
Method Article
トウモロコシ葉肉プロトプラストのスケーラブルトランスフェクション
要約
ここでは、トウモロコシ葉肉細胞におけるプラスミドの大規模なライブラリをテストするために、数百万のトウモロコシプロトプラストを一過性にトランスフェクトするためのハイスループットプロトコルを紹介します。
要約
トウモロコシ葉肉細胞のトランスフェクションでは、多くの場合、植物細胞壁を消化してプロトプラストを作成し、エレクトロポレーションまたはポリエチレングリコール(PEG) を介して DNAを挿入します。以前の方法は、一度に数万個のトランスフェクトプロトプラストを生産するために開発されました。ここでは、トウモロコシ(Zea mays L.)中の数百万の葉葉肉プロトプラストを単離してトランスフェクトする簡単な方法について説明します。この合理化されたプロセスにより、W5での洗浄など、特定の一般的なプロトプラストステップが削除されます。さらに、遠心分離、PEGを介したトランスフェクション、インキュベーションなどのステップは、より多くのプロトプラストで機能するように変更されています。プラスミド構築物の大規模なライブラリを発現する能力は、トウモロコシでの超並列レポーターアッセイなどのゲノムスケールの実験を可能にします。
概要
一過性遺伝子発現は、植物生物学における強力なツールです。しかし、植物細胞は細胞壁に囲まれているため、トランスフェクションが困難です。DNA侵入に対するこの障壁は、哺乳類細胞や昆虫細胞などの他の一般的に研究されている細胞型には存在しません。過去の研究では、プロトプラスト(細胞壁が消化および除去された植物細胞)を採用することで、この課題に対処しました。未処理の葉組織とは対照的に、植物プロトプラストは懸濁液中での取り扱いが容易であり、エレクトロポレーションまたはポリエチレングリコール(PEG)1によって媒介されるトランスフェクション技術に適しています。植物細胞は、細胞壁を除去した後もその機能の多くを保持します。したがって、プロトプラストは、遺伝子およびタンパク質の機能を研究し2,3、シグナル伝達4を理解し、植物育種の可能な標的を特定するために、さまざまな植物で使用されています5。プロトプラストは、コムギやジャガイモなどの多様な作物種のゲノム編集構築物をスクリーニングするための便利な媒体でもあります6,7。要するに、プロトプラストの一過性アッセイは農業バイオテクノロジーに不可欠です。
トウモロコシ(Zea mays L.)は、トン数で世界をリードする穀物です。そのため、基礎および応用植物科学研究の主要な焦点です8。しかしながら、Nicotiana tabacum9のようなモデル植物とは異なり、無傷のトウモロコシ葉における一過性導入遺伝子発現は日常的に行われていない。プロトプラストは、トウモロコシ葉葉肉細胞10,11を取得してトランスフェクトするために使用されています。以前の方法では、エレクトロポレーションを使用して最大75%のトランスフェクション効率を達成し、数万のスケールでトランスフェクションされたプロトプラストを製造していました10,15。
このプロトコルは、何百万ものトウモロコシ葉肉細胞をプロトプラストし、以前の方法に匹敵する効率でそれらをトランスフェクションする方法を詳述しています。主なステップは、エチオレートトウモロコシの苗の成長、葉の組織の収穫、植物の細胞壁の消化、およびPEGを使用したプラスミドDNAの細胞への送達です。このプロトコルの主な貢献は、機能アッセイに必要な細胞生存率を維持しながら、プロトプラストトランスフェクションをスケールアップできることです。これにより、超並列レポーターアッセイなどのゲノムスケールの実験を使用して、トウモロコシゲノム全体の数十万の領域の機能をテストできます。この方法は最近、エンハンサーおよびプロモーターを含むシス調節DNAエレメントの大規模なライブラリを調査するために使用されている12,13,14。
プロトコル
1.植物材料の成長
- トウモロコシの穀粒を水道水で2回すすいでください。カーネルを水道水に室温で一晩浸します。
- 翌日、72セルシードスタータートレイに濡れた1:1バーミキュライト:ピートモスを入れます。カーネルを一度すすぎ、セルごとに1つのカーネルをチップキャップダウンに植えます。
- 長日条件(16時間明、8時間暗)で25°Cで3日間生育する。
- 25°Cの一定の暗所に移し、9〜11日間増殖させる。
2. プラスミド単離
