von Zellpopulationen, um mehrschichtige Strukturen zu erzeugen. ii) Mit diesem Verfahren wurden dreischichtige Zellaggregate aus MCF10As (visualisiert mit Farbstoffen) hergestellt. Maßstabsbalken = 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
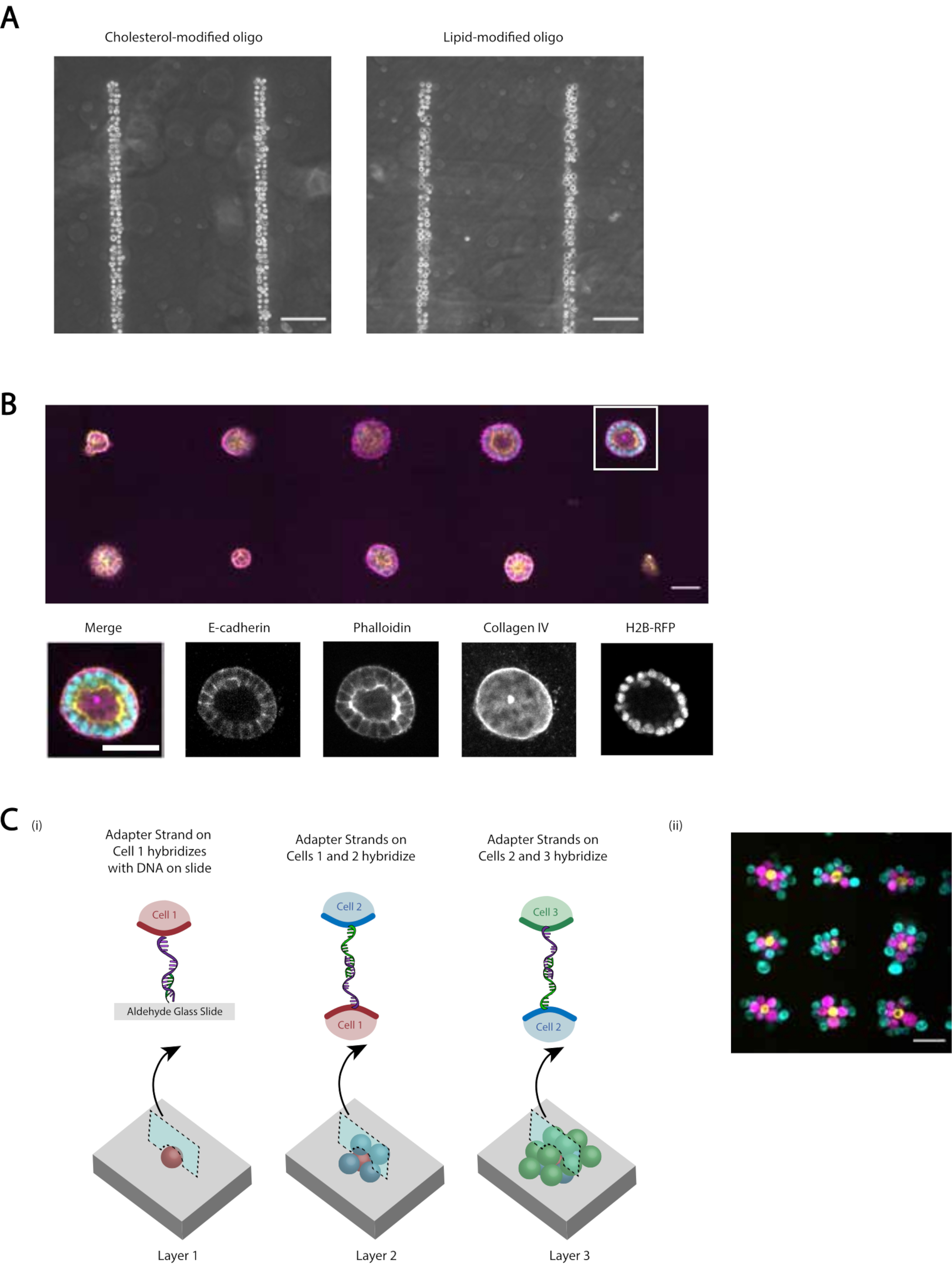{kind=link}

Abbildung 6: Mehrere Zelltypen können ohne Kreuzkontamination oder Adhäsionsverlust strukturiert werden. Mehrere aminmodifizierte DNA-Oligos wurden nacheinander auf einem Aldehydobjektträger strukturiert und unter Verwendung von Metalltreuhandmarkern ausgerichtet. Drei Populationen von MCF10As (Cyan, Magenta, Gelb) wurden mit einzigartigen Farbstoffen gefärbt, die mit komplementären CMOs beschriftet und auf dem Objektträger gemustert wurden, was zu einem Bild der UC Berkeley- und UCSF-Logos führte. Maßstabsleiste 1 mm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergänzende Abbildung 1: Beispielbilder des Benchtop-Photolithographie-Setups. (A) Schieben Sie vor der Schleuderbeschichtung auf Schleuderlack, der mit positivem Fotolack bedeckt ist. (B) Bild der Transparenz Fotomaske. (C) Während der Belichtung wird die Fotomaske zwischen dem mit Fotolack beschichteten Dia und einer Glasscheibe eingeklemmt. (D) Das Gehäuse für UV-Lampen wurde aus einem wiederverwendeten Behälter für scharfe Gegenstände hergestellt. (E) Folie eingetaucht in Entwicklerlösung. (F) Entwickelte Folie. (G) Aminmodifizierte DNA-Lösung, die auf gemusterten Regionen des Objektträgers verteilt ist. (H) PDMS-Durchflusszellen, die auf gemusterten Bereichen des Objektträgers platziert sind. Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
Ergänzende Abbildung 2: Einige Beispiele für häufige Fehler dieses Protokolls. (A) (i) Unterbacken vor UV-Belichtung oder Überentwicklung von Merkmalen nach der Belichtung kann zu Merkmalen führen, die gezackte Kanten aufweisen und unregelmäßig groß sein können. (ii) Ein Beispiel für einen korrekt fotogemusterten Schlitten, der saubere Kanten um Features, eine einheitliche Feature-Größe und keine offensichtlichen Risse im Muster aufweist. Maßstabsbalken = 50 μm. (B) Die Zelldichte ist entscheidend für die Strukturierungseffizienz. Bei der Beobachtung der Zellen auf dem Muster unter dem Mikroskop sollten nur wenige Lücken zwischen den Zellen bestehen, wie das Beispielbild links zeigt. Maßstabsbalken = 50 μm. (C) Gemusterte Zellen können empfindlich auf Flüssigkeitskräfte reagieren, die durch zu starkes Pipettieren entstehen, die die gemusterten Zellen beschädigen und entfernen können. Mehrschichtige Zellaggregate sind besonders anfällig, da eine Zelle am Boden eine Struktur aus mehreren Zellen trägt. (i) Eine Anordnung von Zellaggregaten, die erfolgreich in Matrigel eingebettet sind. (ii) Ein Gitter aus Zellaggregaten, die sich infolge des zu starken Pipettierens von viskosem Matrigel lösten. (D) Verklumpung von Zellen kann auftreten, insbesondere bei Epithelzellen. Diese Klumpen sind in der Regel homotypisch, können aber heterotypisch sein (Zellen, die an bereits gemusterten Zellen eines anderen Typs haften), wenn die Zellen besonders klebrig sind. Das Bild zeigt, dass drei verschiedene Populationen von MCF10As auf einem Array strukturiert wurden, das aus drei verschiedenen einzelligen DNA-Spots (15 μm) besteht. An den meisten DNA-Spots sind 2-4 Zellen gebunden. Verklumpungen können durch EDTA-Behandlung oder durch Herausfiltern der Klumpen vor der Musterung gelöst werden. Maßstabsleiste = 100 μm. Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
Ergänzende Abbildung 3: Überlappende Photomuster führen zur Anwesenheit beider Oligos in reduzierter Konzentration. Zwei orthogonale aminmodifizierte Oligos wurden nacheinander photostrukturiert, zuerst eine vertikale Linie (Strang 1), gefolgt von einer horizontalen Linie, die sie überlappt (Strang 2). Die Oligos wurden dann durch Hybridisierung mit fluoreszierenden komplementären Oligos visualisiert. (A) Fluoreszenzbild von Strang 1. (B) Quantifizierung des Fluoreszenzprofils von Strang 1 über eine vertikale Linie von 100 μm, die die Überlappung überspannt. (C) Fluoreszenzbild von Strang 2. (D) Quantifizierung des Fluoreszenzprofils von Strang 2 über eine horizontale Linie von 100 μm, die die Überlappung überspannt. Maßstabsleiste = 50 μm. Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
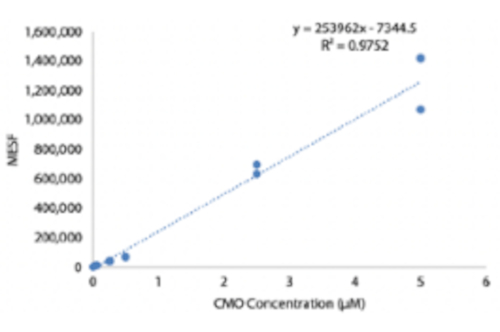
Ergänzende Abbildung 4: Quantifizierung von DNA-Komplexen auf der Zelloberfläche als Funktion der CMO-Markierungskonzentration. HUVECs wurden mit unterschiedlichen Konzentrationen von CMO-Lösung markiert, gewaschen und dann mit einem fluoreszierenden komplementären Strang inkubiert. Ein MESF-Mikrosphärenkit (Molecules of Equivalent Soluble Fluorochrome) wurde verwendet, um quantitative Durchflusszytometrie durchzuführen und die Anzahl der DNA-Komplexe auf der Zelloberfläche als Funktion der CMO-Konzentration während der Markierung abzuschätzen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
Ergänzende Abbildung 5: Die CMO-Kennzeichnung stimuliert die TLR9-Reaktion nicht. Es wurde ein Experiment durchgeführt, um zu sehen, ob die CMO-Markierung den DNA-Nachweismechanismus von TLR9 auslösen würde und ob dies durch CpGs in der Adapterstrangsequenz beeinflusst würde. HEK-Zellen, die Maus-TLR9 exprimieren, wurden über Nacht mit 0,2 μM entweder ODN 1826 (einem CpG-haltigen TLR9-Agonisten), CMO Universal Anchor + Universal Co-Anchor + Adapter Strang, der die gleiche Sequenz wie ODN 1826 (CMO-CpG) enthielt, oder CMO Universal Anchor + Universal Co-Anchor + Adapter Strang, der eine ähnliche Sequenz enthielt, aber mit Ersatz der CpGs durch GpCs (CMO-GpC) inkubiert. Die TLR9-Stimulation würde zur Produktion von SEAP (sezernierte embryonale alkalische Phosphatase) führen. Die SEAP-Sekretion wurde durch einen kolorimetrischen Assay (Absorption) quantifiziert. Die Behandlungsbedingungen wurden mit Ruhezellen verglichen, die nur mit PBS behandelt wurden. Die Inkubation mit CMO-GPC stimulierte die TLR9-Expression nicht. Die Inkubation mit CMO-CpG war etwas höher als bei Ruhezellen, aber viel niedriger als bei ODN-1826. Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
Ergänzende Abbildung 6: Lebensfähigkeit der Zellen nach dem CMO-Markierungsprozess. Um zu beurteilen, wie sich das Protokoll auf die Lebensfähigkeit auswirkt, wurden HUVECs in vier Populationen aufgeteilt: eine blieb 1 h auf Eis, eine wurde mit PBS simuliert, aber ansonsten durch alle Zentrifugen- und Waschschritte geführt, eine wurde mit CMOs markiert und eine wurde mit CMOs markiert und durch einen 40 μm-Filter gefiltert, um Klumpen zu entfernen. Die Zellen wurden dann mit Calcein AM und Ethidium Homodimer gefärbt, um die Anzahl der lebenden und toten Zellen zu beurteilen. Alle Behandlungen führten zu einer signifikant verminderten Lebensfähigkeit als die Eiskontrolle (Einweg-ANOVA mit Tukey-Post-hoc-Analyse), aber die mediane Lebensfähigkeit für die CMO-Kennzeichnung (mit oder ohne Filterung) betrug etwa 94%. Daten aus drei unabhängigen Experimenten. * = p < 0,05. = p < 0.0001 Bitte klicken Sie hier, um diese Datei herunterzuladen.
{kind=link}
| Ergebnis | Mögliche Ursache(n) | Vorgeschlagene Korrekturen |
| Photolithographie – Merkmale werden geknackt | Inkonsistentes oder unzureichendes Soft-Bake | Erhöhen Sie die Zeit des Soft-Bake auf bis zu 3 Minuten; Überprüfen Sie die tatsächliche Temperatur der Kochplatte und erhöhen Sie die Temperatur bei Bedarf |
| Photolithographie - Merkmale sind nicht scharf oder haben Fotolack in ihnen | Unterentwicklung | Erhöhen Sie die Zeit, die die Folie für die Entwicklerlösung aufwendet. sanftes Rühren einbauen |
| Photolithographie – Merkmale auf allen Folien inkonsistent | UV-Licht ist möglicherweise nicht zentriert oder nicht richtig fokussiert | Passen Sie das UV-Licht-Setup an, um kollimiertes Licht mit gleichmäßiger Intensität zu gewährleisten |
| Zellen haften nicht mit hoher Effizienz an gemusterten Stellen | Nicht genug DNA auf der Oberfläche | Bestätigen Sie, dass DNA auf der Oberfläche vorhanden ist, indem Sie den Objektträger mit fluoreszierenden komplementären Oligos hybridisieren und dann unter dem Mikroskop abbilden |
| Zellen sind unzureichend mit CMO markiert | Fügen Sie fluoreszierende komplementäre Oligos zur Zellsuspension hinzu und bestätigen Sie die Fluoreszenz durch Durchflusszytometrie | |
| Nicht genügend Zellen über Muster | Sammeln Sie Zellen, indem Sie sie aus der PDMS-Durchflusszelle auswaschen, zentrifugieren und in geringerem Volumen wieder suspendieren, um die Zellen zu konzentrieren | |
| Zu viel verbleibender CMO in Zellsuspension, Hybridisierung mit DNA auf Objektträgern | Fügen Sie einen weiteren Waschschritt hinzu. Achten Sie darauf, bei jeder Wäsche so viel Überstand wie möglich zu entfernen. | |
| Zu starke Internalisierung von CMO aufgrund von Zeit und Temperatur | Arbeiten Sie schnell nach der Markierung der Zellen mit CMO; Zellen und Gleiten auf Eis halten und eiskalte Reagenzien verwenden | |
| Zellen verklumpen | Zellen wurden während der Trypsinisierung nicht ausreichend getrennt | Verwenden Sie PBS + 0,04% EDTA während der Zellwäsche; Zellsuspension vor der endgültigen Wäsche durch den 35 μm Filter führen |
| Zellen haften unspezifisch | Wenn in einem bestimmten Bereich - könnte auf Kratzer auf dem Objektträger, Fehlausrichtung von PDMS-Flusszellen oder Verschütten von DNA außerhalb der Musterregion zurückzuführen sein | Vermeiden Sie Kratzer, achten Sie darauf, die PDMS-Flusszellen am Musterbereich auszurichten |
| Wenn Zellen überall kleben – unzureichendes Blockieren oder Waschen | Fügen Sie nach dem Mustern der Zellen weitere Waschungen hinzu. Pipett kräftiger während des Waschens; Block mit 1% BSA länger vor Beginn der Zellstrukturierung; Schlitten silanisieren (optionaler Schritt 3) oder bestätigen, dass die Silanisierung erfolgreich war, indem der Kontaktwinkel des Wassertropfens gemessen wurde | |
| Blasen bilden sich innerhalb der Durchflusszelle | Pipettierfehler, unebene hydrophile Oberfläche, die während der Plasmaoxidation entsteht | Wenn die Blasen klein sind, fügen Sie PBS in den Einlass der Durchflusszelle hinzu und sie können ausgewaschen werden. Wenn die Blasen größer sind, üben Sie sanften Druck auf die PDMS-Durchflusszelle aus und stoßen Sie die Blasen in Richtung Einlass oder Auslass an. |
| Zellen haften zunächst an Mustern, werden aber während des Waschens, der Musterung anderer Zelltypen oder der Zugabe des Hydrogel-Vorläufers entfernt | Die Scherkräfte durch zu starkes Pipettieren können dazu führen, dass sich die Zellen von der Oberfläche lösen | Pipettieren Sie sanfter während nachfolgender Wäschen, Zellmusterungen oder Hinzufügen von Hydrogelvorläufern. Da die Hydrogelvorläufer viskos sind, ist es wahrscheinlicher, dass sich das Muster löst, also seien Sie besonders vorsichtig. Mehrschichtige Strukturen neigen dazu, kopflastig zu sein und sind anfälliger dafür, verdrängt zu werden. |
| Gewebe verformt sich beim 3D-Transfer | Hydrogel klebt am Gleiten | Bestätigung der Hydrophobie des Objektträgers mithilfe von Kontaktwinkelmessungen |
| Verwenden Sie eine Rasierklinge, um PDMS an beiden Kanten vollständig anzuheben, so dass PBS unter dem Gewebe schweben kann | ||
| Dies kann mit reinen Kollagenhydrogelen passieren - erwägen Sie, die Proteinkonzentration oder Zusammensetzung von Hydrogel anzupassen | ||
| Zellen übertragen sich nicht mit dem Hydrogel und bleiben auf dem Objektträger | Erhöhen Sie die Turbo DNAse-Konzentration oder erhöhen Sie die Inkubationszeit | |
| Hydrogel ist nicht fest genug | Erhöhen Sie die Inkubationszeit und/oder den Gelierungsmechanismus für das betreffende Hydrogel (z. B. für Kollagen, stellen Sie sicher, dass der pH-Wert korrekt ist) | |
| Hydrogel risse beim Entfernen von PDMS | Machen Sie PDMS-Flusszellen hydrophil mit Plasmaoxidation, bevor Sie mit dem Experiment beginnen, so dass sie sich beim Hinzufügen von Medien leicht lösen können. Verwenden Sie eine Pinzette sehr vorsichtig, um das PDMS zu lösen. |
Tabelle 1: Eine Anleitung zur Problembehandlung zum Identifizieren und Beheben potenzieller Fehler, die durch dieses Protokoll entstehen können. Insbesondere eine schlechte Adhäsion von Zellen an das Muster kann viele Ursachen haben, und dieser Leitfaden sollte bei der Identifizierung und Lösung dieser Probleme helfen.
Ergänzende Datei 1. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 2. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 3. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 4. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
In diesem Artikel stellen wir ein detailliertes Protokoll für die hochauflösende Musterung von Zellen in 2D und 3D für In-vitro-Zellkulturexperimente vor. Im Gegensatz zu zuvor veröffentlichten Versionen dieser Methode konzentriert sich das hier vorgestellte Protokoll auf die Benutzerfreundlichkeit: Es erfordert keine hochspezialisierte Ausrüstung und alle Reagenzien können von Anbietern gekauft werden, anstatt eine kundenspezifische Synthese zu erfordern. Im Gegensatz zu anderen Zellmikrostrukturierungsmethoden ist diese Methode schnell und zelltypunabhängig: Sie erfordert keine spezifische Adhäsion an extrazellulären Matrixproteinen15. Durch CMO-DPAC gemusterte Zellen können in eine extrazelluläre Matrix wie Matrigel oder Kollagen eingebettet werden, was zu 3D-Kulturen mit viel höherer räumlicher Auflösung führt, als dies derzeit mit extrusionsdruckbasierten Verfahren möglich ist22. CMO-DPAC kann verwendet werden, um Hunderte bis Tausende von mikroskopischen Merkmalen pro Objektträger zu erzeugen, so dass viele Replikate gleichzeitig durchgeführt werden können.
Einer der wichtigsten Parameter für den Erfolg dieses Protokolls ist die Dichte der Zellen, die den Flusszellen auf dem gemusterten Objektträger hinzugefügt werden. Idealerweise sollte die Dichte mindestens 25 Millionen Zellen / ml betragen. Beim Laden in die Durchflusszellen führt diese Zelldichte zu einer nahezu dicht gepackten Monoschicht von Zellen über dem Muster (Ergänzende Abbildung 2B). Diese hohen Zelldichten maximieren die Wahrscheinlichkeit, dass sich eine Zelle direkt auf einem DNA-Punkt absetzt und anhaftet. Die Verringerung der Zelldichte verringert die Gesamtstrukturierungseffizienz. Ein weiterer kritischer Schritt in diesem Protokoll ist die gründliche Erneute Suspendierung der Zellen in PBS- oder serumfreien Medien vor Zugabe der CMO-Lösung. Die CMOs teilen sich sehr schnell in Zellmembranen auf und die Zugabe der CMO-Lösung direkt zu einem Zellpellet führt zu einer heterogenen Markierung der Zellen. Nach Zugabe der CMO-Lösung zur Zellsuspension ist es wichtig, gründlich durch Pipettieren zu mischen, damit die Zellen gleichmäßig mit den CMOs gekennzeichnet sind. Nach den Inkubationen ist es notwendig, die überschüssigen CMOs durch mehrere Zentrifugations- und Waschschritte gründlich auszuwaschen. Überschüssiges freies CMO, das in der Zellsuspension vorhanden ist, bindet an die gemusterte aminmodifizierte DNA auf dem Glasobjektträger und blockiert die Hybridisierung und Adhäsion der CMO-modifizierten Zellen in suspension. Zeit ist auch eine wichtige Überlegung für dieses Protokoll. Es ist wichtig, bei der Verwendung von CMOs so schnell wie möglich zu arbeiten und die Zellen auf Eis zu halten, um die Internalisierung der CMOs zu minimieren und die Lebensfähigkeit der Zellen zu maximieren. Durchflusszytometrie-Experimente haben gezeigt, dass CMOs nicht so lange auf der Zelloberfläche persistieren wie LMOs, mit 25% Verlust von CMO-Komplexen über zwei Stunden Inkubation auf Eis36. Darüber hinaus nimmt die Lebensfähigkeit der Zellen mit zunehmender Zellhandhabungszeit ab. Die Lebensfähigkeit kann maximiert werden, indem schnell gearbeitet wird, Zellen auf Eis gehalten werden, eiskalte Reagenzien verwendet werden und serumfreie Medien verwendet werden, um einige Nährstoffe bereitzustellen.
Obwohl CMO-DPAC eine leistungsstarke Möglichkeit sein kann, die Zellbiologie zu untersuchen, indem Zellen mit hoher Präzision strukturiert werden, hat es seine Grenzen. CMO-DPAC-Experimente können eine Herausforderung darstellen, zumal die experimentelle Komplexität mit mehreren Zelltypen, Schichten oder 3D-Zellkulturen hinzugefügt wird (Supplemental File 1). Experimentelle Fehler können beim Starten dieses Protokolls häufig auftreten, wie in Tabelle 1beschrieben. Daher empfehlen wir den Benutzern, Qualitätskontrollen durchzuführen (Bestätigung, dass DNA auf dem Objektträger vorhanden ist, Bestätigung, dass Zellen ausreichend mit DNA markiert sind (Schritt 8), Bestätigung, dass überschüssige Zellen gründlich weggewaschen wurden usw.), um sicherzustellen, dass das Experiment erfolgreich ist und Schritte zu identifizieren, die möglicherweise eine weitere Optimierung erfordern. Wir hoffen, dass die in diesem Manuskript und seinen ergänzenden Dateien enthaltenen Informationen dazu beitragen werden, die erforderliche Fehlerbehebung zu erleichtern.
Cholesterin ist ein bioaktives Molekül, dessen Internalisierung den Zellstoffwechsel, die Genexpression und die Membranfluidität beeinflussen kann37,38. Eine frühere Studie verglich die Auswirkungen auf die Genexpression von CMO- und LMO-markierten Zellen mittels Einzelzell-RNA-Sequenzierung. CMO-markierte HEK-Zellen hatten eine veränderte Genexpression im Vergleich zu nicht markierten und LMO-markierten Zellen36. Die Markierung von Zellen mit CMOs führte zur differentiellen Expression (> 1,5-fachen) von acht Genen im Vergleich zu nicht markierten Kontrollen, einschließlich AP2B1, das mit Cholesterin und Sphingolipidentransport (GeneCards) in Verbindung gebracht wurde, und MALAT1, einer langen nicht-kodierenden RNA, die die Cholesterinakkumulation reguliert39. Obwohl diese transkriptionellen Reaktionen geringfügig sind, können sie dennoch von Belang sein, wenn das betreffende Experiment den Stoffwechsel, die Membrandynamik oder andere cholesterinassoziierte Signalwege in Zellen untersucht.
Dieses Protokoll ist flexibel und kann an die Anforderungen jedes Experiments angepasst werden. Da sich der CMO in die Lipidmembran einfügt, anstatt einen spezifischen Rezeptor zu verwenden, ist die Methode zelltypunabhängig (HUVECs, MCF10As, HEKs und MDCKs wurden hier demonstriert). Obwohl Cholesterin ein anderer hydrophober Anker ist als unsere zuvor veröffentlichten LMOs, haben wir bisher festgestellt, dass sie sich ähnlich verhalten. Daher würden wir erwarten, dass die CMOs mit einer Der Vielzahl von Zelltypen arbeiten, die wir zuvor mit LMOs veröffentlicht haben, einschließlich, aber nicht beschränkt auf neurale Stammzellen, Fibroblasten, periphere mononukleäre Blutzellen, Tumorzellen und primäre Brustepithelzellen6,23,27,29,36 . Die CMO-Markierung stimuliert TLR9 nicht, was darauf hindeutet, dass das Protokoll mit Immunzellen kompatibel ist. Der Membraneinbau des CMO ist eine Funktion der Gesamtzellgröße und des Grades der negativen Ladung in der Zelle Glykokalyx35. Daher haben wir ein Protokoll (Schritt 8) zum Testen des Ausmaßes der Membraneinarbeitung aufgenommen, das für eine schnelle Optimierung geeignet ist. Die spezifischen Merkmale jedes Zellmusters variieren zwangsläufig je nach experimentellem Design (siehe Ergänzende Datei 1 für weitere Anleitungen). Obwohl das oben beschriebene Photomusterungsprotokoll zur Strukturierung der DNA empfohlen wird, sollte jede Methode zur räumlichen Eingrenzung von Tröpfchen der Amin-DNA-Lösung funktionieren, wie z.B. die Verwendung von hochauflösenden Tröpfchendruckern. Die Musterauflösung und der minimale Feature-Abstand variieren je nach verwendeter Methode. Es ist theoretisch auch möglich, die DNA-Photostrukturierungsabschnitte dieses Protokolls mit anderen Methoden zu kombinieren, die verwendet wurden, um Zellen mit DNA zu markieren, wie z.B. mit DNA, die zu membranexprimierten Zinkfingern hybridisiert ist40, unter Verwendung von NHS-konjugierter DNA41und reagierender Azido-Sialinsäurereste auf der Zelloberfläche mit Phosphin-konjugierter DNA42 . CMO-DPAC kann auf eine Vielzahl von Experimenten angewendet werden, die eine strenge Kontrolle über den Zellabstand erfordern, einschließlich Studien der Wechselwirkungen zwischen Zellpaaren, Kokulturexperimente, die die Übertragung von Signalen von "Sender"-Zellen auf "Empfänger"-Zellen untersuchen, und Untersuchungen der Wirkung von nahe gelegenen extrazellulären Hinweisen auf die Stammzelldifferenzierung6,29 . Die Methode kann auch verwendet werden, um Mikrogewebe zu erzeugen, mit denen die Zellmigration in drei Dimensionen, die Selbstorganisation von Zellen in Gewebe23,27und das dynamische Zusammenspiel zwischen Zellen und dem ECM27untersucht werden können. Wir hoffen, dass dieses Protokoll den Forschern eine zugängliche Plattform bietet, um neue Anwendungen der hochauflösenden DNA-basierten Zellstrukturierung in ihren eigenen Labors zu erforschen.
Offenlegungen
Z.J.G. ist Berater und Anteilseigner von Provenance Biosciences.
Danksagungen
Die Autoren danken Jeremy Garcia für das Testen dieses Protokolls und Bhushan Kharbikar für die Schulung der Ausrüstung am UCSF Biomedical Micro and Nanotechnology Core. Diese Forschung wurde zum Teil durch Zuschüsse des Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 und W81XWH-13-1-0221), NIH (U01CA199315, DP2 HD080351-01, 1R01CA190843-01, 1R21EB019181-01A und 1R21CA182375-01A1), der NSF (MCB1330864) und des UCSF Center for Cellular Construction (DBI-1548297), einem NSF Science and Technology Center, unterstützt. O.J.S wurde durch ein NSF Graduate Research Fellowship, ein Siebel-Stipendium und ein P.E.O.-Stipendium finanziert. Z.J.G und A.R.A. sind Chan-Zuckerberg BioHub Investigators.
Materialien
| Name | Company | Catalog Number | Comments |
| 2-well Chambered Coverglass w/ non-removable wells | Thermo Fisher Scientific | 155379 | |
| Acetic Acid | Sigma-Aldrich | A6283 | |
| Adapter with External SM1 Threads and Internal SM3 Thread | ThorLabs | SM3A1 | |
| Aldehyde Functionalized Slides | Schott | Nexterion Slide AL | Store under dry conditions after opening. |
| All Plastic Syringes, 1 mL | Fisher Scientific | 14-817-25 | |
| Amine-Modified DNA Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Aspheric Condenser Lens | ThorLabs | ACL7560 | |
| Borosilicate Disc, 6in Diameter X 1/2in Thick | Chemglass | CG-1906-23 | |
| Cell Culture Dishes 60x15 mm style | Corning | 353002 | |
| Cholesterol-Modified Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Diamond Scribe | Excelta | 475B | |
| DNA Oligonucleotide | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190250 | |
| Isopropyl Alcohol | Sigma-Aldrich | 278475 | |
| Matrigel Matrix, Growth Factor Reduced | Corning | 354230 | |
| Methylene Chloride (Stabilized/Certified ACS) | Fisher Scientific | D37-4 | |
| MF-321 Developer | Kayaku Advanced Materials | n/a | |
| Microposit S1813 Positive Photoresist | Kayaku Advanced Materials | n/a | |
| Ø3" Adjustable Lens Tube, 0.81" Travel | ThorLabs | SM3V10 | |
| Oven | Thermo Scientific | 51-028-112H | |
| PE-50 Compact Benchtop Plasma Cleaning System | Plasma Etch | PE-50 | |
| Photomask (custom) | CAD/Art Services | n/a | Minimum feature size guaranteed by CAD/Art Services is 10 microns. |
| Razor Blades | Fisher Scientific | 12-640 | |
| RCT Basic Hot Plate | IKA | 3810001 | |
| Silicon Wafer (100 mm) | University Wafer | 590 | |
| Sodium Borohydride, 98%, granules | Acros Organics | 419471000 | |
| Spin Coater Kit | Instras | SCK-200 | This is a low cost option, but any spin coater that can maintain a speed of 3000 rpm will suffice. |
| SU-8 2075 | Microchem | Y111074 0500L1GL | |
| SU-8 Developer | Microchem | Y020100 4000L1PE | |
| Sylgard 184 Silicone Elastomer Kit | Dow | 2646340 | |
| Syringe Needles | Sigma-Aldrich | Z192341 | |
| T-Cube LED Driver, 1200 mA Max Drive Current | ThorLabs | LEDD1B | |
| Tridecafluoro-1,1,2,2-tetrahydrooctyl dimethylchlorosilane | Gelest | SIT8170.0 | |
| Triethylamine | Sigma-Aldrich | 90335 | |
| Turbo DNase | Thermo Fisher Scientific | AM2238 | |
| Tweezers Style N7 | VWR | 100488-324 | The curved shape of these tweezers is essential for delicately picking up the PDMS flow cells containing patterned tissues. |
| UV LED (365 nm, 190 mW (Min) Mounted LED, 700 mA) | ThorLabs | M365L2 | |
| Wafer Tweezers | Agar Scientific | T5063 | |
| WHEATON Dry-Seal vacuum desiccator | Millipore Sigma | W365885 |
Referenzen
- Kreeger, P. K., Strong, L. E., Masters, K. S. Engineering approaches to study cellular decision-making. Annual Review of Biomedical Engineering. , 49-72 (2018).
- Goubko, C. a., Cao, X. Patterning multiple cell types in co-cultures: A review. Materials Science and Engineering C. 29 (6), 1855 (2009).
- Sun, W., et al. The bioprinting roadmap. Biofabrication. 12 (2), 022002 (2020).
- Liu, W. F., Chen, C. S. Cellular and multicellular form and function. Advanced Drug Delivery Reviews. 59 (13), 1319-1328 (2007).
- Duffy, R. M., Sun, Y., Feinberg, A. W. Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle. Annals of Biomedical Engineering. 44 (6), 2076-2089 (2016).
- Chen, S., et al. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nature Communications. 7, 10309 (2016).
- Shaya, O., et al. Cell-cell contact area affects notch signaling and notch-dependent patterning. Developmental Cell. 40 (5), 505-511 (2017).
- Rao, N., et al. A co-culture device with a tunable stiffness to understand combinatorial cell-cell and cell-matrix interactions. Integrative Biology. 5 (11), 1344 (2013).
- Sriraghavan, V., Desai, R. A., Kwon, Y., Mrksich, M., Chen, C. S. Micropatterned dynamically adhesive substrates for cell migration. Langmuir. 26 (22), 17733-17738 (2010).
- Wong, L., Pegan, J. D., Gabela-Zuniga, B., Khine, M., McCloskey, K. E. Leaf-inspired microcontact printing vascular patterns. Biofabrication. 9 (2), 021001 (2017).
- Chen, T. H., et al. Directing tissue morphogenesis via self-assembly of vascular mesenchymal cells. Biomaterials. 33 (35), 9019-9026 (2012).
- Laurent, J., et al. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nature Biomedical Engineering. 1 (12), 939-956 (2017).
- Lin, C., Khetani, S. R. Micropatterned co-cultures of human hepatocytes and stromal cells for the assessment of drug clearance and drug-drug interactions. Current Protocols in Toxicology. 2017, 1-23 (2017).
- Hui, E. E., Bhatia, S. N. Micromechanical control of cell-cell interactions. Proceedings of the National Academy of Sciences of the United States of America. 104 (14), 5722-5726 (2007).
- D'Arcangelo, E., McGuigan, A. P. Micropatterning strategies to engineer controlled cell and tissue architecture in vitro. BioTechniques. 58 (1), 13-23 (2015).
- Martinez-Rivas, A., González-Quijano, G. K., Proa-Coronado, S., Séverac, C., Dague, E. Methods of micropatterning and manipulation of cells for biomedical applications. Micromachines. 8 (12), (2017).
- Lee, S., et al. Simple lithography-free single cell micropatterning using laser-cut stencils. Journal of Visualized Experiments. (158), e60888 (2020).
- Strale, P. O., et al. Multiprotein printing by light-induced molecular adsorption. Advanced Materials. 28 (10), 2024-2029 (2016).
- Melero, C., et al. Light-induced molecular adsorption of proteins using the primo system for micro-patterning to study cell responses to extracellular matrix proteins. Journal of Visualized Experiments. (152), e60092 (2019).
- Reid, J. A., Mollica, P. M., Bruno, R. D., Sachs, P. C. Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Research. , 1-13 (2018).
- Wang, Z., Lee, S. J., Cheng, H. -. J., Yoo, J. J., Atala, A. 3D bioprinted functional and contractile cardiac tissue constructs. Acta Biomaterialia. 70, 48-56 (2018).
- Miri, A. K., et al. Effective bioprinting resolution in tissue model fabrication. Lab on a Chip. 19 (11), 2019-2037 (2019).
- Todhunter, M. E., et al. Programmed synthesis of three-dimensional tissues. Nature Methods. 12 (10), 975-981 (2015).
- Todhunter, M. E., Weber, R. J., Farlow, J., Jee, N. Y., Gartner, Z. J. Fabrication of 3D microtissue arrays by DNA programmed assembly of cells. Current Protocols in Chemical Biology. 8 (3), 147-178 (2016).
- Csizmar, C. M., Petersburg, J. R., Wagner, C. R. Programming cell-cell interactions through non-genetic membrane engineering. Cell Chemical Biology. 25 (8), 931-940 (2018).
- Weber, R. J., Liang, S. I., Selden, N. S., Desai, T. A., Gartner, Z. J. Efficient targeting of fatty-acid modified oligonucleotides to live cell membranes through stepwise assembly. Biomacromolecules. 15 (12), 4621-4626 (2014).
- Hughes, A. J., et al. Engineered tissue folding by mechanical compaction of the mesenchyme. Developmental Cell. 44 (2), 165-178 (2018).
- Weber, R. J., et al. Rapid organoid reconstitution by chemical micromolding. ACS Biomaterials Science & Engineering. 2 (11), 1851-1855 (2016).
- Scheideler, O. J., et al. Recapitulating complex biological signaling environments using a multiplexed, DNA-patterning approach. Science Advances. 6 (12), (2020).
- Viola, J. M., et al. Guiding cell network assembly using shape-morphing hydrogels. Advanced materials (Deerfield Beach, Fla.). , 2002195 (2020).
- Mohammad, A., Davis, M., Aprelev, A., Ferrone, F. A. Note: Professional grade microfluidics fabricated simply. Review of Scientific Instruments. 87 (10), 1-4 (2016).
- Lee, O. J., Chuah, H. S., Umar, R., Chen, S. K., Yusra, A. F. I. Construction of cost effective homebuilt spin coater for coating amylose-amylopectin thin films. Journal of Fundamental and Applied Sciences. 9 (2), 279 (2018).
- Webb, K., Hlady, V., Tresco, P. A. Relative importance of surface wettability and charged functional groups on NIH 3T3 fibroblast attachment, spreading, and cytoskeletal organization. Journal of Biomedical Materials Research. 41 (3), 422-430 (1998).
- Processing Guidelines for: SU-8 2025, SU-8 2035, SU-8 2050, SU-8 2075. Microchem SU-8 2000 Permanent Expoxy Negative Photoresist Available from: https://kayakuam.com/wp-content/uploads/2019/09/SU-82000DataSheet2025thru2075Ver4.pdf (2019)
- Palte, M. J., Raines, R. T. Interaction of nucleic acids with the glycocalyx. Journal of the American Chemical Society. 134 (14), 6218-6223 (2012).
- McGinnis, C. S., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods. 16 (7), 619-626 (2019).
- Maxfield, F. R., van Meer, G. Cholesterol, the central lipid of mammalian cells. Current Opinion in Cell Biology. 22 (4), 422-429 (2010).
- Luo, J., Yang, H., Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology. 21 (4), 225-245 (2020).
- Liu, L., Tan, L., Yao, J., Yang, L. Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Molecular Medicine Reports. 21 (4), 1761-1770 (2020).
- Mali, P., Aach, J., Lee, J. H., Levner, D., Nip, L., Church, G. M. Barcoding cells using cell-surface programmable DNA-binding domains. Nature Methods. 10 (5), 403-406 (2013).
- Hsiao, S. C., et al. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir. 25 (12), 6985-6991 (2009).
- Gartner, Z. J., Bertozzi, C. R. Programmed assembly of 3-dimensional microtissues with defined cellular conductivity. Proceedings of the National Academy of Sciences. (17), 1-5 (2009).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten