16S rRNAシーケンシング:細菌種を同定するPCRベースの技術
概要
ソース: エワ・ブコフスカ・ファニバンド1, ティルデ・アンダーソン1, ロルフ・ルード1
1臨床科学ルンド, 感染医学の部門, ルンド大学生物医学センター, 221 00 ルンド, スウェーデン
惑星地球は何百万もの細菌種の生息地であり、それぞれが特定の特性を持っています。細菌種の同定は、感染した患者を診断するために環境試料および医療微生物学の生物多様性を決定するために微生物生態学で広く使用されている。細菌は、顕微鏡検査、特定の培法上の成長、生化学的および血清学的試験、抗生物質感受性アッセイなどの従来の微生物学的方法を使用して分類することができます。ここ数十年で、分子微生物学の方法は細菌の同定に革命を起こしました。一般的な方法は、16SリボソームRNA(rRNA)遺伝子シーケンシングです。この方法は、従来の方法よりも速く、より正確であるだけでなく、実験室の条件で成長することが困難な株の同定を可能にします。さらに、分子レベルでの株の分化は、典型的に同一の細菌(1-4)間の判別を可能にする。
16S rRNAは19個のタンパク質の複合体と結合し、細菌リボソーム(5)の30Sサブユニットを形成する。これは、リボソームアセンブリに不可欠な機能のために存在し、非常に保存されている16S rRNA遺伝子によってコードされています。ただし、特定の種の指紋として機能する可変領域も含まれています。これらの特徴により、16S rRNA遺伝子は、細菌の同定、比較、系統的分類に使用される理想的な遺伝的断片となっています(6)。
16S rRNA遺伝子シーケンシングは、ポリメラーゼ連鎖反応(PCR)(7-8)に基づいてDNAシーケンシング(9)を行う。PCRは、以下を含む一連のサイクルを通じてDNAの特定の断片を増幅するために使用される分子生物学的方法です。
i) 二本鎖DNAテンプレートの脱生
ii) テンプレートを補完するプライマー(短いオリゴヌクレオチド)のアニーリング
iii) 新しいDNA鎖を合成するDNAポリメラーゼ酵素によるプライマーの拡張
このメソッドの概略図を示します。
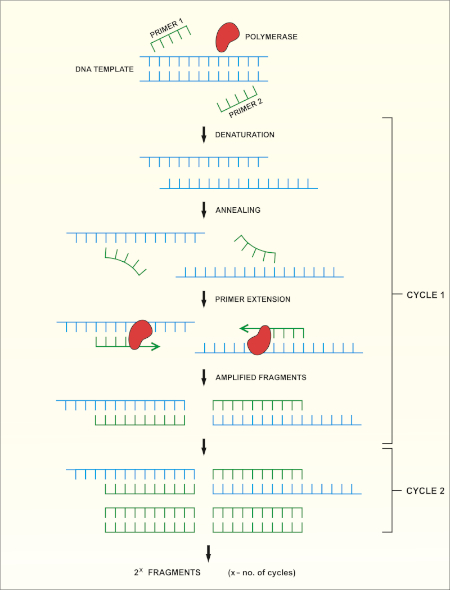
図 1:PCR反応の概略図。この図のより大きなバージョンを表示するには、ここをクリックしてください。
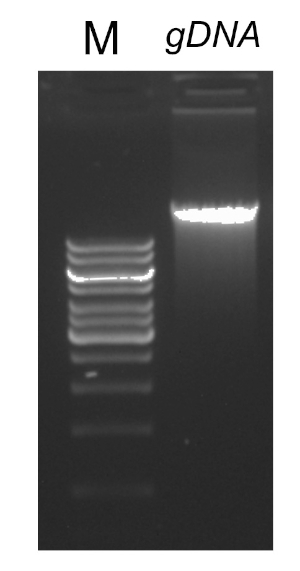
PCR反応を成功させるには重要ないくつかの要因があり、そのうちの1つはDNAテンプレートの品質です。細菌からの染色体DNAの単離は、標準プロトコルまたは市販キットを用いて行うことができる。PCR反応を阻害する汚染物質を含まないDNAを得るためには、特別な注意が必要です。
16S rRNA遺伝子の保存領域は、任意の細菌種の標的領域に結合し、増幅することができるユニバーサルプライマーペア(1つの前方および1逆)の設計を可能にする。ターゲット領域のサイズはさまざまです。プライマーペアの中には、16S rRNA遺伝子のほとんどを増幅できるものもあれば、その一部だけを増幅するものもあります。一般的に使用されるプライマーの例を表 1に示し、その結合部位を図 2 に示します。
| プライマー名 | シーケンス (5'→3') | フォワード/リバース | 参照 |
| 8F b) | アガグットガットックカッグカグ | 転送 | -1 |
| 27F | アガグットガットツクトカッグ | 転送 | -10 |
| 515F | GTGCCAGCMGGCGGTAA | 転送 | -11 |
| 911R | GCCCCCGTCAATTTTGA | 逆 | -12 |
| 1391R | ガグッググググGTRCA | 逆 | -11 |
| 1492R | グッタクットタクタクト | 逆 | -11 |
表 1:16S rRNA遺伝子a)の増幅に用いられる標準的なオリゴヌクレオチドの例。
a)異なるプライマーの組み合わせを使用して生成されたPCR製品の予想長さは、フォワードとリバースプライマーの結合部位間の距離を計算することによって推定できます(図2参照)。プライマーペア8F-1492Rを使用した製品は~1500bp、プライマーペア27F-911R~900bpです。
b) fD1 とも呼ばれます。

図 2:16S rRNA配列およびプライマー結合部位の代表的な図。保存された領域は灰色で色付けされ、可変領域は対角線で塗りつぶされます。最も高い分解能を可能にするために、プライマー8Fおよび1492R(rRNA配列上の位置に基づく名前)を使用して配列全体を増幅し、遺伝子のいくつかの可変領域のシーケンシングを可能にする。この図のより大きなバージョンを表示するには、ここをクリックしてください。
PCRのサイクリング条件(すなわち、DNAが変性し、プライマーでアニールされ、合成されるのに必要な温度と時間)は、使用されるポリメラーゼの種類およびプライマーの特性に依存する。特定のポリメラーゼの製造元のガイドラインに従うことをお勧めします。
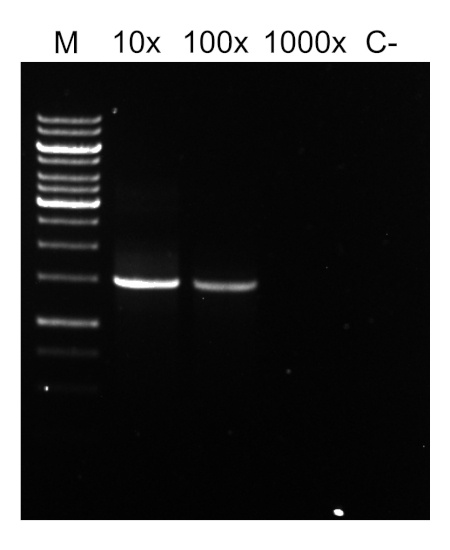
PCRプログラムが完了すると、製品はアガロースゲル電気泳動によって分析されます。正常な PCR は、期待されるサイズの単一バンドを生成します。製品は、PCR反応に存在していた残留プライマー、デオキシリボヌクレオチド、ポリメラーゼ、およびバッファーを除去するために、シーケンシングの前に精製されなければならない。精製されたDNA断片は、通常、商用シーケンシングサービスにシーケンシングのために送られます。しかし、一部の機関は、独自のコア施設でDNAシーケンシングを行います。
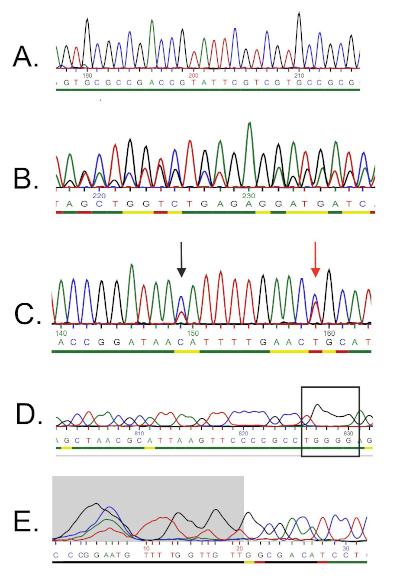
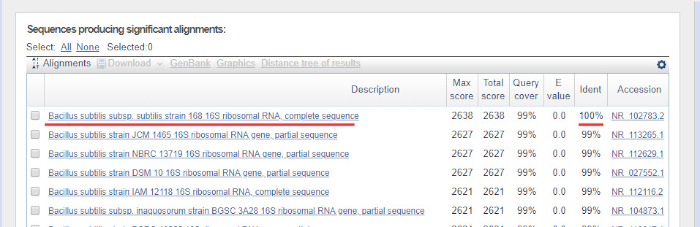
DNA配列は、コンピュータによってDNAクロマトグラムから自動的に生成され、手動編集が必要な場合があるため、品質を慎重にチェックする必要があります。このステップに続いて、遺伝子配列は16S rRNAデータベースに沈着した配列と比較される。類似性の領域が識別され、最も類似したシーケンスが配信されます。
手順
1. 設定
- 微生物を取り扱いながら、良好な微生物の実践に従う必要がある。すべての微生物、特に未知のサンプルは、潜在的な病原体として扱われるべきです。無菌技術に従って、サンプル、研究者、または実験室の汚染を避けてください。細菌の取り扱い前後に手を洗い、手袋を着用し、防護服を着用してください。
- ゲノムDNA単離およびPCR産物精製のための実験プロトコルのリスク評価を行う。一部の試薬は有害である可能性があります!
- 純粋な培養は16S rRNAシーケンシングに不可欠です。ゲノムDNAの単離に進む前に、出発物質が完全に純粋であることを確認してください。これは、個々のコロニーを分離するためにストリークメッキによって行うことができます。これらは、必要に応じて、個別に、またはスープに、さらにストリークすることができます。
- 必要な実験装置:
- PCR用サーマルサイクラー。サーマルサイクラーの機能は、設定されたプログラムに応じて温度を上げたり下げたりすることです。プログラムの作成中に、すべてのPCRステップの温度と時間の値だけでな
申請書と概要
参考文献
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
タグ
スキップ先...
このコレクションのビデオ:

Now Playing
16S rRNAシーケンシング:細菌種を同定するPCRベースの技術
Microbiology
187.9K 閲覧数

ウィノグラツキーカラムの作成:堆積物サンプル中の微生物種を濃縮する方法
Microbiology
128.2K 閲覧数

シリアル希釈とめっき:微生物列挙
Microbiology
313.9K 閲覧数

エンリッチメント培養:選択的および差動媒体における好気性微生物と嫌気性微生物の培養
Microbiology
131.7K 閲覧数

純粋な培養物と縞めっき:混合サンプルからの単一細菌コロニーの単一の分離
Microbiology
165.7K 閲覧数

成長曲線:コロニー形成単位と光学密度測定を用いて成長曲線を生成する
Microbiology
293.0K 閲覧数

抗生物質感受性試験:2つの抗生物質のMIC値を決定し、抗生物質の相乗効果を評価するエプシロメーター試験
Microbiology
93.4K 閲覧数

顕微鏡検査と染色:グラム、カプセル、内胞染色
Microbiology
362.4K 閲覧数

プラークアッセイ:プラーク形成単位(PFU)としてウイルス定数を決定する方法
Microbiology
185.6K 閲覧数

適応塩化カルシウム手順を用いて大腸菌細胞の形質転換
Microbiology
86.3K 閲覧数

結合:アンピシリン耐性をドナーからレシピエント大腸菌に移す方法
Microbiology
38.1K 閲覧数

ファージトランスダクション:アンピシリン耐性をドナーからレシピエント大腸菌に伝達する方法
Microbiology
28.9K 閲覧数
Copyright © 2023 MyJoVE Corporation. All rights reserved