- 市販の化学的に有能な 大腸菌 を、製造元の指示に従って目的のプラスミドに変換します。
- 形質転換 大腸菌 を適切な抗生物質を含む50 mLの液体LB培地で、振とうしながら37°Cで一晩増殖させます。
- 室温で3,000 x gで10分間遠心分離することにより、大腸菌をペレット化します。市販のミディプレッププラスミドDNA単離キットを用いてプラスミドを抽出する。
- マイクロボリューム分光光度計を用いてプラスミドDNA濃度を推定する。
注:トランスフェクションに適した材料は、濃度が≥800 ng/μL、A260/A280が≥1.8である必要があります。
3. プロトプラスト単離
- 20 mLの酵素溶液を調製します。
- 2.18 gのマンニトール、0.30 gのセルラーゼR-10、および0.06 gのマセロザイムR-10を50 mLの遠沈管に加えます。反転して粉末を混ぜます。脱イオンH2Oを15mLに添加する。200 μLの1 M MES、pH 5.7を加えます。混合する渦。
- 溶液を55°Cで10分間加熱します。室温で10分間冷却する。
- 20 μLの1 M CaCl2、0.02 gのウシ血清アルブミン、および1 M β-メルカプトエタノール100 μLを加えます。
- 脱イオンH2Oを20mLまで満たし、ホイルで包んだ250mLビーカーに注ぎます。
- トウモロコシの苗を暗闇から取り出します。新しいかみそりの刃を使って土の上数センチにそれらを切ります。切り口を水に入れます。使用しないときは、室温で暗所に保管してください。
- 茶色または損傷した領域を持つ葉を除外するように注意しながら、各苗の2番目と3番目の葉を選択します。12〜16枚の葉を選択します。各葉の先端から~1cm切り取り、廃棄します。次に、各葉から10 cmのセクションを切り取ります。
注意: より多くの組織が必要な場合は、追加の20mLビーカーにさらに250mLの酵素溶液を使用します。一定量の酵素溶液中の葉の材料が多すぎると、生存率が低下する可能性があります。 - 基底端を一緒にして、葉のセクションを互いに積み重ねます。バインダークリップを使用して、基底端でバンドルを一緒にクランプします。葉の束を一枚の白い紙の上に置きます。
- 遠位端から~1 cmの葉をつかみます。新しいかみそりの刃を使用して、葉脈に垂直な遠位端から葉を薄い(0.5〜1 mm)スライスに切ります。まっすぐに切り刻むのではなく、組織を通して短く前方にスライスします。
- 目に見える残留物が紙に堆積し始めたら、かみそりの刃を交換してください。目に見える残留物は、鈍い刃または貧弱な技術による組織のマッシングを示します。
- 葉束の長さを~2cmに切った後、速やかにスライスを酵素液のビーカーに移します。これは得られるプロトプラストの数を減らすので、材料を乾燥させないでください。酵素溶液を静かに回転させて材料を濡らします。
- 光を遮断するためにビーカーの上にホイルの蓋を置きます。バンドル全体がバインダークリップの近くで切断されるまで、スライスと転送のプロセスを繰り返します。
- 酵素溶液のビーカーをベルジャーに移します。室温で3分間、周囲圧力に対して-50.8 kPaから-67.7 kPaの間で真空浸透させます。
- ビーカーを卓上シェーカーに移します。室温で40RPMで2.5時間振とうします。ビーカーを手でそっと回転させます。完全に消化されると、酵素溶液はわずかに乳白色に見えます。それでも解決策が明確な場合は、40 RPMでさらに最大1時間振とうを続けます。3.5時間を超えないようにしてください。
- 消化中に、マンニトール+ MES + MgCl2 (MMG)溶液、ポリエチレングリコール(PEG)溶液、およびインキュベーション溶液を作ります。
- MMG:50 mLの遠沈管に5.47 gのマンニトール、200 μLの1 M MES、pH 5.7、および750 μLの1 M MgCl2 を加えます。溶解する蒸留H2O.ボルテックスで50mLまで充填する。氷の上に置きます。
- インキュベーション溶液:5.47 gのマンニトール、pH 5.7で200 μLの1 M MES、および200 μLの1 M KClを50 mL遠沈管に加えます。溶解する蒸留H2O.ボルテックスで50mLまで充填する。氷の上に置きます。
- PEG溶液:0.44 gのマンニトール、1.0 gのPEG、および400 μLの1 M CaCl2 を15 mLの遠沈管に加えます。蒸留H2O.ボルテックスを4mL充填して混合する。完全に溶解するまで30分間振とうしながら37°Cでインキュベートします。室温で保管してください。
- 酵素溶液を80RPMで室温で10分間振とうします。
- 酵素溶液を入れたビーカーを氷の上に置きます。ホイルカバーを取り外さないでください。20 mLの氷冷MMGを酵素溶液に加えます。40 μmのセルストレーナーを通して50 mLの遠沈管にろ過します。
注意: PEGが追加されるまで、次の手順のためにすべてのソリューションを氷上に保管してください。光への露出を最小限に抑えるために、プロトプラストをホイルで覆ってください。 - ろ液をそれぞれ10mLの等量に分割します。各ボリュームを丸底ガラス遠沈管に注ぎ、室温で100 x g で4分間スピンダウンします。
- 上清を廃棄し、各チューブに1 mLの氷冷MMGを加えます。チューブを穏やかに回転させて軽くたたくことにより、ペレットを再懸濁します。サンプルを1本の丸底ガラス製遠心チューブに結合します。MMGを総容量5 mLまで添加します。室温で100 x g で3分間スピンダウンします。
- 5 mLのMMGで洗浄し、室温で100 x g で3分間スピンダウンします。
- 5 mLのMMGで洗浄を繰り返し、室温で100 x g で3分間スピンダウンします。2回目の洗浄をスピンダウンした後、上清が透明かどうかを確認します。曇ったままの場合は、3回目の洗浄を行います。
- プロトプラストを1 mLのMMGに再懸濁します。.ホイルでゆるく覆い、氷の上に置いておきます。
- プロトプラスト収量を決定し、生存率を確認します。
- 1.5 mLの微量遠心チューブに、4 μLのプロトプラストと72 μLのMMGを一緒に加えます。
- 1.5 mLの微量遠心チューブに、1 μLの0.5%フルオレセインジアセテート(FDA)ストック溶液を49 μLのMMGに加え、20倍の作業溶液を作ります。希釈したプロトプラストと共に4 μLのFDA作動溶液をチューブに加えます。室温で暗所で5分間インキュベートします。
- 11 μLのプロトプラスト混合物を血球計算盤にロードします。明視野光学顕微鏡の下に置きます。細胞が細胞壁を欠いていることを視覚的に確認します。次に、プロトプラストの数を数えます。この数を使用して、得られたプロトプラストの総数を推定します。次に、FDAで染色したときにUV光下で緑色に蛍光を発するプロトプラストの数を数えます。プロトプラスト単離が成功すると、70%〜90%の生存率が得られます。
4. PEGを介したトランスフェクション
- ステップ3.17の推定プロトプラスト濃度から始めて、~10,000プロトプラスト/μLに調整します。濃度が低い場合は、100 x gで3分間遠心分離し、MMGに~10,000プロトプラスト/μLの濃度まで再懸濁します。
- ~1,000,000個のプロトプラストを1.5 mLの微量遠心チューブで15 μgのDNAと混合します。MMGを114.4 μLまで補充し、氷上で30分間インキュベートします。
- チューブの側面をタップしてプロトプラストを再懸濁します。105.6 μLの25%PEG溶液をプロトプラストに加え、最終的な重量/容量12%PEGに到達します。5〜10回反転させて穏やかに混ぜます。PEG溶液中のプロトプラストは壊れやすい。チューブを振らないでください。室温で暗所で10分間インキュベートします。
注:トランスフェクションは、ステップ 4.2 およびステップ 4.3 で示した容量の倍数でスケールアップできます。例えば、1,000万個のプロトプラストと150 μgのDNAを1,444 μLのMMGに懸濁し、50 mLの遠沈管で1,056 μLのPEG溶液で処理することができます。 - 5容量のインキュベーション溶液で希釈する。反転して穏やかに混ぜます。室温で100 x gで4分間スピンダウンします。
注:スケールアップする場合は、プロトプラストをそれぞれ10mL以下の複数の丸底ガラス管に分割して、完全なペレット化を確保します。洗浄に使用するインキュベーション液の量を比例的にスケールアップします。 - ピペッティングにより上清を除去する。1〜5 mLのインキュベーション溶液で洗浄し、室温で100 x g で3分間スピンダウンします。
- プロトプラストをインキュベーション溶液に500〜1,000細胞/μLの濃度に再懸濁します。
- プロトプラストを暗所で室温で一晩(~16時間)インキュベートします。
5. プロトプラスト回収と発現検証
- プロトプラストを室温で100 x gで4分間スピンダウンします。注:スケールアップする場合は、プロトプラストをそれぞれ10mL以下の複数の丸底ガラス管に分割します。
- 1〜5 mLのインキュベーション溶液で洗浄し、室温で100 x g で3分間スピンダウンします。
- 再懸濁し、1〜5 mLのインキュベーション溶液に混ぜ合わせます。
- プロトプラストに蛍光レポーター遺伝子(GFPなど)をトランスフェクトする場合は、アリコートを採取してトランスフェクション効率を確認します。血球計算盤を使用して、まず明視野光学顕微鏡を使用してプロトプラストの総数を数えます。次に、UV光下で蛍光プロトプラストの割合をカウントします。
- プロトプラストを室温で100 x g で3分間遠心分離します。
注:プロトプラストは、フェノールやグアニジンイソチオシアネートによるRNA抽出などのダウンストリームアプリケーションに対応できました。
結果
プロトプラスト単離に最も適した組織は、10〜11日齢の苗の2番目と3番目の葉です(図1)。この研究では、それぞれ長さ10 cmの16枚の葉の切片から約1,000万個のプロトプラストが得られました。単離されたプロトプラストの数は、葉材料の質量および酵素溶液中で消化される葉スライスの幅に依存する。
明視野光学顕微鏡では、損傷を受けていないプロトプラストはほぼ球形で見え、色素体が表面に斑点を付けています(図2A)。トランスフェクションに進む前に、プロトプラストの少量のアリコートをフルオレセインジアセテート(FDA)で染色することにより、生存率を確認できます。紫外線下で蛍光を発するFDA染色プロトプラストは生存可能と見なされます(図2B、C)。損傷を受けていないように見えるすべてのプロトプラストがまだ生存可能であるわけではなく、いくつかの生存可能なプロトプラストは避難によって死滅の過程にある可能性があります(図2C)。これらの細胞では、液胞は腫れており、最終的には細胞膜から排出されます。
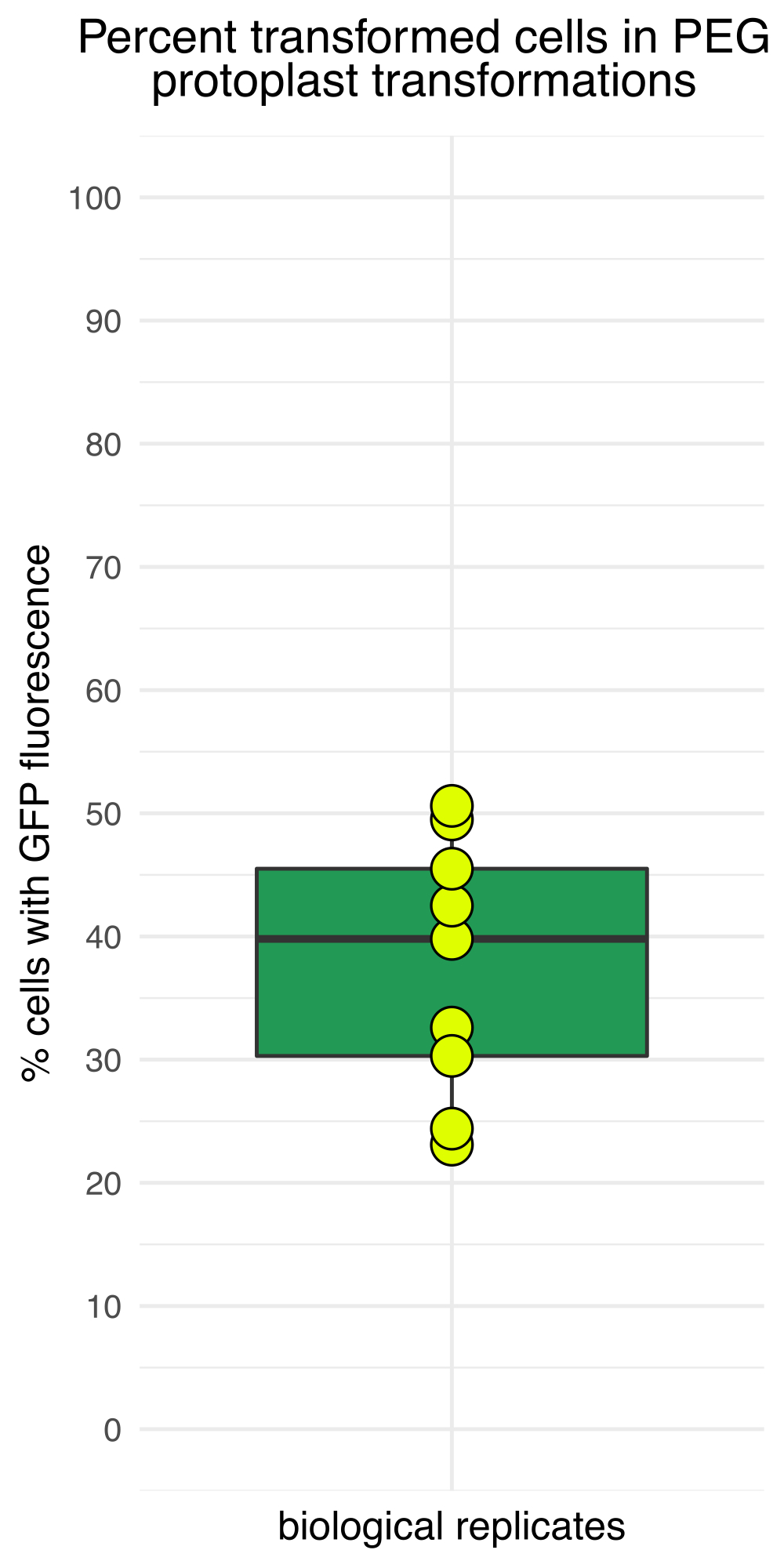
図3は、CD3-911(ABRC)15に由来する4.3 kbのトウモロコシ発現プラスミドであるpJT01で形質転換されたプロトプラストを示しています。トランスフェクションは、c-MycからのC末端核局在シグナルでタグ付けされたsGFPの発現をもたらします(図3A)。sGFP発現プロトプラストの割合を、血球計算盤を用いた複数の実験にわたって測定した。この方法によって達成されたトランスフェクション効率の中央値は、一晩インキュベーション後に40%であり(n = 9、 図4、 表1)、これは以前に公開された方法15に匹敵します。実験用途に応じて、プラスミドまたはプラスミドライブラリに蛍光レポーターがない場合は、トランスフェクションを確認するためにポジティブコントロールを含めることをお勧めします。

図1:発芽後10日間暗所で生育した代表的なトウモロコシ苗。 矢印は、プロトプラストに最も適した2番目と3番目の葉を示しています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:単離後のプロトプラスト生存率の蛍光顕微鏡。 (A)単離直後のプロトプラストの明視野像。(B)FDA染色プロトプラストの蛍光シグナル。(C)生存可能なプロトプラストを示す明視野と蛍光の重なり。矢印は、排気を受けているプロトプラストを示す。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:sGFPを導入したプロトプラストの蛍光顕微鏡 。 (A)トランスフェクション後のプロトプラストの明視野画像。(B)GFPチャネルにトランスフェクトされたプロトプラストの蛍光シグナル。(C)形質転換されたプロトプラストを示す明視野と蛍光の重なり。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:インキュベーションの16時間後にGFP蛍光を示すプロトプラストの割合。 トランスフェクション効率は独立した試験間で異なり、最も低い効率は約20%、最も高い効率は50%近くでした。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| 裁判 | GFP細胞カウント | カウントされた合計セル数 | GFPの割合 |
| 1 | 104 | 261 | 39.8 |
| 2 | 148 | 298 | 49.5 |
| 3 | 84 | 258 | 32.6 |
| 4 | 137 | 271 | 50.6 |
| 5 | 127 | 299 | 42.5 |
| 6 | 107 | 235 | 45.5 |
| 7 | 83 | 360 | 23.1 |
| 8 | 134 | 549 | 24.4 |
| 9 | 61 | 201 | 30.3 |
| 意味する | 109 | 304 | 36 |
| 標準偏差 | 29 | 102 | 10 |
表1:代表的なプロトプラストトランスフェクション効率。 著者らの研究室での最近の9つのプロトプラスト試験のデータ。
補足ファイル1:pJT01のシーケンスを含むGenBank形式のテキストファイル。 4.3 kbのsGFP発現プラスミドは、トランスフェクトされたトウモロコシプロトプラストのポジティブコントロールとして機能します。 このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
以前に報告されたように、プロトプラストに使用される植物材料の品質は、高品質のプロトプラストを得るために不可欠である16,17,18。健康で傷のない葉を選ぶことが重要です。この作品は、人気のある研究用トウモロコシ品種B73を使用しました。他の栽培品種はテストしていません。このプロトコルに使用されたトウモロコシの苗は、緑葉のプロトプラストと比較して、トランスフェクション後16時間でより高い生存率のプロトプラストを生成するため、暗闇で栽培されました。一晩のインキュベーションを必要としないアプリケーションでは、緑または緑化葉の材料を使用することが可能です。
重要なステップは、酵素消化の前に葉の材料を適切に薄切りに切断することです。このプロセスは、葉肉細胞が酵素溶液にさらされるための表面積を増加させ、それは回収されたプロトプラストの数を増加させる。遠位の葉の先端は捨てられ、薄い(≤1 mm)スライスは新しいかみそりの刃で切られます。一定量の組織に対して、葉の粉砕を最小限に抑えるために正しい技術が採用されていれば、より薄いスライスはより多くのプロトプラストを生成します。
プロトプラストは細胞壁除去を受けた後は非常に繊細であるため、プロトプラストを正しく洗浄することも重要です。消化後、すべての溶液は氷上に保たれ、暗く成長したプロトプラストは光から保護されます。浸透圧およびpHは、MMG溶液中で洗浄を行うことによって緩衝される。密度の低い細胞破片からプロトプラストを単離するために、遠心分離は100 x gで行われます。200 x g で遠心分離したプロトプラストは、単離後に同様の生存率を示しますが、翌日には生存率が低下し、約半分も形質転換します。単離直後にFDA(フルオレセインジアセテート)で染色することにより、プロトプラストの生存率を確認することをお勧めします。通常の生存率は70%〜90%の範囲です。単離後の生存率が40%未満のプロトプラストは、形質転換体をほとんど生み出しません。
多くのプロトプラストを扱う場合、トランスフェクション後にはっきりと見えるペレットが得られるため、ペレット化と洗浄が容易になります。このため、トランスフェクションは100万個以上のプロトプラストを用いて行われます。プロトコルをスケールアップするときは、インキュベーションステップ(1.5 mL、15 mL、または50 mLの遠沈管)に適したサイズの容器を選択し、洗浄ステップに使用する溶液の量をそれに応じてスケーリングする必要があります。いずれの場合も、プロトプラストが上清中に残らないようにするために、≤10 mLアリコートで遠心分離することが有益です。このプロトコルの1つの革新は、他のプロトコル14,19に記載されているW5溶液の1時間のインキュベーションおよび洗浄ステップの削除であり、これは私たちの手には不要であることが証明されました。
他の研究で見られるように、DNAの量と質はどちらもトランスフェクションを成功させるために重要です19。4〜6 kbの範囲のプラスミドの場合、100万個のプロトプラストあたり15 μgのDNAが使用されます。DNA調製の質が低いと、トランスフェクション後の生存率が低下する可能性があるため、細菌からプラスミドを単離する場合は市販のDNA抽出キットを使用することをお勧めします。プロトプラストを一晩インキュベートすることは、ストレスを最小限に抑えながらプラスミドまたはプラスミドライブラリの転写を可能にするために、暗所で室温で行われます。一晩のインキュベーション後、最初にトランスフェクトされたプロトプラストの数の約半分が回収されます。プロトプラストを500〜1,000細胞/μLの濃度に希釈して一晩インキュベートします。プロトプラストを1,000細胞/μLを超える濃度で一晩インキュベートすると、回収率が低く、生存率も低くなります。より高い濃度では、損傷したプロトプラストおよび死んだプロトプラストからの破片が隣接するプロトプラストの死に寄与する可能性がある。一晩インキュベーションした後にプロトプラストを洗浄することは、この破片を除去することおよび過剰なプラスミドを除去することの両方に役立つ。
このようなプロトコルには、すべての植物種または組織タイプがプロトプラストに適しているわけではないという制限があります。さらに、遺伝子発現差は、プロトプラストプロセスによって誘導され得る。
要約すると、主食農作物Z. maysから何百万もの生存可能なプロトプラストを単離およびトランスフェクトするためのシンプルでスケーラブルなプロトコルを詳述しました。この方法は、PEGを使用してさらに多くのプロトプラストをトランスフェクションすることができ、エレクトロポレーションベースの方法とほぼ同じくらい高い形質転換効率が得られます15。形質転換体の正味数が多いほど、超並列レポーターアッセイなどの大規模実験において数百または数千の構築物の試験が可能になります12,13。トウモロコシでの将来のゲノムスケールの実験により、科学者はトウモロコシの遺伝子発現と遺伝子調節をよりよく理解できるようになります。
開示事項
著者は競合する利益を宣言しません。
謝辞
この研究は、国立ヒトゲノム研究所のゲノム科学における学際的トレーニング(T32トレーニング助成金番号HG000035からJ.T.)および国立衛生研究所(NIGMS 1R35GM139532からC.Q.)、および国立科学財団(RESEARCH-PGR IOS-1748843およびPlantSynBio 2240888からCQ)によってサポートされました。トウモロコシの種を贈ってくれたラトガース大学のアンドレア・ギャロヴァッティに感謝します。
資料
| Name | Company | Catalog Number | Comments |
| 1.5 mL graduated microcentrifuge tubes | VWR | 490004-444 | |
| 15 mL Falcon conical centrifuge tube | Corning | 05-527-90 | |
| 250 mL Pyrex glass beaker | Fisher | 50-121-5005 | |
| 40 um nylon mesh cell strainer | Fisher | 22363547 | |
| 50 mL Falcon tube | Corning | 14-432-22 | |
| 72 cell propagation tray | T.O. Plastics | 725607C | |
| Aluminum foil | VWR | 89107-732 | |
| Bell jar | Bel-Art | F42022-0000 | |
| Benchtop centrifuge | Eppendorf | 5424-R | |
| Beta-mercaptoethanol | Sigma-Aldrich | M6250-250ML | Dissolve in ultrapure H2O to make a 1M stock solution. Store at room temperature. |
| Bovine serum albumin | Sigma-Aldrich | A2153 | |
| Calcium chloride dihydrate (CaCl2*2H2O) | PhytoTech Labs | C135 | Dissolve in ultrapure H2O to make a 1M stock solution. Store at room temperature. |
| Cellulase Onozuka R-10 | Duchefa Biochemie | C8001.0010 | |
| D-Mannitol | PhytoTech Labs | M562 | |
| Dissecting scope | Reichert | Microstar IV | |
| Fluorescein Diacetate (FDA) | Thermofischer Scientific | F1303 | Dissolve in acetone at 0.5% weight/volume to make a 1000X stock solution. Protect from light and store at -20 °C. |
| Fluorescence Illumination Systems | Prior | Lumen200 | |
| Fluorescence microscope | Leica | MDG36 | |
| High-capacity centrifuge | Eppendorf | 5810 R | |
| Macerozyme | Duchefa Biochemie | M8002.0005 | |
| Magnesium chloride (MgCl2) | Sigma-Aldrich | M292-1KG | Dissolve in ultrapure H2O to make a 1M stock solution. Store at room temperature. |
| Maize seeds | B73 | USDA-ARS | https://npgsweb.ars-grin.gov/gringlobal/accessiondetail?accid=PI+550473 |
| Medium binder clip | Uline | S-24649 | |
| MES | Sigma-Aldrich | M5287-250G | Dissolve in ultrapure H2O to make a 1M stock solution. Adjust pH to 5.7 using KOH. Protect from light and store at room temperature. |
| Micropipette 20-200 uL | Gilson | F123601G | |
| Nanodrop microvolume spectrophotometer | Thermofischer Scientific | ND-1000 | |
| NEB 5-alpha competent E. coli | New England BioLabs | C2987H | |
| Neubauer hemocytometer | Daigger Scientific | EF16034F | |
| No. 9 Single-edge razor blade | Excel | 20009 | |
| Orbital shaker | VWR | 89032-104 | |
| Poly(ethylene) glycol 4,000 | Sigma-Aldrich | 81240-1KG | |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541-1KG | Dissolve in ultrapure H2O to make a 1M stock solution. Store at room temperature. |
| Professional Grade Vermiculite | NK Lawn & Garden | G208 | |
| Round-bottom glass centrifuge tube 30 mL | Kimble Chase | 45500-30 | |
| Rubber adapter for Corex centrifuge tubes | Corning | CLS8445AO | |
| Sphagnum peat moss | Premier Horticulture | 0128P | |
| TRIzol Reagent | Thermofischer Scientific | 15596018 | |
| Tygon vacuum tubing | VWR | 76336-844 | |
| Vacuum gauge | Wika | 4269978 |
参考文献
- Jiang, F., Zhu, J., Liu, H. -L. Protoplasts: A useful research system for plant cell biology, especially dedifferentiation. Protoplasma. 250 (6), 1231-1238 (2013).
- Wang, S., Tiwari, S. B., Hagen, G., Guilfoyle, T. J. AUXIN RESPONSE FACTOR7 restores the expression of auxin-responsive genes in mutant Arabidopsis leaf mesophyll protoplasts. The Plant Cell. 17 (7), 1979-1993 (2005).
- Hirner, A., et al. Arabidopsis LHT1 is a high-affinity transporter for cellular amino acid uptake in both root epidermis and leaf mesophyll. The Plant Cell. 18 (8), 1931-1946 (2006).
- Hwang, I., Sheen, J. Two-component circuitry in Arabidopsis cytokinin signal transduction. Nature. 413 (6854), 383-389 (2001).
- Tan, M. -L. M. C., Rietveld, E. M., van Marrewijk, G. A. M., Kool, A. J. Regeneration of leaf mesophyll protoplasts of tomato cultivars (L. esculentum): Factors important for efficient protoplast culture and plant regeneration. Plant Cell Reports. 6 (3), 172-175 (1987).
- Brandt, K. M., Gunn, H., Moretti, N., Zemetra, R. S. A streamlined protocol for wheat (Triticum aestivum) protoplast isolation and transformation with CRISPR-Cas ribonucleoprotein complexes. Frontiers in Plant Science. 11, 769(2020).
- Nadakuduti, S. S., et al. Evaluation of methods to assess in vivo activity of engineered genome-editing nucleases in protoplasts. Frontiers in Plant Science. 10, 110(2019).
- OECD, Food and Agriculture Organization of the United Nations. OECD-FAO Agricultural Outlook 2021-2030. OECD, Food and Agriculture Organization of the United Nations. , Paris, France. (2021).
- Gallois, P., Marinho, P. Leaf disk transformation using Agrobacterium tumefaciens-expression of heterologous genes in tobacco. Plant Gene Transfer and Expression Protocols. 49, 39-48 (1995).
- Schäffner, A. R., Sheen, J. Maize rbcS promoter activity depends on sequence elements not found in dicot rbcS promoters. The Plant Cell. 3 (9), 997-1012 (1991).
- Sheen, J. Molecular mechanisms underlying the differential expression of maize pyruvate, orthophosphate dikinase genes. The Plant Cell. 3 (3), 225-245 (1991).
- Jores, T., et al. Identification of plant enhancers and their constituent elements by STARR-seq in tobacco leaves. The Plant Cell. 32 (7), 2120-2131 (2020).
- Jores, T., et al. Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nature Plants. 7 (6), 842-855 (2021).
- Ricci, W. A., et al. Widespread long-range cis-regulatory elements in the maize genome. Nature Plants. 5 (12), 1237-1249 (2019).
- Sheen, J. Signal transduction in maize and Arabidopsis mesophyll protoplasts. Plant Physiology. 127 (4), 1466-1475 (2001).
- Jeon, J. M., et al. Efficient transient expression and transformation of PEG-mediated gene uptake into mesophyll protoplasts of pepper (Capsicum annuum L.). Plant Cell, Tissue and Organ Culture. 88 (2), 225-232 (2007).
- Pindel, A. Optimization of isolation conditions of Cymbidium protoplasts. Folia Horticulturae. 19, 79-88 (2007).
- Ren, R., et al. Highly efficient leaf base protoplast isolation and transient expression systems for orchids and other important monocot crops. Frontiers in Plant Science. 12, 626015(2021).
- Yoo, S. -D., Cho, Y. -H., Sheen, J. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nature Protocols. 2 (7), 1565-1572 (2007).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved